
INTRODUCTION
Prostate cancer is one of the most common malignancies in men worldwide [1]. When the cancer is localized within the prostate gland, it can usually be treated with surgery or radiotherapy with good prognosis. However, the mortality rate increases significantly when the cancer cells metastasize beyond the gland. Androgen deprivation therapy with medical or surgical castration is themainstay treatment for metastatic prostate. However, the treatment becomes ineffective when castration-resistant prostate cancer (CRPC) eventually developed. Currently effective treatment options for CRCP are quite limited and development of new therapeutic agents will greatly benefit the CRPC patients [2].
Ginsenosides are a class of natural components extracted from the plant ginseng which has been used in traditional medicine for long time. The ginsenosides have also been known for their medicinal effects such as anti-inflammatory and anti-proliferative activities [3, 4]. Ginsenoside Rh2 was isolated and identified as an anti-tumor constituent by Chen et al. [5]. Thereafter, its anti-cancer activities have been reported in various malignant diseases including ovarian cancer, breast cancer and melanoma [6–8]. It was demonstrated that Rh2 could induce cell apoptosis through activation of caspase-3 protease [9]. For prostatic cancer, Rh2 inhibited proliferation of androgen-dependent and -independent prostate cancer cells [10]. It has also been shown that Rh2 could inhibit growth of prostatic cancer both in vivo and in vitro. Moreover, the treatment effect was due to a combined inhibitory action on tumor cell proliferation and invasiveness [11].
Peroxisome proliferator-activated receptors (PPARs) are a group of nuclear receptors that function as transcription factors for the regulation of gene expression. There are three PPAR isoforms: PPAR-gamma, PPAR-alpha and PPAR-delta (also named as PPAR-beta and PPAR-beta/delta) [12] The PPARs are involved in a wide range of cellular functions including proliferation, differentiation, development and metabolism. Pathologically, the receptors are related to atherosclerosis, inflammation, cancer, infertility, and degenerative diseases. Recent evidence has shown the relationship between PPAR-delta expression and apoptosis in prostate cancer DU145 cells [13]. The DU145 cell line was derived from human prostate adenocarcinoma metastatic to the brain [14]. The cells are hormone-insensitive and thus a useful model for studying CRPC. In the present study, the apoptotic effect of Rh2 on DU145 cells and the possible underlying mechanism(s) involving PPAR-delta were investigated.
RESULTS
Effect of Rh2 on DU145 cell survival
Rh2 incubation in culture medium reduced DU145 cell viability significantly in a dose-dependent fashion (Figures 1, 2). At a concentration of 1 × 10–4 M, the percentage of viable DU145 cells fell to about 50% (Figure 3). On the other hand, flow cytometry showed that Rh2 induced significant increase in the percentage of apoptotic DU145 cells (Figure 4). Co-incubation with GSK0660 or pre-treatment with PPAR-delta small interfering RNA (siRNA) could reverse these changes.
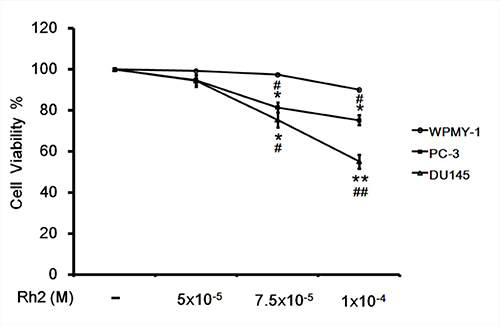
Figure 1: Cell viability assay showing the inhibitory effect of Rh2 on DU145 cell viability. DU145 cells were incubated with Rh2 (5 × 10–5 to 1 × 10–4 M) for 24 h. Human prostate cancer PC-3 cells and prostate stromal myofibroblast cell WPMY-1 were used as controls. Rh2 significantly reduced cell viability of DU145 and PC-3 cells in a concentration-dependent manner but not the WPMY-1 cell. The data are expressed as the means ± S.E.M. (n = 8 for each group). *P < 0.05 and **P < 0.01 compared with control DU145 cells; #P < 0.05 and ##P < 0.01 compared with WPMY-1 cells treated with Rh2.
