
INTRODUCTION
The human tumor microenvironment of non-malignant cells and stroma is molecularly and immunologically heterogeneous. It is now abundantly clear that growth, metastasis and therapeutic response is regulated by multiple interactive pathways that make modulating specific targets necessary but which by itself is not always sufficient in advancing long-term survival and remission in cancer patients. With better understanding of various mechanisms in cancer biology and recent progress in molecular and immunological targeted therapies, there have been some significant advances in treatment outcome. Yet durable responses are achieved only in a small fraction of patients with specific types of cancers, such as melanoma, lung, and renal.
To build upon the knowledge gained and the clinical advances achieved to date, it is imperative that mechanism-based combinations of targeted and cytotoxic agents be implemented and validated clinically with a focus on dose, sequence, route and duration of administration. The focus of this review is to identify small molecules that can effectively and selectively modulate the in vivo expression of biomarkers commonly expressed in a majority of cancers that are implicated in angiogenesis and drug resistance. Hypoxia-inducible and constitutively-expressed 1α and 2α (HIFs) factors and vascular endothelial growth factor (VEGF) have been shown to be targets of methylselenocysteine (Se-methylselenocysteine [MSC]) and selenomethionine (SLM) in xenograft models [1–19]. Therapeutic doses of MSC have been found to downregulate expression of oncogenic miRNAs, and upregulate tumor suppressor miRNAs in clear cell renal cell carcinoma (ccRCC) and xenografts. This modulation of biomarkers by SLM or MSC was associated with an enhanced antitumor activity of a wide range of anticancer drugs [1, 3-8, 13-19].
Selenium (Se) is a trace element present in high concentrations in Brazil nuts [20, 21], fish, and in plants grown in soil with high Se content. Se is classified as an antioxidant that regulates cell metabolism, oxidative stress, and DNA- and RNA-protein-synthesis. Se exists either in organic forms such as SLM and MSC, or in inorganic forms such as selenide and selenite (Table 1) [22]. Inorganic Se is converted in plants to organic Se, and is retained as such in animals and humans. The daily recommended dose for adults is approximately 50μg. The circulating Se blood levels in adult populations worldwide vary considerably, influenced by dietary and supplement intake, and Se soil levels where consumable vegetation is grown. Published reports suggest an inverse relationship between Se status and risk for colon, prostate, lung, and bladder cancer, among others [23–26]. Se toxicity has been reported in patients with Se intake approximately 200 times the doses used in prevention trials, and at least 70 times the SLM doses used in therapeutic trials [27, 28]
Table 1: Selenium compounds for cancer prevention and therapy
Forms of Se |
Active metabolites |
Remarks |
|---|---|---|
Sodium selenite |
Hydrogen selenide |
50% absorbed and retained. High toxicity – genotoxic, induces single strand DNA breaks in vitro. Conversion to methylselenol is a rate-limiting step and occurs when selenite is present in excess, selenite do not regulate the expression of NKG2D ligand that trigger immune activation [22] |
Sodium selenate |
Hydrogen selenide |
Low toxicity - almost completely absorbed but most gets excreted in urine before being incorporated into protein. Activator of PP2A phosphatase |
Methyl selininic acid |
Methyl selenol |
High toxicity with low dose tolerance and of little nutritional value. Regulates expression of NKG2D ligand that trigger immune activation [22] |
Selenocysteine |
Hydrogen selenide, Methyl selenol |
Toxic in higher concentration similar to selenite [57]. Chemically unstable and difficult to handle. |
Selenomethionine |
Methyl selenol Hydrogen selenite, |
Well tolerated, low toxicity but binds to plasma components. |
Methylselenocysteine |
Methyl selenol |
90% absorbed, accumulates in a free pool post ingestion. Lowest toxicity amongst Se compounds and more bioactive making it the most ideal candidate for supplementation and therapeutic usage. Bind relatively less to plasma components. A single step conversion β-lyase leads to methylselenol. It regulate the expression of NKG2D ligand that trigger immune activation [22] |
The aim of this review is to identify targets associated with the observed therapeutic benefit achieved by the sequential combination of SLM or MSC with anticancer drugs to provide the basis for expanded preclinical and clinical therapeutic use.
Chemical structure of investigated se
The biochemistry and functional role of Se has been extensively reviewed [20–58]. Briefly, Se-containing molecules have been found to exert pleiotropic effects against multiple targets associated with tumor growth, metastasis and drug resistance. The chemical structures of SLM and MSC used in in vivo studies and their presumed active metabolite, MSA, used in in vitro studies, are shown in Figure 1. SLM and MSC are antioxidant pro-drugs with relatively low in vitro cytotoxic effects and are activated in vivo to the active moiety methylselenol by β-lyase. While SLM requires multiple enzymatic activations, MSC has one-step activation, is less toxic and less protein bound. Sodium selenite and selenized yeast are molecules metabolized to selenide with subsequent conversion to methylselenol (Table 1) [31]. Methylselenol derived from MSC, but not selenide derived from SLM, has been reported to regulate the expression of ligands that trigger immune activation [30, 31]. Differences in the structural-based metabolic activation of Se are likely contributors to the differences in their mechanisms of action and efficacy in prevention and therapeutic trials. There is an inverse correlation between the basal-levels of plasma Se and cancer incidence and mortality [41, 59]. Investigators at Pennsylvania State University are currently developing interesting and potentially promising Se-containing molecules [42]. Selenocompounds have been reported to exert epigenetic effects, in part by interfering with the one-carbon metabolism that provides the methyl donor for DNA methylation [58, 60].
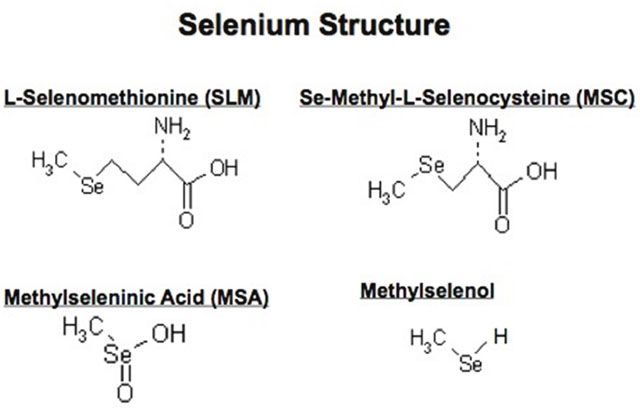
Figure 1: Chemical structure of L-Selenomethionine, Se-methyl-L-selenocysteine (MSC), methylseleninic acid (MSA) and methylselenol.
The importance of Se as a nutritional supplement and its’ potential as a chemopreventive agent have also been evaluated [30, 31]. In contrast, the therapeutic potential of specific types of Se, their doses, schedule and sequence in the selective modulation of antitumor activity in vivo of standard and newly developed targeted agents in advanced cancers has not yet been fully addressed.
Se-containing molecules in cancer prevention
A number of clinical prevention trials have utilized SLM, sodium selenite, and selenized yeast [34, 37, 38]. Table 1 summarizes the different forms of Se that have been evaluated in in vitro and in vivo models. SLM is better absorbed than selenite, and plasma concentrations derived from SLM dosing are significantly higher than those derived from the other Se molecules, with no dose-dependent modulation of glutathione peroxidase or selenoprotein P1 [33]. The low doses of SLM used in the SELECT trials [36, 40] and sodium selenite and selenized yeast used in other prevention trials were based on an initial trial conducted over 30 years ago by Clark et al [35]. Recently, the use of SLM as a cancer preventive agent in the SELECT trial did not demonstrate favorable clinical results [36, 40].
It is reasonable to assume that the lack of demonstrable clinical benefit is multifactorial and could include the type of Se used. SLM, rather than high-selenium brewer’s yeast with more than 20 Se-containing species used in other trials [35], the dose of Se administered; gender dependent metabolism of Se; pre-existing circulating basal levels of Se and uncontrolled use of other supplementary agents such as vitamins, non-steroidal anti-inflammatory drugs, and trace elements. For example, supplements with zinc which is needed for the maintenance of immune function, reportedly protect cells in the early steps of the apoptotic pathway [44]. Further, circulating levels of Se in certain population such as in New Zealand is lower than those in the western countries [32] and supplementation may benefit such population more than those with a higher baseline. In the SELECT trials, only men with high Se baseline level were enrolled. The SELECT and the New Zealand trials [35, 40] were conducted in male patients utilizing different types of Se. Our preclinical data confirms a greater therapeutic efficacy from MSC or SLM in combination with anticancer drugs than when used in combination with selenized yeast or MSA.
Selective modulation of the antitumor activity and toxicity of anticancer drugs by MSC and SLM is dose and schedule dependent
Studies in FaDu head and neck squamous cell carcinoma cells expressing hypoxia-inducible HIF1α confirmed that HIF is a Se target and its downregulation enhances drug effects. Under normoxia, FaDu cells with no detectable HIF1α protein are relatively less sensitive to MSA, the active metabolite of MSC and SLM, but more sensitive to SN38, the active metabolite of irinotecan (Figure 2A). In contrast, under hypoxia, FaDu cells expressing HIF1α protein become exquisitely more sensitive to MSA, but less sensitive to SN38. The observed enhanced SN38 cytotoxicity by MSA is associated with a pronounced inhibition of HIF1α and an enhanced level of apoptosis induced by SN38 (Figure 2B). Thus, chemoresistance of FaDu tumor cells expressing HIF1α to SN38 can be reversed as a consequence of HIF1α inhibition by MSA.
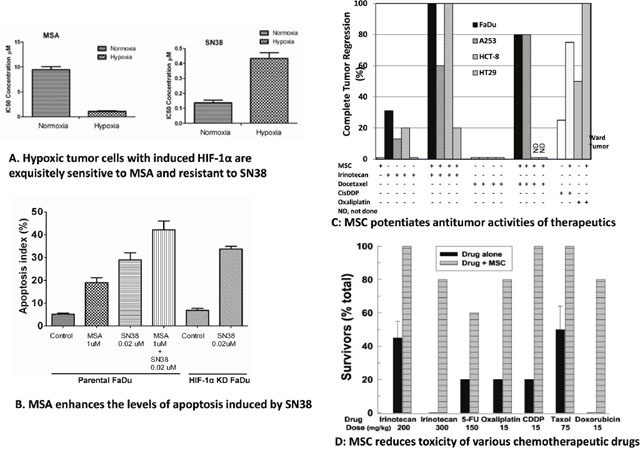
Figure 2: Differential in vitro effects of MSA and SN38 in hypoxic cells expressing HIF-1α and in normoxic cells with no detectable HIF1α protein expression (A). Effects of MSA on the level of apoptosis induced by MSA and SN38 in hypoxic FaDu tumor cells expressing HIF1α, and, in HIF-1α shRNA knockdown FaDu cells treated with SN38 alone (B). Effects of MSC on the antitumor activity of anticancer drugs in head and neck FaDu and A253; colorectal HCT8 and HT29 xenografts, and, in rats bearing Ward colon tumors (C). Effects of MSC on the toxicity induced by lethal doses of anticancer drugs (D). CR, complete tumor response with no evidence of tumor relapse at 3 months.
Saifo et al demonstrated that β-catenin, a molecule associated with drug resistance, is a target of Se and its inhibition is associated with increased multiple drug cytotoxicity [13]. Further, degradation of β-catenin induced by GSK-3β phosphorylation is not a general mechanism but is cell type-dependent [61]. Studies in prostate tumor cell lines have demonstrated that growth of hormone refractory prostate cancer cells can be controlled by treatment with MSA and these effects are associated with the downregulation of HIF-1α, possibly through stabilization and/or increase in prolyl hydroxylase activity, and with the downregulation of VEGF, and Glut1. Beppu et al [62] and Puppo et al [63] reported that inhibition of HIFs by low doses of topotecan resulted in dose-dependent inhibition of VEGF in neuroblastoma tumor cells. Recently, Sun et al also demonstrated that Se-enriched extracts from pyracantha fortuneana inhibit the growth and proliferation of ovarian cancer cell lines by enhancing apoptosis and inhibiting β-catenin signaling [54] which has been found to be associated with transcriptional upregulation of HIF-1α [64] and colorectal tumorigenesis [65].
The role of Se in the in vivo modulation of anticancer drugs has been evaluated in several human tumor xenografts, including head and neck FaDu and A253, colorectal HT29 and HCT8 models, ward rat colon tumors (Figure 2C) and 786.0 ccRCC xenografts [19]. The observed therapeutic augmentation between Se and anticancer drugs was associated with PHD-dependent and VHL-independent degradation of HIFs, downregulation of cyclooxygenase-2, and nitric oxide synthase, with enhanced pericyte recruitment and vascular normalization [3, 5, 16, 18]. In contrast to the relatively poorly differentiated FaDu and HCT8 xenografts which were sensitive to the MTD dose of irinotecan, the well-differentiated tumors of A253 and HT-29 xenografts were relatively resistant (Figure 2C). Optimal efficacy was achieved only when MSC was administered in sequential combination with two to three times the MTD of irinotecan, a dose that is lethal but can be administered safely in combination with MSC [15]. Thus, protection of normal tissues by MSC against treatment-induced toxicities allows drug dose administration higher than their respective MTDs. This therapeutic outcome from MSC and anticancer drugs administered in combination setting in several preclinical xenografts was achieved only when MSC was orally administered daily for a minimum of seven days prior to and continued daily during the duration of anticancer drug treatment. Concurrent treatment, i.e., without MSC pretreatment, did not achieve similar therapeutic outcome. The MSC or SLM dose administered to achieve a plasma Se concentration comparable to those achieved with SLM dose during SELECT prevention trial was insufficient in modulating anti-cancer efficacy of chemotherapeutic drugs in the same xenorgrafts. Similar therapeutic effects have been reported between MSC and VEGF/VEGFR targeted agents in combination with chemotherapy [16, 19].
The potential role of MSC and SLM in protecting normal tissues from drug-induced toxicity has been evaluated in xenografts (Figure 2D). Oral administration of 8 mg/kg Se offers selective protection against toxicity induced by lethal doses of various chemotherapeutic agents, with different mechanisms of action and different target organs. In these studies selective protection of normal mouse bone marrow and epithelial gut cells by SLM and its active metabolite MSA from cisplatin and radiation-induced toxicity was associated with an enhanced level of the XPC DNA repair gene [9, 12]. SLM induces XPC/Gadd45, thioredoxin reductase and p53 in normal bone marrow and gut epithelial cells that expresses wild type p53.
In summary, Se-containing molecules used in vivo at their optimal doses and schedules in sequential combination with varying chemotherapeutic agents are a highly selective and effective treatment resulting in durable cure in several tumor xenografts. Enhancement of antitumor activity and protection against drug-induced toxicity are unique properties of MSC and SLM that are defined by a therapeutically effective and non-toxic dose and schedule.
Se targets
Data generated in our laboratories and by others have demonstrated multiple targets affected by Se [1–19]. We have found that the observed enhanced therapeutic efficacy of anticancer drugs by Se is associated with a collective modulation of several targets found altered in preclinical models. However, it is unclear at this time whether such modulation of a single target by Se would be necessary and sufficient for tumor cell sensitization to anticancer drugs. It is likely, however, that the pleiotropic role of MSC and SLM in modulating multiple targets would be essential for the selective and effective sensitization of tumor cells to subsequent treatment with anticancer drugs. The potential Se targets presented in chart 1 are discussed below
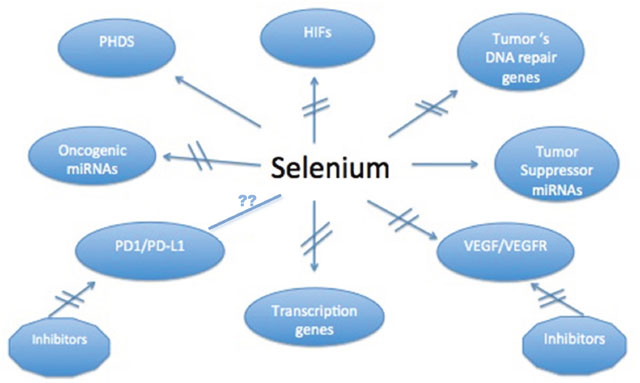
Chart 1: Proposed biomarkers modulated by therapeutic doses of MSC in several human tumor xenografts.
HIF1α and 2α
HIFs as critical therapeutic targets have been extensively evaluated [43, 52, 53, 57, 64-96]. A study by Zhao et al [76] indicated that while there is homology between HIF1α and HIF2α, HIF2α might play an important role in the growth, metastasis and resistance to chemotherapy in digestive tract cancers. Further studies are required to confirm the role of HIF2α in tumor biology and as a predictor of drug response in digestive tract cancers and other solid tumors. HIF1α downregulates C-Myc and mTOR while activating P53 while HIF2α upregulates C-Myc and downregulates P53 and mTOR [69, 74]. In renal cancers, HIF1α and -2α seem to play an opposing role, with HIF1α acting as a tumor suppressor and HIF2α behaving as an oncogene [75]. A report by Imamura et al demonstrated the divergent roles of HIF1α and HIF2α [72]. Cui et al found that HIF1α/2α overexpression in 244 pancreatic cancer specimens was associated with the activation of lactate dehydrogenase A [67].
Cellular accumulation of HIFs are determined by the rate of protein synthesis and its degradation. Under normoxia, oxygen-dependent hydroxylation of proline in HIFs by prolyl hydroxylases is a key step that leads to the recognition of HIFs by VHL protein, followed by its degradation through the ubiquitin-proteasome pathway. Therefore, under normoxia, prevalent in most normal organs, HIF protein is rapidly degraded and thus is undetectable. Under hypoxia, the enzymatic activity of PHDs decreases, resulting in a decreased hydroxylation and degradation of HIFs. Unlike in many solid tumors, ccRCC tumors, at initial diagnosis, are normoxic, highly vascular and express high levels of HIFs [8, 16, 17]. With the recognition that HIFs are upregulated by both hypoxia-dependent and independent pathways and play a crucial role in dug resistance and angiogenesis, a number of HIFs inhibitors are currently under clinical development. However, many of these agents, such as topotecan, are toxic and offer limited efficacy [92, 97–100].
As an alternative approach, our laboratory was the first to demonstrate that therapeutic doses and schedules of MSC and SLM are potent and selective in vivo inhibitors of hypoxia-induced and constitutively-expressed HIFs [8, 16]. However, the efficacy of HIF-targeted agents is likely to be dependent on its ability to reach the intended targets in sufficient enough concentrations in tumor in order to lower the levels and function of the intended targets while the incidence, intensity, and distribution of the targets are also equally critical. For example, in treatment of ccRCC xenografts that are often characterized by a VHL loss of function and results in constitutive expression of HIF1α and HIF2α [66], therapeutic doses of MSC caused a partial downregulation of HIFs with limited antitumor activity. Enhanced inhibition of HIF by MSC in combination with topotecan, an inhibitor of HIFs synthesis, results in a significant increase in treatment efficacy. In contrast, treatment with therapeutic doses of MSC alone (without topotecan) of colorectal and head and neck xenografts expressing lower incidence of HIFs resulted in a pronounced inhibition of HIF1α and sensitizes cancer cells to treatment with a variety of anticancer drugs [8, 16].