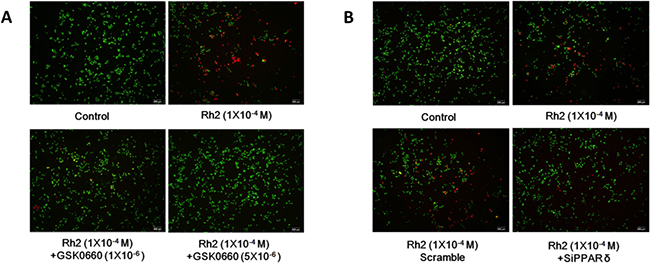
Figure 2: Live/dead cell staining showing GSK0660 and siRNA inhibition on Rh2 apoptotic effect. DU145 cells were incubated with Rh2 (1 × 10–4 M) with/without GSK0060 (1–5 × 10–6 M) for 24 h or transfected with PPAR-delta siRNA 48 h prior to Rh2 treatment. Live cells were stained green, whereas dead cells are stained red. (A) Co-incubation with GSK0660 (1–5 × 10–6 M) inhibited the Rh2 apoptotic effect. (B) PPAR-delta siRNA (SiPPARδ) but not scramble siRNA (Scramble) inhibited the Rh2 apoptotic effect on DU145 cells.
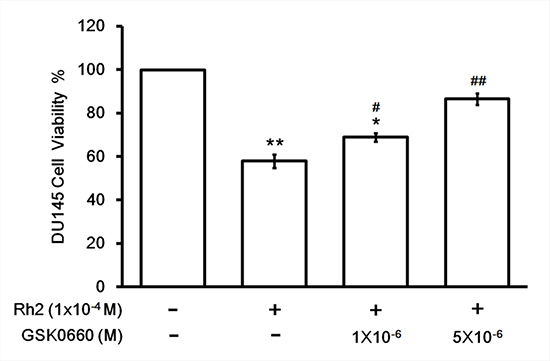
Figure 3: Cell viability assay showing GSK0660 inhibition on Rh2 apoptotic effect. DU145 cells were incubated with Rh2 (1 × 10–4 M) with/without GSK0060 (1–5 × 10–6 M) for 24 hours. The bars depict the quantitative data of cell viability assay on DU145 cells. The data are expressed as the means ± S.E.M. (n = 8 for each group). *P < 0.05 and **P < 0.01 compared with control DU145 cells; #P < 0.05 and ##P < 0.01 compared with DU145 cells treated with Rh2 only.
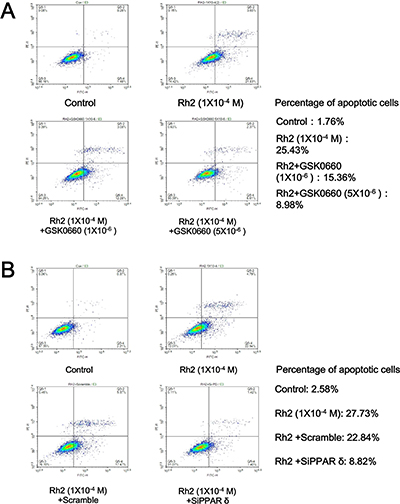
Figure 4: Flow cytometry showing GSK0660 and siRNA inhibition on Rh2 apoptotic effect. DU145 cells were incubated with Rh2 (1 × 10–4 M) with/without GSK0060 (1–5 × 10–6 M) for 24 h or transfected with PPAR-delta siRNA 48 h prior to Rh2 treatment. or PPAR-delta siRNA for 24 hours. (A) Rh2 significantly increased the percentage of apoptotic cells; the effect was inhibited by GSK0660. (B) Rh2 significantly increased the percentage of apoptotic cells; the effect was inhibited by PPAR-delta siRNA (SiPPARδ) but not scramble siRNA (Scramble).
Effect of Rh2 on PPAR-delta, p-STAT/STAT3 protein expression
Western blots showed that Rh2 significantly increased DU 145 cell PPAR-delta protein expression. On the contrary the activated form of signal transducer and activator of transcription 3 (STAT3), phosphorylated-STAT3 (p-STAT3), was significantly decreased (Figure 5). Treatment with PPAR-delta siRNA inhibited these changes (Figure 6).
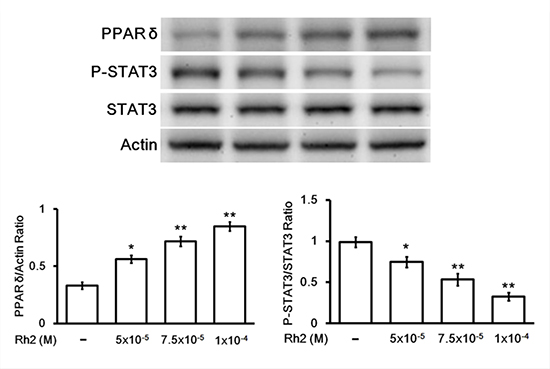
Figure 5: Western blot showing Rh2 effects on PPAR-delta and p-STAT3/STAT3 protein expression. DU145 cells were incubated with Rh2 (5 × 10–5 to 1 × 10–4 M) for 24 h. Rh2 significantly increased PPAR-delta and decreased P-STAT3/STAT3 expression in a concentration-dependent manner. The data are expressed as the means ± S.E.M. (n = 8 for each group). *P < 0.05 and **P < 0.01 compared with control DU145 cells.
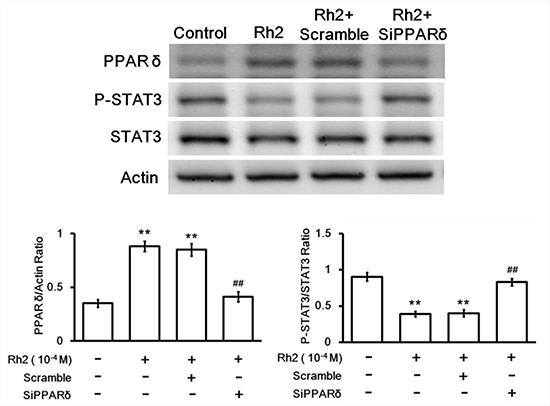
Figure 6: Western blot showing siRNA inhibition on Rh2-induced PPAR-delta and p-STAT3/STAT3 changes. DU145 cells were transfected with PPAR-delta siRNA 48 h prior to incubation with Rh2 (1 × 10–4 M). Rh2 significantly increased PPAR-delta and decreased P-STAT3/STAT3 expression. PPAR-delta siRNA (SiPPARδ) but not scramble siRNA (Scramble) inhibited the Rh2-induced changes. The data are expressed as the means ± S.E.M. (n = 8 for each group). **P < 0.01 compared with control DU145 cells; ##P < 0.01 compared with DU145 cells treated with Rh2 only.
Rh2 induction of intracellular ROS and superoxide
Figure 7 showed that DU145 intracellular ROS and superoxide were significantly increased after incubation with Rh2. Treatment with GSK0660 (Figure 7A) or PPAR-delta siRNA (Figure 7B) could both reverse these effects.
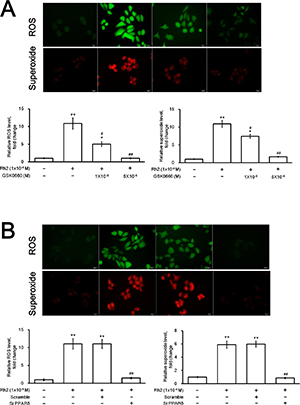
Figure 7: Fluorescence microscopy showing intracellular ROS and superoxide formation. DU145 cells were incubated with Rh2 (1 × 10–4 M) with/without GSK0060 (1–5 × 10–6 M) for 24 h or transfected with PPAR-delta siRNA 48 h prior to Rh2 treatment. or PPAR-delta siRNA for 24 hours. (A) Rh2 significantly increased intracellular ROS (green) and superoxide (red); the effect was inhibited by GSK0660. (B) Rh2 significantly increased intracellular ROS and superoxide; the effect was inhibited by PPAR-delta siRNA (SiPPARδ) but not scramble siRNA (Scramble). The data are expressed as the means ± S.E.M. (n = 8 for each group). The ROS and superoxide level in the control DU145 cells are taken as 1 arbitrary unit. *P < 0.05 and **P < 0.01 compared with control DU145 cells; #P < 0.05 and ##P < 0.01compared with DU145 cells treated with Rh2 only.
DISCUSSION
Our study has demonstrated that ginsensodie Rh2 could effectively control prostate cancer growth by its apoptotic effect on cancer cells. Both DU145 and PC-3 cell viability were significantly reduced by Rh2 in our experiment. PC-3 cell line was originally established from human grade-4 prostate cancer metastatic to the brain and has a greater metastatic potential than DU145 cells [15]. On the contrary, Rh2 did not exhibit apoptotic effect towards the benign control WPMY-1 cells which was cultured from prostate stromal myofibroblast. Therefore Rh2 may be potentially useful in the treatment of metastatic or hormonal refractory prostatic cancer.