HIF1α regulated VEGF
Studies in our laboratory have demonstrated that HIF1α and HIF2α proteins are overexpressed in 92% of primary ccRCC, 27% in colorectal, and 38% in head and neck tumors [16, 17]. In contrast, VEGF-A was expressed in 54%, 79%, and 97% of ccRCC, colorectal, and, head and neck tumors, respectively. The average immunoscore of VEGF-A in ccRCC, colorectal, and head and neck tumors were 2.3, 5.7, and 4.2, respectively [16, 17]. ccRCC is known to respond to VEGF-targeted agents, but colorectal or head and neck cancers do not. Since the dose and schedule of the VEGF-targeted agents are generally kept constant irrespective of cancer types, it is possible that the documented responses to VEGF/VEGFR inhibitors of ccRCC patients are due, in part, to the lower levels of VEGF-A target agents. Our investigations have shown that levels of VEGF-A are higher in tumor cells expressing HIF1α than in cells expressing HIF2α [16, 18]. In in vivo studies, therapeutic doses of MSC partially downregulated VEGF-A in cells expressing HIF1α but not in cells expressing HIF2α. Significant levels of VEGF remained stably expressed even though HIF1α and HIF2α were similarly inhibited by MSC. These results suggest that VEGF levels are differentially regulated by HIF1α and HIF2α, and a complete inhibition of VEGF may require administration of a combination of agents that inhibit VEGF regulated by HIF1α and those that target VEGF-regulated by HIF-independent mechanisms. This concept was confirmed by use of MSC, an enhancer of HIF degradation, in sequential combination with topotecan, a cytotoxic anticancer drug that also inhibits HIF synthesis, and, sunitnib, an inhibitor of VEGFR [19]. Validation of this concept in additional relevant preclinical models with similar targets of interest could provide the basis for clinical translation of this approach.
Tumor microenvironment (TME)
TME is comprised of heterogeneous cell types, including stromal cells, tumor infiltrating lymphocytes, macrophages, and multiple immune cells [101–110]. TME regulates neoangiogenesis, cancer metastasis, and response to anti-cancer treatments. Stromal-modulating growth factors act in a paracrine fashion to disrupt normal tissue homeostasis such as neoangiogenesis and inflammatory responses. Se is known to modulate expression levels and function of immune cells within the TME milieu [5]. Unstable, chaotic and immature tumor vasculature leads to heterogeneity in tumor blood flow, a higher tumor interstitial fluid pressure (10-100 mm Hg compared to around zero in normal tissue), tumor hypoxia and a low extracellular pH within TME. Along with the presence of extracellular matrix such as collagen and mucin within TME, this contributes to a poor drug delivery while facilitating clonal selection of resistant cancer cells [1, 5, 103, 104]. Unlike normal tissues, TME is unstable with leaky vasculature, and expresses multiple molecular and immunological biomarkers associated with angiogenesis and drug resistance [102, 103, 106, 108]. Cells comprising TME support growth and development of cancer cells while representing a potential barrier to intratumoral drug delivery in effective cytotoxic concentrations sufficient enough for tumor cell kill. TME is, therefore, recognized as a potential target for drug and therapy development. TME stabilization may be achieved by treatment with agents that selectively target drug resistance biomarkers. Thus, if tumor cells are the ultimate target, TME is the initial gatekeeper modulating drug delivery and response.
The potential in vivo role of therapeutic doses and schedules of various forms of Se in targeting tumor cells and surrounding TME has not been extensively reported. Our group was the first to report that therapeutic doses of MSC cause an antivascular effect, leading to decreased microvessel density, lower tumor interstitial fluid pressure, optimal vascular permeability through an increased pericyte coverage of blood vessels (or vessel normalization), and a selective increase in drug distribution within tumors (Figure 3). The antiangiogenic effects of MSC are similar to that of bevacizumab in human cancer cells [103, 111]. Figure 3A-3D depicts the antivascular effects of therapeutic doses of MSC, resulting in enhanced intratumoral drug accumulation. These effects followed an inhibition of HIF1α and led to an enhancement of the antitumor activity of irinotecan (Figure 3E, 3F). Therapeutic augmentation was achieved only when irinotecan was administered after optimal inhibition of HIF1α and stabilization of TME by MSC. Paolicchi et al demonstrated that activation of HIF1α in the hypoxic TME causes activation of gene regulating glucose transport, glycolysis, angiogenesis, and changes in mitochondrial functions [71]. A study by Ribiero and Okamoto supported the concept that tumor vascular pericyte coverage contributes to TME stabilization [108].
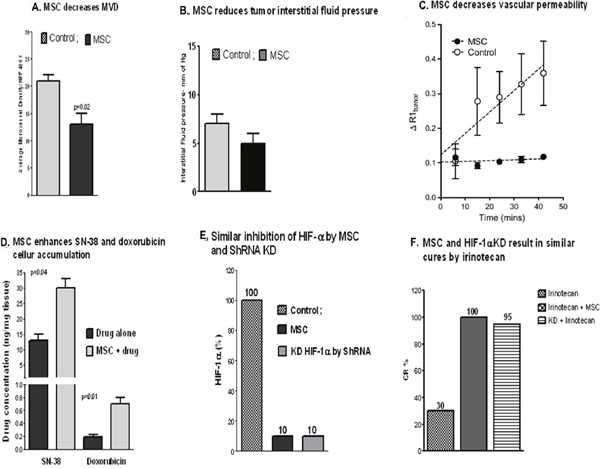
Figure 3: Pleiotropic effects of MSC. Effects of MSC on TME in FaDu xenografts: a reduced microvessel density (A, MVD) along with a reduced tumor interstitial pressure (B) and an increased vessel normalization effect as reflected by a reduced vascular permeability [3] compared with the untreated (C) resulting in an increased drug accumulation in tumors (D). Inhibition of HIF-1α by MSC was similar to the one seen with HIF-1α knockdown ShRNA (E) and the effect of MSC in the combination therapy with irinotecan mimicked the treatment response seen with irinotecan and HIF1α knockdown shRNA (F) [8].
Although current data suggests that HIF is a critical therapeutic target of MSC, observed TME stabilization and enhanced efficacy of anticancer drugs are likely due to additional effects by Se on other targets. This may include altered expressions of miRNAs known to play a key role in the crosstalk between TME and tumor cells.
DNA repair and transcription genes
Induction and maintenance of DNA double-strand break is generally associated with positive tumor response to therapy. In contrast, repair of DNA damage is associated with therapeutic resistance. Tumors that overexpress HIF are associated with a higher expression of miRNAs such as miRNA-210, which in turn regulates factors implicated in DNA repair pathways [70, 112]. Smith et al demonstrated in mouse bone marrow cells and human head and neck cancer cell lines A253 and FaDu that while Se offered a selective protective effect against drug-induced DNA damage in normal bone marrow cells, not in tumor cells. Se was found to selectively upregulate XPC, a DNA repair gene, in normal cells but not in tumor cells. The protective effects of Se were p53-dependent [9, 14] and the observed DNA damage involved a Ref1/p53/Brca1 protein complex. Collectively, these effects of MSC were associated with an enhanced efficacy of radiation and antitumor activity of cisplatin.
Peroxiredoxin-1 (Prx1) is frequently elevated in many solid tumors and TME and is often associated with tumor growth and drug resistance. Transcription factor Nrf2, over-expressed in many human solid tumors, also reportedly regulates miRNA-155 expression [113, 114]. Increased translation of Nrf2 leads to an upregulation of Prx1. These transcription factors play a dual role in that they protect normal tissues against oxidative damage while promoting tumor growth, metastasis, and chemoresistance. Park et al used the NCI-60 panel of NSCLC cell lines to demonstrate Prx1 over-expression in nine non-small cell lung cancer (NSCLC) cells [10, 11]. Data generated by Kim and others suggests that Nrf2/Prx1, which are upregulated in primary human lung cancer, are potentially critical therapeutic targets [11] (Table 2). Therapeutic doses of Se can selectively downregulate Nrf2/Prx1 levels induced by hypoxia in tumor cells while up-regulating these markers in several normal mouse tissues [11]. Thus, selective inhibition of transcription factors by MSC was found to enhance antitumor activity of chemo and radiation therapies in lung cancer A549 and colorectal cancer HT29 while protecting the normal healthy tissues.
Table 2: MSC differentially activate Nrf2 in A549 lung tumor vs normal lung tissue [11]
Relative Intensity |
||||
|---|---|---|---|---|
TUMOR |
LUNG |
|||
C |
MSC |
C |
MSC |
|
Prx1 |
5.4 |
1.2 |
1.2 |
3.8 |
Nrf2 |
6.0 |
1.0 |
1.0 |
4.0 |
In brief, therapeutic doses of MSC are effective and selective inhibitors of specific DNA repair gene and specific types of transcription factors. Modulation of these markers by MSC results in an enhanced antitumor activity of chemo and radiation therapies in preclinical models.
Altered oncogenic and tumor suppressor miRNA expression
miRNAs are single-stranded, small noncoding RNAs, approximately 22 nucleotides long, that function as post-transcriptional regulators of multiple genes. There are two types of altered miRNAs: 1) oncogenic miRNAs are upregulated in advanced cancers and in unstable TMEs, where they are associated with enhanced angiogenesis, metastasis and drug resistance; and, 2) tumor suppressor miRNAs, which are downregulated in tumor cells and TMEs while being linked to drug efficacy [116].
The altered expression of specific types of miRNAs is associated with therapeutic resistance and thus may serve as cancer biomarkers. Redova et al reported that in 35 ccRCC tumors, compared with 10 non-tumor kidney tissues, miRNA 210 and 155 levels were 5.99 and 4.57 fold higher, respectively [115]. miRNAs are master regulators of cell stemness, cancer metastasis and drug resistance [116]. miRNA 210 is inversely correlated with disease-free survival and overall survival of patients with NSCLC. miRNA 155 and 210 have been identified as VHL-regulated miRNA, activated by HIF1α and associated with drug resistance [117], and as prognostic and diagnostic biomarkers for overall survivor in glioblastoma following surgery [118]. miRNA 155 also represses a ubiquitin ligase that promotes degradation of NF-k-β family transcription factor c-Rel. An upregulated miRNA 155 modulates T-cell proliferation responses by targeting cytotoxic T lymphocyte-associated antigen 4 [119]. Based on published data, oncogenic miRNA 155 and 210 seem to function as pleiotropic regulators of immunologic and molecular biomarkers associated with therapeutic resistance in cancer cells.