The possible role of PPAR-delta in prostate cancer cells apoptosis was reported recently. Similar to our current finding, it has been shown the telmisartan, an angiotensin II receptor blocker, induced DU145 cell apoptosis through PPAR-delta up-regulation [13]. Previous evidence has demonstrated the relationship between PPARs and various kinds of malignant diseases [16–18]. PPAR-gamma activation in cultured breast cancer cells induced lipid accumulation and gene expression changes associated with a more differentiated, less malignant state [19]. It was therefore postulated that PPAR-gamma transcriptional pathway could induce differentiation of malignant breast epithelial cells and provide a potential therapy for breast cancer. As for the prostate, the expression of PPAR-gamma was found significantly higher in high-risk prostate cancer than in low-risk prostate cancer and benign prostatic hyperplasia [20]. In addition, PPAR agonists including troglitazone, rosiglitazone, ciglitazone and pioglitazone could inhibit proliferation of castration-sensitive (LNCaP) as well as castration-resistant (C4-2, PC-3 and DU145) prostate cancer cell lines [21–23]. Regarding PPAR-delta, it was reported that genetic knockdown of the receptor promotes colon cancer growth by decreasing differentiation, increasing proliferation and vascular endothelial growth factor expression in cancer cells [24]. The receptor could also promote breast cancer and leukemic cell survival cultured under unfavorable metabolic conditions [25, 26]. Contrary to these reported PPAR-delta oncogenic effects, studies have also shown possible tumor suppression actions by PPAR-delta. Inhibition of cell proliferation by PPAR-delta ligands were demonstrated in a number of tumors including lung cancer, colon cancer and neuroblastoma [27–29]. Therefore the role of PPAR-delta in cancer treatment is still controversial. It has been hypothesized that PPAR-delta may have tumor-promotion and tumor-suppression actions depending on the tissue, cell-type or organ in question [30].
Our results showed that p-STAT3/STAT3 down-regulation and ROS/ superoxide production are down-stream events associated with the Rh2-induced PPAR-delta up-regulation. STAT3 belongs to the STAT protein family which are intracellular transcription factors that mediate a variety of cellular functions including proliferation, apoptosis and differentiation. When stimulated by cytokines and growth factors, STAT3 is phosphorylated to p-STAT3 by the action of membrane-associated Janus kinases (JAK). The activated p-STAT3 is translocated into the cell nucleus and acts as a transcription activator that regulates gene expression. In the study of lung adenocarcinoma, it was shown that JAK/STAT3 signaling pathway participated in the apoptosis of PC-9 cells induced by icotinib. It has been postulated that icotinib inhibits the gene expression levels of JAK2 and STAT3 leading to up-regulation of the apoptosis regulator BAX gene [31]. The functional linkage between STAT3 and ROS has also been reported. In the A-431 carcinoma cell, platelet-derived growth factor activated STAT3 by using intracellular ROS as a second messenger [32]. Moreover, ROS regulated heat-shock protein 70 via the JAK/STAT pathway in vascular smooth muscle cells [33]. Research evidence demonstrates that ROS play an important role in malignant transformation through various cellular signaling pathways, STAT3 being one of the important ones [34]. On the other hand, it was shown that ROS is an important signaling intermediate leading to Rh2-induced apoptosis in HeLa, MCF10A-ras, and MCF7 cells [35]. In human leukemia Jurkat cells, Rh2 could induce mitochondrial-associated apoptosis by increasing mitochondrial ROS [36]. With relationship to the mitochondria-mediated apoptosis pathway, Rh2 exhibited the actions of release of mitochondrial cytochrome c, activation of caspase-3 and Bax protein, inhibition of Bcl-2 protein as well as production of intracellular reactive oxygen species in Hep3B cells. These findings suggest that Rh2 may induce apoptosis by direct activation of the mitochondrial pathway [37]. Taken together, our findings support previous studies of the inhibitory effect of Rh2 on prostate cancer growth [10, 11]. In addition, it is the first study showing the role of PPAR-delta in the Rh2-induced prostate cancer cell apoptosis. However, the exact underlying mechanism of this process is still unclear and will need further investigation.
In conclusion, our findings have shown that ginsenoside Rh2 induces prostate cancer DU145 cells apoptosis through up-regulation of PPAR-delta which leads to down-regulation of p-STAT3/STAT3 and intracellular oxidative stress. Rh2 may be potentially useful for the clinical treatment of prostate cancer.
MATERIALS AND METHODS
Culture of DU145 cells
DU145 cells (purchased from the Culture Collection and Research Center of the Food Industry Institute, Hsin-Chiu City, Taiwan) were incubated in α-MEM medium (Hyclone, Logan, UT, USA) with 10% heat-inactivated fetal bovine serum (FBS) (Gibco, Life Technologies, Kaohsiung, Taiwan). A humidified atmosphere of 95% air and 5% CO2 was kept at 37° C.
Pharmacological study with Rh2 and GSK0660
When the cells reached 60% confluence, the culture was replaced with serum-free cell medium containing Rh2 (5 × 10–5 to 1 × 10–4 M; Tauto Biotech, Shanghai, China) with/without GSK0660 (a PPAR-delta antagonist, 1 × 10–6 to 5 × 10–6 M; Sigma-Aldrich, St. Louis, MO, USA). After drug incubation for 24 hours the cells were harvested by treatment with 0.25% trypsin and 0.2 g/L EDTA.
PPAR-delta siRNA study
This method was previously reported [13]. In brief, duplexed RNA oligonucleotides for human PPAR-delta (siGENOME SMARTpool TM) were obtained from Thermo Fisher Scientific Inc., Taipei, Taiwan. DU145 cells were transfected with 40 pmol of PPAR-delta siRNA or scramble siRNA (scramble) using transfection reagents (TransIT-TKO, Mirus) as directed in the manufacturer’s protocols. The adequacy of PPAR-delta silencing was tested by immunoblot to optimize the experimental conditions (siRNA dose and time after transfection) according to previous reports [38–40].
DU145 cell viability assay
The method employed for measuring DU145 cell viability by the 3- (4,5-cimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay was previously reported [41]. Briefly, 104 cells were seeded on 96-well plates in 100 μl/well of fresh medium and 10 μl of MTT (final concentration of 0.5 mg/ml). The plates were incubated at 37° C for 4 h. During this time, viable cells were able to reduce the yellow tetrazolium salt into dark blue formazan crystals. The formazan crystals were dissolved using a solution of 0.01 M HCl/10% sodium docecyl sulfate (SDS). The absorbance of each well was by Synergy HT Multi-Mode Microplate Reader at 595 nm (BioTek, Winooski, VT, USA).
Live/dead cell staining
Our method was previously reported [13]. A LIVE/DEAD viability assay kit (Molecular Probes; Eugene, OR, USA) was used according to the manufacturer’s instructions. DU145 cells were incubated with two probes, calcein-AM (green color) and ethidium homodimer-1 (EtdD-1, red color), for intracellular esterase activity and plasma membrane integrity respectively. The cells were then visualized under a fluorescence microscope (Olympus IX71; Olympus, Japan).
Flow cytometry analysis
DU145 apoptotic cells measurement was performed by flow cytometry with Annexin V- propidium iodide staining [42]. FITC Apoptosis Detection Kit (BD Bioscience, San Diego, CA, USA) was used. DU145 cells were seeded in 12-well plates at a density of 5 × 105 cells/well and the experiment procedure was performed according to the manufacturer’s protocol. Staining was analyzed by fluorescence-activated cell sorting (FACS).
Western blot analysis
Our method was previously reported [13]. Extraction of protein from cell lysates was performed using ice-cold radio-immuno-precipitation assay (RIPA) buffer supplemented with phosphatase and protease inhibitors (50 mM sodium vanadate, 0.5 mM phenylmethylsulphonyl fluoride, 2 mg/mL aprotinin, and 0.5 mg/mL leupeptin). The protein concentrations were determined via the Bio-Rad protein assay (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Total protein samples (30 μg) were separated via SDS/polyacrylamide gel electrophoresis (10% acrylamide gel) using the Bio-Rad Mini-Protein II system. The proteins were then transferred to polyvinylidene difluoride membranes (PerkinElmer, Waltham, MA, USA) using a Bio-Rad Trans-Blot system. The membrane was blocked with 5% non-fat milk in Tris-Buffered saline containing 0.1% Tween 20 (TBS-T). After incubation for two hours, the membrane was washed in TBS-T and hybridized with primary antibodies specific to PPAR-delta, STAT3 and p-STAT3 in TBS-T for 16 hours. β-actin was used as an internal control. Incubation with secondary antibodies and detection of the antigen–antibody complex were performed using an enhanced chemiluminescence (ECL) kit (Amersham Biosciences, Buckinghamshire, UK). The immunoblots of β-actin (43 kDa), PPAR-delta (55 kDa), STAT3/p-STAT3 (88 kDa) were quantified with a laser densitometer (Avegene Life Science, Taipei, Taiwan).