In view of the identification of altered specific oncogenic and tumor suppressor miRNAs as potential therapeutic targets, it is a critical and unmet need to identify agents that can selectively and effectively target such miRNAs. Several approaches currently under evaluation are aimed at downregulating oncogenic miRNAs and/or upregulating tumor suppressor miRNAs. This includes chemotherapeutic drugs, antisense oligonucleotides and viruses [77, 94-96, 120]. To date, in vivo instability of TME, poor drug uptake, dose-limiting toxicity and the need for better evaluation of the effects of the clinical agent under development on miRNA targets with its consequences in therapeutic outcomes continue to represent a major challenge. Our published data indicates that effective dosing and scheduling of MSC and perhaps SLMs that are capable of inhibiting HIFs are also associated with the downregulation of 28 oncogenic miRNAs and the upregulation of 12 tumor suppressor miRNAs expressed in ccRCC xenografts. The miRNAs modulated by MSC include the oncogenic miRNA 155, 106b, and 210, and the tumor suppressor miRNA-Let7b, -85, and 328 which have also been found in primary ccRCC tumor biopsies.
In brief, altered expression of various miRNAs seem to play a pivotal role in the regulation of different immunological and molecular biomarkers implicated in tumor drug resistance and TME heterogeneity.
Immune response cells and immune response checkpoint
Programmed death 1 (PD-1), an immune inhibitory receptor expressed on several immune cells including cytotoxic T cells, interacts with two ligands—programmed death ligand (PD-L) 1 and PD-L2. PD-L2 is expressed primarily on macrophages and dendritic cells and PD-L1 is expressed on tumor cells and other immune cells. Interaction of these ligands with PD-1 inhibits T-cell activation and cytokine production and is important in maintaining homeostasis of the immune response, thus preventing autoimmunity during infection or inflammation in normal tissue. The same interaction in TMEs provides an immune escape mechanism for tumor cells by turning off cytotoxic T cells. Blocking these interactions may enable the cytotoxic T cells in attacking the tumor cells. PD-L1 is a novel transcription target of HIF2α and HIF1α in tumor cell deficient in VHL [89, 90]. Blockade of HIF1α enhances myeloid derived suppressor cells (MDSC)-mediated T-cell activation. An unstable TME is associated with an increased expression of PD-L1 in tumor cells [93]. Inhibition of PD-L1 leads to an improved treatment outcome [87, 121]. PD-1 promotes ERK and mTOR pathways, and inhibition of the PD-1/PD-L1 axis leads to an enhanced antitumor activity of Docetaxel and Doxorubicin in an orthotopic metastatic mouse model [122]. PD-1/PD-L1 and HIFs are also associated with altered expression of specific types of oncogenic and tumor suppressor miRNAs. PD-L1 and FoxP3 regulatory T-cell infiltration of tumor cells are independent prognostic factors associated with poor prognosis in cancer patients [123, 124]. HIF1α mediates immune adaptation through the AKT/ERK/VEGF axis [125]. miRNA 155 and miRNA 210 target multiple pathways involved in the regulation of the immune response [126–130].
Collectively, current data suggests that PD-L1 expression is a result of an altered expression of miRNA and HIF, biomarkers that regulate multiple pathways including those that result in an unstable TME and cause tumor drug resistance. It is currently unclear whether the collective or individual modulation of these biomarkers by Se is necessary for tumor cell sensitization to targeted and cytotoxic drug therapies.
Plasma Se concentrations derived from MSC or SLM determined therapeutic in xenograft models can be achieved clinically without dose-limiting toxicity
Enhancement of antitumor activity of multiple anticancer drugs by MSC/SLM in several xenograft models have been found to be dependent on Se dose and schedule. In several studies, oral administration of 10 mg/kg/day MSC or SLM were sufficient in inhibiting HIF by greater than 80% and in enhancing anti-tumor activity of anticancer drugs [15–18] To verify that the optimal plasma Se levels achieved in xenografts treated with therapeutic doses and schedule of SLM can be achieved clinically without host toxicities, cancer patients were treated with escalating doses of SLM.
The data in Figure 4A represents the total plasma Se levels achieved at 2h following the oral administration of different doses of MSC and SLM to xenografts. Minimal and optimal therapeutic doses for MSC and SLM were 0.05 mg, and 0.2 mg, respectively. Se plasma concentrations were dose dependent, with higher levels seen with SLM. Plasma Se concentrations after administration of 0.2 mg MSC and SLM orally were approximately 15 and 45 μM, respectively. Tumor tissue Se concentrations were also found to be Se dose-dependent (Figure 4B). As with plasma, A253 and FaDu tumors accumulated higher levels of Se derived from SLM than from MSC. Human studies were carried out in colorectal cancer patients treated with escalating doses of SLM in sequential combination with a fixed dose of irinotecan at a dose of 125 mg/m2/week for 4weeks, [27, 28]. The data in Figure 4C shows that the total plasma Se concentrations were dose-dependent. The dose of SLM in the prevention clinical trial, 200 μg/pt/d, yielded 3-4μM total Se plasma concentrations, slightly higher than the circulating levels in “normal” U.S. population. The dose of SLM at 7,200 μg/BIDx7 followed by 7200 μg/d concurrently with irinotecan for at least 28 days yielded plasma Se concentrations in the range of 30-40μM (Figure 4D), without toxicity. This concentration was similar to what was achieved with therapeutic doses of SLM in mice bearing human tumor xenografts.
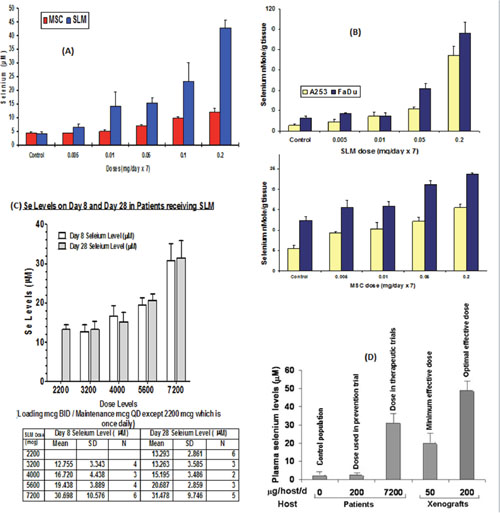
Figure 4: Plasma and tumor cells Se concentrations derived from the administration of therapeutic doses of MSC or SLM in mice and in plasma of patients treated with SLM. (A) Total plasma Se level in mice after oral doses of MSC or SLM. (B) Intra tumor Se concentration 2h after 7 days of SLM or MSC [131]. (C) Plasma Se levels on day 8 and day 28 in patients receiving SLM [27, 28]. (D) Plasma Se levels in patients treated with SLM in the therapeutic trial reached levels higher than the minimal effective dose needed in mice for therapeutic augmentation with anticancer drugs and is significantly higher than the Se levels in untreated population or with SLM dose used in the prevention trial [35, 40].
The data in Figure 4D represents comparative levels of total plasma Se in a “control” population, patients in prevention trials, colorectal cancer patients treated with SLM, and in mice bearing tumor xenografts treated with the minimal and optimal therapeutic doses of SLM. Toxicity was limited to garlic breath in a few patients and minor dyspepsia [27, 29]. The clinical use of the high doses of SLM determined clinically safe is now under evaluation in sequential combination with axitinib, a VEGFR inhibitor, in heavily pretreated patients with advanced ccRCC.
Data presented in Table 3 is an attempt to correlate plasma and tumor tissue levels of Se with treatment outcome of xenografts with MSC or SLM in sequential combination with 100 mg/kg/wkX4 irinotecan (MTD) The data in Table 3 indicates that at a mouse plasma Se concentration of 10 μM derived post-administration of MSC, the tumor concentrations of SN38, the active metabolite of irinotecan, is higher in FaDu than in A253 xenografts. Cures, defined by the 90-day survival of xenografts without any evidence of cancer at the site of tumor transplant, seem to correlate with tumor levels of SN38. FaDu tumors with higher SN38 accumulation than A253, achieved a 100% cure rate compared with 60% cures seen in A253. Protection of xenografts from drug-induced toxicity by MSC allowed the administration of higher doses of irinotecan than the MTD that resulted in higher cures. While plasma and tumor tissue Se levels achieved with SLM were higher than those derived from MSC, a similar augmentation of the antitumor activity of irinotecan was seen in FaDu and A253 xenografts. The data in Table 3 also indicate that the higher cure rates of FaDu xenografts were associated with greater tumor cell concentrations of Se and SN38.
Table 3: Plasma and tumor Se levels achieved with oral aministration of 0.2gm/day MSC or SLM; correlation with “cure”, and tumor SN38 concentrations in A253, and FaDU xengrafts treated with irinotecan, 100 mg/kg/wkx4
Se (0.2 mg) |
Plasma Cmax (μM) |
Tumor Se (nmol/gm) |
“Cures” (%) |
SN38 (nmol/gm) |
|||
|---|---|---|---|---|---|---|---|
A253 |
FaDU |
A253 |
FaDU |
A253 |
FaDU |
||
MSC |
15 |
16 |
22 |
60 |
100 |
13.1 |
29.4 |
SLM |
48 |
80 |
100 |
60 |
100 |
ND |
ND |
In six patients with advanced colorectal cancer treated with 2200 μg SLM in combination with 125 mg/m2 irinotecan, no grade 3 diarrhea was observed and one out of six patients developed grade 4 neutropenia [27] with no grade 3 diarrhea. Normally, with the 125 mg/m2 per week irinotecan treatment, 20-30% of patients develop grade 3/4 diarrhea.
The optimal 0.2 mg/kg SLM dose delivered to xenografts was about 4000- and 57-fold higher than that achieved with 200 dose delivered to patients in the prevention clinical trial, and the 7,200 μg dose administered twice daily to cancer patients (assumed patient weight was averaged to approximately 85 kg), respectively. The SLM dose administered in our therapeutic clinical trial was about 70-fold higher than the dose used in the prevention trials. The high SLM dose utilized in our clinical trials, however, was 6-fold lower than the 40,800 μg dose of inorganic selenite that was determined to be toxic [25]. It is important to note that oral administration of 0.2 mg/kg MSC or SLM alone for several weeks resulted in minimal antitumor activity of less than 50% tumor growth inhibition. Doses lower than 0.05 mg/kg did not alter significantly the efficacy or the toxicity of irinotecan in the xenografts evaluated in the preclinical studies.
In summary, plasma Se concentrations in xenografts and in patients are Se dose-dependent. Similarly, enhanced antitumor activity of anticancer drugs in xenografts was MSC and SLM dose-dependent. Plasma Se concentrations associated with enhanced antitumor activity of anticancer drugs seen in mice bearing human tumor xenografts can be achieved clinically in human patients without toxicity. Enhanced tumor accumulation of Se and SN38 results in an enhanced antitumor activity of irinotecan
Phase I clinical trial development
We have provided the rationale for use of a defined dose and schedule of SLM that can effectively enhance the therapeutic efficacy and selectivity of anticancer drugs [6-8, 15, 16], including axitinib, a tyrosine kinase inhibitor [19] in ccRCC xenografts. While MSC is under development, SLM is FDA-approved for clinical trials in the U.S. Patients with metastatic ccRCC who progressed on a prior line of treatment are being enrolled on 3 + 3 standard clinical trial design, combining escalating high doses of SLM at 2500, 3000, 4000, and 5000 μg in sequential combination with a standard of care axitinib dose. SLM is being given twice daily for 14 days, followed by once daily in combination with axitinib until disease progression or toxicity (NCT02535533). The plan is to develop a phase 2 clinical trial once the desired optimal plasma Se concentration has been achieved without toxicity as defined in the ongoing phase 1 trial
Future directions
In vivo drug resistance is regulated by multiple molecular and immunological biomarkers and pathways expressed in tumor cells, the surrounding TME and cross-talk between them. Cells in the TME are functionally interactive, expressing different drug resistant biomarkers including specific types of oncogenic miRNAs, HIFs, VEGF/VEGFR, and PD-1/PD-L1, among others. While TME acts as a gatekeeper, tumor cells are the ultimate therapeutic targets. The molecular and immunological biomarkers implicated in angiogenesis and drug resistance can be modulated by therapeutic doses and schedules of MSC and SLM in several in vitro and in vivo models. The pleiotropic effects of SLM and MSC led to a significant enhancement of the antitumor activity of multiple anticancer drugs in several xenograft models. In addition to the demonstrated therapeutic benefits and protective effects against drug toxicities, MSC shares two unique properties: it downregulates expression levels of oncogenic miRNAs, include miRNA-155, -106b, and -210, and upregulates expression levels of tumor suppressor miRNAs, include miRNA-let 7b, -185, and -328; 2.
MSC also enhances degradation of hypoxia- induced and constitutively expressed HIF1α and HIF2α through PHD-dependent mechanism [8, 16]. Altered miRNAs and HIFs represent master regulators of angiogenesis, tumor growth and drug resistance. Based on the data generated in our laboratory, primarily in ccRCC tumors, together with published reports by others, the schema in Figure 5 represents the potential role of miRNA-155, and -210 in the modulation of multidrug resistance targets of Se. While the oncogenic miRNA regulates HIFs, these two miRNAs may interact and possibly regulate each other. From the therapeutic viewpoint, agents under development to target these markers have fallen short of clinical expectations and their limited clinical benefits have been attributed in part to their in vivo instability, toxicity, and inability to effectively modulate their intended targets [88]. Our laboratory was the first to demonstrate that the in vivo expression of HIFs and specific types of miRNAs altered in ccRCC tumor can be selectively modulated by a defined dose and schedule of Se. Recent data suggests that specific types of miRNAs in ccRCC tumors are regulated by HIF through VHL–dependent and independent pathways while PD-L1 is a target of the VHL-HIFs axis [121]. Thus, the reported stable expression of HIFs in ccRCC tumors may be a consequence of VHL inactivation by miRNA-155 and or miRNA-210.
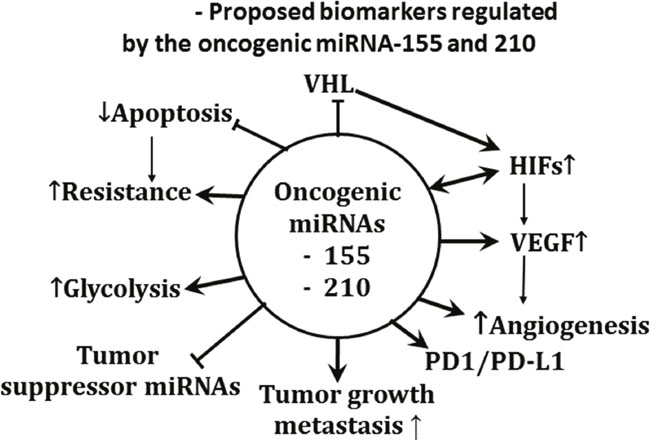
Figure 5: Pleiotropic effects of oncogenic miRNA-210, and miRNA-155. The proposed biomarkers modulated by the oncogenic miRNAs were identified from results generated primarily in RC2, 786.0, and primary ccRCC tumors, and, FaDu head and neck tumors
To validate the concept that HIFs and specific types of miRNAs are critical therapeutic targets of Se whose modulation can impact treatment outcome, a pilot phase 1 clinical trial with SLM in sequential combination with axitinib is underway in pretreated patients with advanced ccRCC. Based on the results obtained in the pilot trial, a phase 2 clinical trial with SLM in sequential combination with other targeted agents will be developed.
Overall summary
With the knowledge gained in our understanding of the pharmacology and the mechanism of action of cytotoxic drugs combined with the specific information and identification of biomarkers associated with angiogenesis and drug resistance, we have a unique opportunity to design new and novel mechanism-bases combinations. To this end, it is important to recognize that the dose, schedule and sequence of the drugs used in combination settings are critical in the design of future clinical trials. The unique profile of ccRCC tumors expressing mutant VHL stable expression of HIFs, and upregulated oncogenic miRNAs, and downregulated tumor suppressor miRNAs make it an excellent model for proof-of-concept that modulation of the expression levels and functions of these markers by therapeutically effective doses and schedules of Se may offer the potential to circumvent drug resistance, and may offer a new and novel clinical approach for the treatment of cancer
Author contributions
All authors edited and approved the final manuscript.
ACKNOWLEDGMENTS AND FUNDING
This work was supported by the National cancer institute grant P30CA016056 involving the use of Roswell Park Cancer Institute's Pathology Network, Genomic, and Clinical Data Network Shared Resources.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Azrak RG, Cao S, Durrani FA, Toth K, Bhattacharya A, Rustum YM. Augmented therapeutic efficacy of irinotecan is associated with enhanced drug accumulation. Cancer letters. 2011; 311:219-229.
2. Bhattacharya A. Methylselenocysteine: a promising antiangiogenic agent for overcoming drug delivery barriers in solid malignancies for therapeutic synergy with anticancer drugs. Expert opinion on drug delivery. 2011; 8:749-763.
3. Bhattacharya A, Seshadri M, Oven SD, Toth K, Vaughan MM, Rustum YM. Tumor vascular maturation and improved drug delivery induced by methylselenocysteine leads to therapeutic synergy with anticancer drugs. Clinical cancer research: an official journal of the American Association for Cancer Research. 2008; 14:3926-3932.
4. Bhattacharya A, Toth K, Sen A, Seshadri M, Cao S, Durrani FA, Faber E, Repasky EA, Rustum YM. Inhibition of colon cancer growth by methylselenocysteine-induced angiogenic chemomodulation is influenced by histologic characteristics of the tumor. Clinical colorectal cancer. 2009; 8:155-162.
5. Bhattacharya A, Turowski SG, San Martin ID, Rajput A, Rustum YM, Hoffman RM, Seshadri M. Magnetic resonance and fluorescence-protein imaging of the anti-angiogenic and anti-tumor efficacy of selenium in an orthotopic model of human colon cancer. Anticancer research. 2011; 31:387-393.
6. Cao S, Durrani FA, Rustum YM. Selective modulation of the therapeutic efficacy of anticancer drugs by selenium containing compounds against human tumor xenografts. Clinical cancer research: an official journal of the American Association for Cancer Research. 2004; 10:2561-2569.
7. Cao S, Durrani FA, Toth K, Rustum YM, Seshadri M. Bevacizumab enhances the therapeutic efficacy of Irinotecan against human head and neck squamous cell carcinoma xenografts. Oral oncology. 2011; 47:459-466.
8. Chintala S, Toth K, Cao S, Durrani FA, Vaughan MM, Jensen RL, Rustum YM. Se-methylselenocysteine sensitizes hypoxic tumor cells to irinotecan by targeting hypoxia-inducible factor 1alpha. Cancer chemotherapy and pharmacology. 2010; 66:899-911.
9. Fischer JL, Mihelc EM, Pollok KE, Smith ML. Chemotherapeutic selectivity conferred by selenium: a role for p53-dependent DNA repair. Molecular cancer therapeutics. 2007; 6:355-361.
10. Kim JH, Bogner PN, Baek SH, Ramnath N, Liang P, Kim HR, Andrews C, Park YM. Up-regulation of peroxiredoxin 1 in lung cancer and its implication as a prognostic and therapeutic target. Clinical cancer research: an official journal of the American Association for Cancer Research. 2008; 14:2326-2333.