Immunostaining for reactive oxygen species (ROS) and superoxide
For the detection of the intracellular ROS and superoxide, the Oxidative Stress Detection Kit from Thermo Fisher Scientific Inc. (Rockford, IL, USA) was used. The procedures were performed according to the manufacturer’s manual. The staining was visualized using a fluorescence microscope connected to an imaging system (IX71 Olympus, Tokyo, Japan).
Statistical analysis
The data are presented as the mean ± S.E.M. for the number (n) of individual experiments. One-way analysis of variance (ANOVA) followed by a post hoc test was used to compare protein expression level changes and other parameters. Differences resulting in a P-value of 0.05 or less were considered to be statistically significant.
Abbreviations
CRPC: castration-resistant prostate cancer; JAK: Janus kinases; PPAR: peroxisome proliferator-activated receptor; p-STAT3: phosphorylated signal transducer and activator of transcription 3; ROS: reactive oxygen species; STAT3: signal transducer and activator of transcription 3.
Author contributions
Tony Tong-Lin Wu: Conception and design; Obtaining funding; Acquisition of data; Drafting of the manuscript. Yat-Ching Tong: Conception and design; Obtaining funding; Analysis and Interpretation of data; Drafting of the manuscript. I-Hung Cheng: Analysis and Interpretation of data; Drafting of the manuscript. Ho-Shan Niu: Acquisition of data; Statistical analysis. Yingxiao Li: Statistical analysis, Drafting of the manuscript and revision. Juei-Tang Cheng: Conception and design; Obtaining funding; Supervision; Administrative, technical or material support.
ACKNOWLEDGMENTS
We thank Miss Pei-Lin Jou, Pei-Ru Liao and Mr. Yu-Chi Chen for their technical assistance in the experiments.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
The present study was in part supported by grants from the Ministry of Science and Technology (MOST 105-2314-B-006-024) of the Republic of China (Taiwan).
REFERENCES
1. Monn MF, Tatem AJ, Cheng L. Prevalence and management of prostate cancer among East Asian men: Current trends and future perspectives. Urologic oncology. 2016; 34:58 e51–59.
2. Pant MK, Abughaban A, Aragon-Ching JB. Advances in systemic therapies for metastatic castration-resistant prostate cancer. Future oncology. 2014; 10:2213–2226.
3. Li QY, Chen L, Fu WH, Li ZD, Wang B, Shi XJ, Zhong MK. Ginsenoside Rb1 inhibits proliferation and inflammatory responses in rat aortic smooth muscle cells. Journal of agricultural and food chemistry. 2011; 59:6312–6318.
4. Hu X, Chen X, Xiong L. Effects of Ginsenoside Rb1 on proliferation of Schwann cells in culture. Chinese journal of traumatology. 2002; 5:365–368.
5. Chen YJ, Xie H, Pei YP, Xu SX, Yao XS. [Isolation and identification of the anti-tumor constituent, ginsenoside-Rh2]. Zhong yao tong bao. 1988; 13:40–42, 64. [Article in Chinese].
6. Kikuchi Y, Sasa H, Kita T, Hirata J, Tode T, Nagata I. Inhibition of human ovarian cancer cell proliferation in vitro by ginsenoside Rh2 and adjuvant effects to cisplatin in vivo. Anti-cancer drugs. 1991; 2:63–67.
7. Ota T, Maeda M, Odashima S. Mechanism of action of ginsenoside Rh2: uptake and metabolism of ginsenoside Rh2 by cultured B16 melanoma cells. Journal of pharmaceutical sciences. 1991; 80:1141–1146.
8. Oh M, Choi YH, Choi S, Chung H, Kim K, Kim SI, Kim DK, Kim ND. Anti-proliferating effects of ginsenoside Rh2 on MCF-7 human breast cancer cells. International journal of oncology. 1999; 14:869–875.
9. Park JA, Lee KY, Oh YJ, Kim KW, Lee SK. Activation of caspase-3 protease via a Bcl-2-insensitive pathway during the process of ginsenoside Rh2-induced apoptosis. Cancer letters. 1997; 121:73–81.
10. Liu J, Shimizu K, Yu H, Zhang C, Jin F, Kondo R. Stereospecificity of hydroxyl group at C-20 in antiproliferative action of ginsenoside Rh2 on prostate cancer cells. Fitoterapia. 2010; 81:902–905.