11. Kim YJ, Baek SH, Bogner PN, Ip C, Rustum YM, Fakih MG, Ramnath N, Park YM. Targeting the Nrf2-Prx1 pathway with selenium to enhance the efficacy and eelectivity of cancer therapy. Journal of Cancer Molecules. 2007; 3:7.
12. Kumar MS, Pollok KE, Smith ML. Selenomethionine or methylseleninic acid inhibits mutagenesis of a reporter gene in mouse bone marrow. Anticancer research. 2010; 30:291-293.
13. Saifo MS, Rempinski DR Jr, Rustum YM, Azrak RG. Targeting the oncogenic protein beta-catenin to enhance chemotherapy outcome against solid human cancers. Molecular cancer. 2010; 9:310.
14. Vaughan MM, Toth K, Chintala S, Rustum YM. Double immunohistochemical staining method for HIF-1alpha and its regulators PHD2 and PHD3 in formalin-fixed paraffin-embedded tissues. Applied immunohistochemistry & molecular morphology: AIMM. 2010; 18:375-381.
15. Cao S, Durrani FA, Toth K, Rustum YM. Se-methylselenocysteine offers selective protection against toxicity and potentiates the antitumour activity of anticancer drugs in preclinical animal models. British journal of cancer. 2014; 110:1733-1743.
16. Chintala S, Najrana T, Toth K, Cao S, Durrani FA, Pili R, Rustum YM. Prolyl hydroxylase 2 dependent and Von-Hippel-Lindau independent degradation of Hypoxia-inducible factor 1 and 2 alpha by selenium in clear cell renal cell carcinoma leads to tumor growth inhibition. BMC cancer. 2012; 12:293.
17. Toth K, Chintala S, Rustum YM. Constitutive expression of HIF-alpha plays a major role in generation of clear-cell phenotype in human primary and metastatic renal carcinoma. Applied immunohistochemistry & molecular morphology: AIMM. 2014; 22:642-647.
18. Yin MB, Li ZR, Toth K, Cao S, Durrani FA, Hapke G, Bhattacharya A, Azrak RG, Frank C, Rustum YM. Potentiation of irinotecan sensitivity by Se-methylselenocysteine in an in vivo tumor model is associated with downregulation of cyclooxygenase-2, inducible nitric oxide synthase, and hypoxia-inducible factor 1alpha expression, resulting in reduced angiogenesis. Oncogene. 2006; 25:2509-2519.
19. Durrani FA, Chintala S, Toth K, Cao S, Rustum YM. Mechanism-based drug combination targeting HIF-2α and VEGF in renal cancer xenografts Trends in Cell and Molecular Biology. 2015; 10:12.
20. Thomson CD. Assessment of requirements for selenium and adequacy of selenium status: a review. European journal of clinical nutrition. 2004; 58:391-402.
21. Thomson CD, Chisholm A, McLachlan SK, Campbell JM. Brazil nuts: an effective way to improve selenium status. The American journal of clinical nutrition. 2008; 87:379-384.
22. Hagemann-Jensen M, Uhlenbrock F, Kehlet S, Andresen L, Gabel-Jensen C, Ellgaard L, Gammelgaard B, Skov S. The selenium metabolite methylselenol regulates the expression of ligands that trigger immune activation through the lymphocyte receptor NKG2D. The Journal of biological chemistry. 2014; 289:31576-31590.
23. Dennert G, Zwahlen M, Brinkman M, Vinceti M, Zeegers MP, Horneber M. Selenium for preventing cancer. The Cochrane database of systematic reviews. 2011; (5):CD005195.
24. Duffield AJ, Thomson CD, Hill KE, Williams S. An estimation of selenium requirements for New Zealanders. The American journal of clinical nutrition. 1999; 70:896-903.
25. MacFarquhar JK, Broussard DL, Melstrom P, Hutchinson R, Wolkin A, Martin C, Burk RF, Dunn JR, Green AL, Hammond R, Schaffner W, Jones TF. Acute selenium toxicity associated with a dietary supplement. Archives of internal medicine. 2010; 170:256-261.
26. Micke O, Schomburg L, Buentzel J, Kisters K, Muecke R. Selenium in oncology: from chemistry to clinics. Molecules. 2009; 14:3975-3988.
27. Fakih MG, Pendyala L, Brady W, Smith PF, Ross ME, Creaven PJ, Badmaev V, Prey JD, Rustum YM. A Phase I and pharmacokinetic study of selenomethionine in combination with a fixed dose of irinotecan in solid tumors. Cancer chemotherapy and pharmacology. 2008; 62:499-508.
28. Fakih MG, Pendyala L, Smith PF, Creaven PJ, Reid ME, Badmaev V, Azrak RG, Prey JD, Lawrence D, Rustum YM. A phase I and pharmacokinetic study of fixed-dose selenomethionine and irinotecan in solid tumors. Clinical cancer research: an official journal of the American Association for Cancer Research. 2006; 12:1237-1244.
29. Fakih M, Cao S, Durrani FA, Rustum YM. Selenium protects against toxicity induced by anticancer drugs and augments antitumor activity: a highly selective, new, and novel approach for the treatment of solid tumors. Clinical colorectal cancer. 2005; 5:132-135.
30. Ip C, Thompson HJ, Zhu Z, Ganther HE. in vitro and in vivo studies of methylseleninic acid: evidence that a monomethylated selenium metabolite is critical for cancer chemoprevention. Cancer research. 2000; 60:2882-2886.
31. Li GX, Lee HJ, Wang Z, Hu H, Liao JD, Watts JC, Combs GF Jr, Lu J. Superior in vivo inhibitory efficacy of methylseleninic acid against human prostate cancer over selenomethionine or selenite. Carcinogenesis. 2008; 29:1005-1012.
32. Patrick L. Selenium biochemistry and cancer: a review of the literature. Alternative medicine review: a journal of clinical therapeutic. 2004; 9:239-258.
33. Burk RF, Norsworthy BK, Hill KE, Motley AK, Byrne DW. Effects of chemical form of selenium on plasma biomarkers in a high-dose human supplementation trial. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2006; 15:804-810.
34. Chen J, Clark LC. Proposed supplemental dosages of selenium for a phase I trial based on dietary and supplemental selenium intakes and episodes of chronic selenosis. International Journal of Toxicology. 1986; 5:8.
35. Clark LC, Combs GF Jr, Turnbull BW, Slate EH, Chalker DK, Chow J, Davis LS, Glover RA, Graham GF, Gross EG, Krongrad A, Lesher JL Jr, Park HK, et al. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. A randomized controlled trial. Nutritional Prevention of Cancer Study Group. Jama. 1996; 276:1957-1963.
36. Hatfield DL, Gladyshev VN. The Outcome of Selenium and Vitamin E Cancer Prevention Trial (SELECT) reveals the need for better understanding of selenium biology. Molecular interventions. 2009; 9:18-21.
37. Jaworska K, Gupta S, Durda K, Muszynska M, Sukiennicki G, Jaworowska E, Grodzki T, Sulikowski M, Waloszczyk P, Wojcik J, Lubinski J, Cybulski C, Debniak T, et al. A low selenium level is associated with lung and laryngeal cancers. PloS one. 2013; 8:e59051.
38. Karamali M, Nourgostar S, Zamani A, Vahedpoor Z, Asemi Z. The favourable effects of long-term selenium supplementation on regression of cervical tissues and metabolic profiles of patients with cervical intraepithelial neoplasia: a randomised, double-blind, placebo-controlled trial. The British journal of nutrition. 2015; 114:2039-2045.
39. Karunasinghe N, Han DY, Zhu S, Duan H, Ko YJ, Yu JF, Triggs CM, Ferguson LR. Effects of supplementation with selenium, as selenized yeast, in a healthy male population from New Zealand. Nutrition and cancer. 2013; 65:355-366.
40. Lippman SM, Klein EA, Goodman PJ, Lucia MS, Thompson IM, Ford LG, Parnes HL, Minasian LM, Gaziano JM, Hartline JA, Parsons JK, Bearden JD 3rd, Crawford ED, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). Jama. 2009; 301:39-51.
41. Lopez-Saez JB, Senra-Varela A, Pousa-Estevez L. Selenium in breast cancer. Oncology. 2003; 64:227-231.
42. Madhunapantula SV, Desai D, Sharma A, Huh SJ, Amin S, Robertson GP. PBISe, a novel selenium-containing drug for the treatment of malignant melanoma. Molecular cancer therapeutics. 2008; 7:1297-1308.
43. Weekley CM, Aitken JB, Vogt S, Finney LA, Paterson DJ, de Jonge MD, Howard DL, Musgrave IF, Harris HH. Uptake, distribution, and speciation of selenoamino acids by human cancer cells: X-ray absorption and fluorescence methods. Biochemistry. 2011; 50:1641-1650.
44. Wolf CM, Morana SJ, Eastman A. Zinc inhibits apoptosis upstream of ICE/CED-3 proteases rather than at the level of an endonuclease. Cell death and differentiation. 1997; 4:125-129.
45. Caffrey PB, Frenkel GD. Selenium compounds prevent the induction of drug resistance by cisplatin in human ovarian tumor xenografts in vivo. Cancer chemotherapy and pharmacology. 2000; 46:74-78.
46. Chen YC, Prabhu KS, Mastro AM. Is selenium a potential treatment for cancer metastasis? Nutrients. 2013; 5:1149-1168.
47. Fernandes AP, Gandin V. Selenium compounds as therapeutic agents in cancer. Biochimica et biophysica acta. 2015; 1850:1642-1660.
48. Hoefig CS, Renko K, Kohrle J, Birringer M, Schomburg L. Comparison of different selenocompounds with respect to nutritional value vs. toxicity using liver cells in culture. The Journal of nutritional biochemistry. 2011; 22:945-955.