11. Zhang Q, Hong B, Wu S, Niu T. Inhibition of prostatic cancer growth by ginsenoside Rh2. Tumour biology. 2015; 36:2377–2381.
12. Berger J, Moller DE. The mechanisms of action of PPARs. Annual review of medicine. 2002; 53:409–435.
13. Wu TT, Niu HS, Chen LJ, Cheng JT, Tong YC. Increase of human prostate cancer cell (DU145) apoptosis by telmisartan through PPAR-delta pathway. European journal of pharmacology. 2016; 775:35–42.
14. Stone KR, Mickey DD, Wunderli H, Mickey GH, Paulson DF. Isolation of a human prostate carcinoma cell line (DU 145). International journal of cancer. 1978; 21:274–281. https://doi.org/10.1002/ijc.2910210305.
15. Kaighn ME, Narayan KS, Ohnuki Y, Lechner JF, Jones LW. Establishment and characterization of a human prostatic carcinoma cell line (PC-3). Investigative urology. 1979; 17:16–23.
16. Nunez NP, Liu H, Meadows GG. PPAR-gamma ligands and amino acid deprivation promote apoptosis of melanoma, prostate, and breast cancer cells. Cancer letters. 2006; 236:133–141.
17. Zuo X, Xu M, Yu J, Wu Y, Moussalli MJ, Manyam GC, Lee SI, Liang S, Gagea M, Morris JS, Broaddus RR, Shureiqi I. Potentiation of colon cancer susceptibility in mice by colonic epithelial PPAR-delta/beta overexpression. Journal of the National Cancer Institute. 2014; 106:052.
18. Xiong X, Ye Y, Fu L, Dai B, Liu J, Jia J, Tang J, Li L, Wang L, Shen J, Mei C. Antitumor activity of a novel series of alpha-aryloxy-alpha-methylhydrocinnamic acid derivatives as PPAR gamma agonists against a panel of human cancer cell lines. Investigational new drugs. 2009; 27:223–232.
19. Mueller E, Sarraf P, Tontonoz P, Evans RM, Martin KJ, Zhang M, Fletcher C, Singer S, Spiegelman BM. Terminal differentiation of human breast cancer through PPAR gamma. Molecular cell. 1998; 1:465–470.
20. Rogenhofer S, Ellinger J, Kahl P, Stoehr C, Hartmann A, Engehausen D, Wieland WF, Muller SC, Hofstadter F, Walter B. Enhanced expression of peroxisome proliferate-activated receptor gamma (PPAR-gamma) in advanced prostate cancer. Anticancer research. 2012; 32:3479–3483.
21. Ban JO, Oh JH, Son SM, Won D, Song HS, Han SB, Moon DC, Kang KW, Song MJ, Hong JT. Troglitazone, a PPAR agonist, inhibits human prostate cancer cell growth through inactivation of NFkappaB via suppression of GSK-3beta expression. Cancer biology & therapy. 2011; 12:288–296.
22. Kubota T, Koshizuka K, Williamson EA, Asou H, Said JW, Holden S, Miyoshi I, Koeffler HP. Ligand for peroxisome proliferator-activated receptor gamma (troglitazone) has potent antitumor effect against human prostate cancer both in vitro and in vivo. Cancer research. 1998; 58:3344–3352.
23. Panigrahy D, Singer S, Shen LQ, Butterfield CE, Freedman DA, Chen EJ, Moses MA, Kilroy S, Duensing S, Fletcher C, Fletcher JA, Hlatky L, Hahnfeldt P, et al. PPARgamma ligands inhibit primary tumor growth and metastasis by inhibiting angiogenesis. The Journal of clinical investigation. 2002; 110:923–932.
24. Yang L, Zhou J, Ma Q, Wang C, Chen K, Meng W, Yu Y, Zhou Z, Sun X. Knockdown of PPAR delta gene promotes the growth of colon cancer and reduces the sensitivity to bevacizumab in nude mice model. PLoS One. 2013; 8:e60715.
25. Wang X, Wang G, Shi Y, Sun L, Gorczynski R, Li YJ, Xu Z, Spaner DE. PPAR-delta promotes survival of breast cancer cells in harsh metabolic conditions. Oncogenesis. 2016; 5:e232.
26. Li YJ, Sun L, Shi Y, Wang G, Wang X, Dunn SE, Iorio C, Screaton RA, Spaner DE. PPAR-delta promotes survival of chronic lymphocytic leukemia cells in energetically unfavorable conditions. Leukemia. 2017; 31:1905–1914.