49. Hu YJ, Chen Y, Zhang YQ, Zhou MZ, Song XM, Zhang BZ, Luo L, Xu PM, Zhao YN, Zhao YB, Cheng G. The protective role of selenium on the toxicity of cisplatin-contained chemotherapy regimen in cancer patients. Biological trace element research. 1997; 56:331-341.
50. Karavelioglu E, Boyaci MG, Simsek N, Sonmez MA, Koc R, Karademir M, Guven M, Eser O. Selenium protects cerebral cells by cisplatin induced neurotoxicity. Acta cirurgica brasileira. 2015; 30:394-400.
51. Martin JL, Hurlbut JA. Tissue selenium levels and growth responses of mice fed selenomethioine, Se-methylselenocysteine or sodium selenite. Phosphorous and Sulfur and the Related Elements. 1976; 1:6.
52. Rezvanfar MA, Rezvanfar MA, Shahverdi AR, Ahmadi A, Baeeri M, Mohammadirad A, Abdollahi M. Protection of cisplatin-induced spermatotoxicity, DNA damage and chromatin abnormality by selenium nano-particles. Toxicology and applied pharmacology. 2013; 266:356-365.
53. Satoh M, Naganuma A, Imura N. Effect of coadministration of selenite on the toxicity and antitumor activity of cis-diamminedichloroplatinum (II) given repeatedly to mice. Cancer chemotherapy and pharmacology. 1992; 30:439-443.
54. Sun Q, Dong M, Wang Z, Wang C, Sheng D, Li Z, Huang D, Yuan C. Selenium-enriched polysaccharides from Pyracantha fortuneana (Se-PFPs) inhibit the growth and invasive potential of ovarian cancer cells through inhibiting beta-catenin signaling.Oncotarget. 2016; 7:28369-28383. https://doi.org/10.18632/oncotarget.8619.
55. Weekley CM, Aitken JB, Musgrave IF, Harris HH. Methylselenocysteine treatment leads to diselenide formation in human cancer cells: evidence from X-ray absorption spectroscopy studies. Biochemistry. 2012; 51:736-738.
56. Zeng H, Cheng WH, Johnson LK. Methylselenol, a selenium metabolite, modulates p53 pathway and inhibits the growth of colon cancer xenografts in Balb/c mice. The Journal of nutritional biochemistry. 2013; 24:776-780.
57. Zwolak I, Zaporowska H. Selenium interactions and toxicity: a review. Selenium interactions and toxicity. Cell biology and toxicology. 2012; 28:31-46.
58. de Miranda JX, Andrade FO, Conti AD, Dagli ML, Moreno FS, Ong TP. Effects of selenium compounds on proliferation and epigenetic markers of Breast cancer. Journal of trace elements in medicine and biology: organ of the Society for Minerals and Trace Elements. 2014; 28:6.
59. Duffield-Lillico AJ, Dalkin BL, Reid ME, Turnbull BW, Slate EH, Jacobs ET, Marshall JR, Clark LC, Nutritional Prevention of Cancer Study G. Selenium supplementation, baseline plasma selenium status and incidence of prostate cancer: an analysis of the complete treatment period of the Nutritional Prevention of Cancer Trial. BJU international. 2003; 91:608-612.
60. Speckmann B, Grune T. Epigenetic effects of selenium and their implications for health. Epigenetics. 2015; 10:179-190.
61. Ikeda S, Kishida S, Yamamoto H, Murai H, Koyama S, Kikuchi A. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. The EMBO journal. 1998; 17:1371-1384.
62. Beppu K, Nakamura K, Linehan WM, Rapisarda A, Thiele CJ. Topotecan blocks hypoxia-inducible factor-1alpha and vascular endothelial growth factor expression induced by insulin-like growth factor-I in neuroblastoma cells. Cancer research. 2005; 65:4775-4781.
63. Puppo M, Battaglia F, Ottaviano C, Delfino S, Ribatti D, Varesio L, Bosco MC. Topotecan inhibits vascular endothelial growth factor production and angiogenic activity induced by hypoxia in human neuroblastoma by targeting hypoxia-inducible factor-1alpha and -2alpha. Molecular cancer therapeutics. 2008; 7:1974-1984.
64. Yoshida T, Hashimura M, Mastumoto T, Tazo Y, Inoue H, Kuwata T, Saegusa M. Transcriptional upregulation of HIF-1alpha by NF-kappaB/p65 and its associations with beta-catenin/p300 complexes in endometrial carcinoma cells. Laboratory investigation; a journal of technical methods and pathology. 2013; 93:1184-1193.
65. Giles RH, Lolkema MP, Snijckers CM, Belderbos M, van der Groep P, Mans DA, van Beest M, van Noort M, Goldschmeding R, van Diest PJ, Clevers H, Voest EE. Interplay between VHL/HIF1alpha and Wnt/beta-catenin pathways during colorectal tumorigenesis. Oncogene. 2006; 25:3065-3070.
66. Carroll VA, Ashcroft M. Role of hypoxia-inducible factor (HIF)-1alpha versus HIF-2alpha in the regulation of HIF target genes in response to hypoxia, insulin-like growth factor-I, or loss of von Hippel-Lindau function: implications for targeting the HIF pathway. Cancer research. 2006; 66:6264-6270.
67. Cui XG, Han ZT, He SH, Wu XD, Chen TR, Shao CH, Chen DL, Su N, Chen YM, Wang T, Wang J, Song DW, Yan WJ, et al. HIF1/2alpha mediates hypoxia-induced LDHA expression in human pancreatic cancer cells. Oncotarget. 2017; 8:24840-24852. https://doi.org/10.18632/oncotarget.15266.
68. Lauer V, Schodel J. Hypoxia-inducible Factor Crosses the Checkpoint. European urology. 2016; 70:633-634.
69. Masson N, Ratcliffe PJ. Hypoxia signaling pathways in cancer metabolism: the importance of co-selecting interconnected physiological pathways. Cancer & metabolism. 2014; 2:3.
70. Meng AX, Jalali F, Cuddihy A, Chan N, Bindra RS, Glazer PM, Bristow RG. Hypoxia down-regulates DNA double strand break repair gene expression in prostate cancer cells. Radiotherapy and oncology: journal of the European Society for Therapeutic Radiology and Oncology. 2005; 76:168-176.
71. Paolicchi E, Gemignani F, Krstic-Demonacos M, Dedhar S, Mutti L, Landi S. Targeting hypoxic response for cancer therapy.Oncotarget. 2016; 7:13464-13478. https://doi.org/10.18632/oncotarget.7229.
72. Imamura T, Kikuchi H, Herraiz MT, Park DY, Mizukami Y, Mino-Kenduson M, Lynch MP, Rueda BR, Benita Y, Xavier RJ, Chung DC. HIF-1alpha and HIF-2alpha have divergent roles in colon cancer. International journal of cancer. 2009; 124:763-771.
73. Kaelin WG Jr. The von Hippel-Lindau tumor suppressor protein and clear cell renal carcinoma. Clinical cancer research: an official journal of the American Association for Cancer Research. 2007; 13:680s-684s.
74. Keith B, Johnson RS, Simon MC. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nature reviews Cancer. 2011; 12:9-22.
75. Nanus DM, Gudas LJ. The Tale of Two Hypoxia-Inducible Factors in Renal Cell Carcinoma. European urology. 2016; 69:658-659.
76. Zhao J, Du F, Shen G, Zheng F, Xu B. The role of hypoxia-inducible factor-2 in digestive system cancers. Cell death & disease. 2015; 6:e1600.
77. Deng H, Zhou J, Sundersingh F, Messer JA, Somers DO, Ajakane M, Arico-Muendel CC, Beljean A, Belyanskaya SL, Bingham R, Blazensky E, Boullay AB, Boursier E, et al. Discovery and Optimization of Potent, Selective, and in vivo Efficacious 2-Aryl Benzimidazole BCATm Inhibitors. ACS medicinal chemistry letters. 2016; 7:379-384.
78. Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG Jr. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001; 292:464-468.
79. Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001; 292:468-472.
80. Mahon PC, Hirota K, Semenza GL. FIH-1: a novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes & development. 2001; 15:2675-2686.
81. Martinez-Saez O, Gajate Borau P, Alonso-Gordoa T, Molina-Cerrillo J, Grande E. Targeting HIF-2 alpha in clear cell renal cell carcinoma: A promising therapeutic strategy. Critical reviews in oncology/hematology. 2017; 111:117-123.
82. Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proceedings of the National Academy of Sciences of the United States of America. 1995; 92:5510-5514.
83. Yu F, White SB, Zhao Q, Lee FS. HIF-1alpha binding to VHL is regulated by stimulus-sensitive proline hydroxylation. Proceedings of the National Academy of Sciences of the United States of America. 2001; 98:9630-9635.
84. Zhang H, Qian DZ, Tan YS, Lee K, Gao P, Ren YR, Rey S, Hammers H, Chang D, Pili R, Dang CV, Liu JO, Semenza GL. Digoxin and other cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proceedings of the National Academy of Sciences of the United States of America. 2008; 105:19579-19586.
85. Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D, Buechler P, Isaacs WB, Semenza GL, Simons JW. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer research. 1999; 59:5830-5835.
86. Ghorpade DS, Leyland R, Kurowska-Stolarska M, Patil SA, Balaji KN. MicroRNA-155 is required for Mycobacterium bovis BCG-mediated apoptosis of macrophages. Molecular and cellular biology. 2012; 32:2239-2253.
87. Massari F, Santoni M, Ciccarese C, Santini D, Alfieri S, Martignoni G, Brunelli M, Piva F, Berardi R, Montironi R, Porta C, Cascinu S, Tortora G. PD-1 blockade therapy in renal cell carcinoma: current studies and future promises. Cancer treatment reviews. 2015; 41:114-121.
88. Neal CS, Michael MZ, Rawlings LH, Van der Hoek MB, Gleadle JM. The VHL-dependent regulation of microRNAs in renal cancer. BMC medicine. 2010; 8:64.
89. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, Bronte V, Chouaib S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. The Journal of experimental medicine. 2014; 211:781-790.
90. Noman MZ, Janji B, Hu S, Wu JC, Martelli F, Bronte V, Chouaib S. Tumor-Promoting Effects of Myeloid-Derived Suppressor Cells Are Potentiated by Hypoxia-Induced Expression of miR-210. Cancer research. 2015; 75:3771-3787.
91. Peng Y, Dong W, Lin TX, Zhong GZ, Liao B, Wang B, Gu P, Huang L, Xie Y, Lu FD, Chen X, Xie WB, He W, et al. MicroRNA-155 promotes bladder cancer growth by repressing the tumor suppressor DMTF1. Oncotarget. 2015; 6:16043-16058. https://doi.org/10.18632/oncotarget.3755.
92. Rapisarda A, Zalek J, Hollingshead M, Braunschweig T, Uranchimeg B, Bonomi CA, Borgel SD, Carter JP, Hewitt SM, Shoemaker RH, Melillo G. Schedule-dependent inhibition of hypoxia-inducible factor-1alpha protein accumulation, angiogenesis, and tumor growth by topotecan in U251-HRE glioblastoma xenografts. Cancer research. 2004; 64:6845-6848.
93. Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet AL, Latreche S, Bergaya S, Benhamouda N, Tanchot C, Stockmann C, Combe P, Berger A, et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. The Journal of experimental medicine. 2015; 212:139-148.
94. Aagaard L, Rossi JJ. RNAi therapeutics: principles, prospects and challenges. Advanced drug delivery reviews. 2007; 59:75-86.
95. Schmidt MF. Drug target miRNAs: chances and challenges. Trends in biotechnology. 2014; 32:578-585.
96. Shah MY, Ferrajoli A, Sood AK, Lopez-Berestein G, Calin GA. microRNA Therapeutics in Cancer - An Emerging Concept. EBioMedicine. 2016; 12:34-42.
97. Kummar S, Raffeld M, Juwara L, Horneffer Y, Strassberger A, Allen D, Steinberg SM, Rapisarda A, Spencer SD, Figg WD, Chen X, Turkbey IB, Choyke P, et al. Multihistology, target-driven pilot trial of oral topotecan as an inhibitor of hypoxia-inducible factor-1alpha in advanced solid tumors. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011; 17:5123-5131.
98. Onnis B, Rapisarda A, Melillo G. Development of HIF-1 inhibitors for cancer therapy. Journal of cellular and molecular medicine. 2009; 13:2780-2786.
99. Wigerup C, Pahlman S, Bexell D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacology & therapeutics. 2016; 164:152-169.
100. Yu T, Tang B, Sun X. Development of Inhibitors Targeting Hypoxia-Inducible Factor 1 and 2 for Cancer Therapy. Yonsei medical journal. 2017; 58:489-496.
101. Frediani JN, Fabbri M. Essential role of miRNAs in orchestrating the biology of the tumor microenvironment. Molecular cancer. 2016; 15:42.
102. Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, Jain RK. Normalization of the vasculature for treatment of cancer and other diseases. Physiological reviews. 2011; 91:1071-1121.
103. Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005; 307:58-62.
104. Jain RK. Normalizing tumor microenvironment to treat cancer: bench to bedside to biomarkers. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2013; 31:2205-2218.
105. Kharaishvili G, Simkova D, Bouchalova K, Gachechiladze M, Narsia N, Bouchal J. The role of cancer-associated fibroblasts, solid stress and other microenvironmental factors in tumor progression and therapy resistance. Cancer cell international. 2014; 14:41.
106. Klemm F, Joyce JA. Microenvironmental regulation of therapeutic response in cancer. Trends in cell biology. 2015; 25:198-213.
107. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nature medicine. 2013; 19:1423-1437.
108. Ribeiro AL, Okamoto OK. Combined effects of pericytes in the tumor microenvironment. Stem cells international. 2015; 2015:868475.
109. Bussard KM, Mutkus L, Stumpf K, Gomez-Manzano C, Marini FC. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast cancer research: BCR. 2016; 18:84.
110. Wessler S, Aberger F, Hartmann TN. The sound of tumor cell-microenvironment communication - composed by the Cancer Cluster Salzburg research network. Cell communication and signaling: CCS. 2017; 15:20.
111. Willett CG, Boucher Y, di Tomaso E, Duda DG, Munn LL, Tong RT, Chung DC, Sahani DV, Kalva SP, Kozin SV, Mino M, Cohen KS, Scadden DT, et al. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nature medicine. 2004; 10:145-147.
112. Crosby ME, Kulshreshtha R, Ivan M, Glazer PM. MicroRNA regulation of DNA repair gene expression in hypoxic stress. Cancer research. 2009; 69:1221-1229.
113. Shah NM, Rushworth SA, Murray MY, Bowles KM, MacEwan DJ. Understanding the role of NRF2-regulated miRNAs in human malignancies. Oncotarget. 2013; 4:1130-1142. https://doi.org/10.18632/oncotarget.1181.
114. Syu JP, Chi JT, Kung HN. Nrf2 is the key to chemotherapy resistance in MCF7 breast cancer cells under hypoxia. Oncotarget. 2016; 7:14659-14672. https://doi.org/10.18632/oncotarget.7406.
115. Redova M, Poprach A, Besse A, Iliev R, Nekvindova J, Lakomy R, Radova L, Svoboda M, Dolezel J, Vyzula R, Slaby O. MiR-210 expression in tumor tissue and in vitro effects of its silencing in renal cell carcinoma. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine. 2013; 34:481-491.
116. Raza U, Zhang JD, Sahin O. MicroRNAs: master regulators of drug resistance, stemness, and metastasis. Journal of molecular medicine. 2014; 92:321-336.
117. Osugi J, Kimura Y, Owada Y, Inoue T, Watanabe Y, Yamaura T, Fukuhara M, Muto S, Okabe N, Matsumura Y, Hasegawa T, Yonechi A, Hoshino M, et al. Prognostic Impact of Hypoxia-Inducible miRNA-210 in Patients with Lung Adenocarcinoma. Journal of oncology. 2015; 2015:316745.
118. Lai NS, Wu DG, Fang XG, Lin YC, Chen SS, Li ZB, Xu SS. Serum microRNA-210 as a potential noninvasive biomarker for the diagnosis and prognosis of glioma. British journal of cancer. 2015; 112:1241-1246.
119. Sonkoly E, Janson P, Majuri ML, Savinko T, Fyhrquist N, Eidsmo L, Xu N, Meisgen F, Wei T, Bradley M, Stenvang J, Kauppinen S, Alenius H, et al. MiR-155 is overexpressed in patients with atopic dermatitis and modulates T-cell proliferative responses by targeting cytotoxic T lymphocyte-associated antigen 4. The Journal of allergy and clinical immunology. 2010; 126:581-589 e581-520.
120. Garzon R, Marcucci G, Croce CM. Targeting microRNAs in cancer: rationale, strategies and challenges. Nature reviews Drug discovery. 2010; 9:775-789.
121. Messai Y, Gad S, Noman MZ, Le Teuff G, Couve S, Janji B, Kammerer SF, Rioux-Leclerc N, Hasmim M, Ferlicot S, Baud V, Mejean A, Mole DR, et al. Renal Cell Carcinoma Programmed Death-ligand 1, a New Direct Target of Hypoxia-inducible Factor-2 Alpha, is Regulated by von Hippel-Lindau Gene Mutation Status. European urology. 2016; 70:623-632.
122. Black M, Barsoum IB, Truesdell P, Cotechini T, Macdonald-Goodfellow SK, Petroff M, Siemens DR, Koti M, Craig AW, Graham CH. Activation of the PD-1/PD-L1 immune checkpoint confers tumor cell chemoresistance associated with increased metastasis. Oncotarget. 2016; 7:10557-10567. https://doi.org/10.18632/oncotarget.7235.
123. Barbi J, Pardoll DM, Pan F. Ubiquitin-dependent regulation of Foxp3 and Treg function. Immunological reviews. 2015; 266:27-45.
124. Li Z, Dong P, Ren M, Song Y, Qian X, Yang Y, Li S, Zhang X, Liu F. PD-L1 Expression Is Associated with Tumor FOXP3(+) Regulatory T-Cell Infiltration of Breast Cancer and Poor Prognosis of Patient. Journal of Cancer. 2016; 7:784-793.
125. Lee YH, Bae HC, Noh KH, Song KH, Ye SK, Mao CP, Lee KM, Wu TC, Kim TW. Gain of HIF-1alpha under normoxia in cancer mediates immune adaptation through the AKT/ERK and VEGFA axes. Clinical cancer research: an official journal of the American Association for Cancer Research. 2015; 21:1438-1446.
126. Boosani CS, Agrawal DK. Epigenetic Regulation of Innate Immunity by microRNAs. Antibodies. 2016; 5:15.
127. Jia S, Zhai H, Zhao M. MicroRNAs regulate immune system via multiple targets. Discovery medicine. 2014; 18:237-247.
128. Kuninty PR, Schnittert J, Storm G, Prakash J. MicroRNA Targeting to Modulate Tumor Microenvironment. Frontiers in oncology. 2016; 6:3.
129. Li L, Zhang J, Diao W, Wang D, Wei Y, Zhang CY, Zen K. MicroRNA-155 and MicroRNA-21 promote the expansion of functional myeloid-derived suppressor cells. Journal of immunology. 2014; 192:1034-1043.
130. Tsitsiou E, Lindsay MA. microRNAs and the immune response. Current opinion in pharmacology. 2009; 9:514-520.
131. Azrak RG, Cao S, Pendyala L, Durrani FA, Fakih M, Combs GF Jr, Prey J, Smith PF, Rustum YM. Efficacy of increasing the therapeutic index of irinotecan, plasma and tissue selenium concentrations is methylselenocysteine dose dependent. Biochemical pharmacology. 2007; 73:1280-1287.