27. Fukumoto K, Yano Y, Virgona N, Hagiwara H, Sato H, Senba H, Suzuki K, Asano R, Yamada K, Yano T. Peroxisome proliferator-activated receptor delta as a molecular target to regulate lung cancer cell growth. FEBS letters. 2005; 579:3829–3836.
28. Matthiessen MW, Pedersen G, Albrektsen T, Adamsen S, Fleckner J, Brynskov J. Peroxisome proliferator-activated receptor expression and activation in normal human colonic epithelial cells and tubular adenomas. Scandinavian journal of gastroenterology. 2005; 40:198–205.
29. Yao PL, Chen L, Dobrzanski TP, Zhu B, Kang BH, Muller R, Gonzalez FJ, Peters JM. Peroxisome proliferator-activated receptor-beta/delta inhibits human neuroblastoma cell tumorigenesis by inducing p53- and SOX2-mediated cell differentiation. Molecular carcinogenesis. 2017; 56:1472–1483.
30. Beyaz S, Yilmaz OH. Molecular Pathways: Dietary Regulation of Stemness and Tumor Initiation by the PPAR-delta Pathway. Clinical cancer research. 2016; 22:5636–5641.
31. Zhang Y, Meng X, Shi H, Li W, Ming Z, Zhong Y, Deng W, Zhang Q, Fan N, Niu Z, Chen G, Yang S. The role of JAK/STAT3 signaling pathway on apoptosis of lung adenocarcinoma cell line PC-9 induced by icotinib. American journal of translational research. 2016; 8:1730–1737.
32. Simon AR, Rai U, Fanburg BL, Cochran BH. Activation of the JAK-STAT pathway by reactive oxygen species. The American journal of physiology. 1998; 275:C1640–1652.
33. Madamanchi NR, Li S, Patterson C, Runge MS. Reactive oxygen species regulate heat-shock protein 70 via the JAK/STAT pathway. Arteriosclerosis, thrombosis, and vascular biology. 2001; 21:321–326.
34. Prasad S, Gupta SC, Tyagi AK. Reactive oxygen species (ROS) and cancer: Role of antioxidative nutraceuticals. Cancer letters. 2017; 387:95–105.
35. Ham YM, Lim JH, Na HK, Choi JS, Park BD, Yim H, Lee SK. Ginsenoside-Rh2-induced mitochondrial depolarization and apoptosis are associated with reactive oxygen species- and Ca2+-mediated c-Jun NH2-terminal kinase 1 activation in HeLa cells. The Journal of pharmacology and experimental therapeutics. 2006; 319:1276–1285.
36. Xia T, Wang YN, Zhou CX, Wu LM, Liu Y, Zeng QH, Zhang XL, Yao JH, Wang M, Fang JP. Ginsenoside Rh2 and Rg3 inhibit cell proliferation and induce apoptosis by increasing mitochondrial reactive oxygen species in human leukemia Jurkat cells. Molecular medicine reports. 2017; 15:3591–3598.
37. Park HM, Kim SJ, Kim JS, Kang HS. Reactive oxygen species mediated ginsenoside Rg3- and Rh2-induced apoptosis in hepatoma cells through mitochondrial signaling pathways. Food and chemical toxicology. 2012; 50:2736–2741.
38. Rujitanaroj PO, Jao B, Yang J, Wang F, Anderson JM, Wang J, Chew SY. Controlling fibrous capsule formation through long-term down-regulation of collagen type I (COL1A1) expression by nanofiber-mediated siRNA gene silencing. Acta biomaterialia. 2013; 9:4513–4524.
39. Metwally AA, Blagbrough IS. Self-Assembled Lipoplexes of Short Interfering RNA (siRNA) Using Spermine-Based Fatty Acid Amide Guanidines: Effect on Gene Silencing Efficiency. Pharmaceutics. 2011; 3:406–424.
40. Andersen MO, Howard KA, Paludan SR, Besenbacher F, Kjems J. Delivery of siRNA from lyophilized polymeric surfaces. Biomaterials. 2008; 29:506–512.
41. Scudiero DA, Shoemaker RH, Paull KD, Monks A, Tierney S, Nofziger TH, Currens MJ, Seniff D, Boyd MR. Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines. Cancer research. 1988; 48:4827–4833.
42. Calastretti A, Gatti G, Quaresmini C, Bevilacqua A. Down-modulation of Bcl-2 sensitizes PTEN-mutated prostate cancer cells to starvation and taxanes. Prostate. 2014; 74:1411–1422.