
INTRODUCTION
Hodgkin lymphoma (HL) and primary mediastinal large B-cell lymphoma (PMBL) are two of the most common malignancies among adolescents and young adults (AYA). HL and PMBL share similar molecular features [1, 2]. HL has a bimodal age distribution, with a first peak at 15–35 years of age and second peak after 50 years of age [3]. Although HL represents approximately 4–5% of all cancers in children younger than 15 years of age, HL is the most common cancer in the 15–35 years AYA group, with an incidence of about 16% [4]. The prognosis is excellent in AYA with HL, with 10 year overall survival rates approximately 85–95% [5]. However, patients with an early relapse (≤12 months from diagnosis) and/or have metastatic extra-nodal disease at relapse/progression have a dismal prognosis [6, 7]. Furthermore, there are significant late effects secondary to chemo-radiotherapy in patients with HL, including cardiac and pulmonary toxicity, infertility and increased risk of secondary malignancy, which affect the long-term event-free survival (EFS) especially in AYA [8, 9]. Therefore, new targeted agents are needed to avoid late effects in HL and improve the outcomes in patients with high risk of relapse/progression.
PMBL was previously considered a subtype of diffuse large B-cell lymphoma (DLBCL) and is now classified as distinct mature B-cell lymphoma [10]. PMBL is a rare malignancy that, although more common among adults in their third and fourth decades of life, is also commonly seen in AYA, with a female predominance [4, 11–13]. PMBL represents approximately 4% of mature B-cell non-Hodgkin lymphoma in patients ≤18 years of age [14–17]. Although considered a mature B-cell lymphoma, we have identified that children and adolescents with newly diagnosed PMBL have a significantly decreased EFS compared to children and adolescents with other forms of DLBCL treated with similar therapy [16]. HL and PMBL also share similar cytogenetic abnormalities, namely chromosome 9p and 2p gains [18, 19] including rearrangement of chromosome 13 [20, 21]. Gain in 9p has been demonstrated to be associated with an upregulation in the expression of the Janus Kinase (JAK) 2 (JAK2) gene in about 50% of patients, leading to phosphorylation and activation of transcription factor signal transducer and activator of transcription (STAT) 6 [2, 22, 23]. Gain in 2p16 region has been associated with duplication of REL proto-oncogene that encodes for nuclear factor kappa B (NFKB) transcription factor [22]. PTPN1 mutations have also been shown recently in both HL and PMBL, leading to hyper-phosphorylation in JAK-STAT pathway [24]. HL and PMBL thus exhibit higher JAK2 transcript levels with increased JAK2 activity [25], suggesting aberrant activity of JAK2 and STAT pathways may in part play an important role in the pathogenesis and/or resistance in AYA HL and PMBL. Inhibition of JAK2 by fedratinib in HL and mediastinal large B-cell lymphoma (MLBL) has been significantly associated with an inhibition of cell proliferation and decreased growth in MLBL xenografted non-obese diabetic severe combined immunodeficiency gamma (NSG) mice, further establishing the importance of JAK2 activation in these tumors [26].
The role of JAK2 inhibitors like ruxolitinib and fedratinib has been studied in patients with myelofibrosis and myeloproliferative neoplasms (MPNs) that consistently exhibit dysregulation of the JAK1/JAK2 pathway [27–29]. Ruxolitinib is a potent and selective ATP-competitive inhibitor of JAK1 and JAK2 kinases against MPNs including those with a JAK2V617F mutation. Ruxolitinib also inhibits JAK2/STAT5 signaling in vitro and in murine models of MPNs [30]. It is worthy to note that ruxolitinib is associated with marked and durable clinical benefits in patients with myelofibrosis [31]. Interestingly the clinical benefit in MPN patients was achieved irrespective of JAK2V617F status, which suggests that the pathophysiological consequence of hyperactivity of the JAK/STAT pathway can be downregulated with ruxolitinib therapy [32].
We hypothesize that ruxolitinib may potentially be an effective therapeutic agent, in part by inducing targeted programmed cell death in both HL and PMBL. Therefore, we investigated the in vitro and in vivo efficacy of ruxolitinib against HL and PMBL cells in an immunodeficient mouse model (NSG) xenografted with human HL and PMBL and its in-vitro effects on downstream protein signaling pathways.
RESULTS
Effect of ruxolitinib on the JAK2/STAT signaling pathway in HL cells
The effect of ruxolitinib on the JAK2/STAT signaling pathways was examined measuring the phosphorylation status of JAK2 and its downstream substrates in HL cell lines. First, we observed that increasing concentrations of ruxolitinib (10-100 nM) for 24 h significantly inhibited downstream active phosphorylated STAT3 (p-STAT3, p < 0.005 at 10 nM, and p < 0.0005 at 25 - 100 nM) and phosphorylated STAT5 (p-STAT5, p < 0.005 at 10 nM, p < 0.0005 at 25 nM, and p < 0.0001 at 50 and 100 nM) in a dose-dependent manner in HDLM-2 cells (Figure 1A and 1C), whereas, total STAT3 and STAT5 levels remained unchanged at the concentrations up to 100 nM (Figure 1B and 1D). Similarly, no differences in expression of p-STAT3 and p-STAT5 were observed at different time points up to 100 nM ruxolitinib treated HDLM-2 cells at 48 and 72 hours (Data not shown). The dose escalation of ruxolitinib demonstrated an increase of the level of phosphorylated JAK2 (p-JAK2) in HDLM-2 cells. Conversely, we found no inhibitory effects of ruxolitinib at concentrations up to 100 nM in another HL cell line, L-540, which contains constitutively-active forms of JAK3, but not JAK2. In contrast, the pan-JAK inhibitor AG490 non-selectively inhibited the phosphorylation levels of these p-JAK2, p-STAT3 and p-STAT5 tested in both of HDLM-2 cells and L-540 cells. These results suggest that ruxolitinib inhibits JAK2/STAT signaling by blocking of downstream of the phosphorylation of STAT3 and STAT5 and that ruxolitinib shows selective activity of JAK2 against JAK3 in HL cell lines which is consistent with other studies suggesting that ruxolitinib is a JAK1/JAK2 inhibitor with marked selectivity over JAK3.
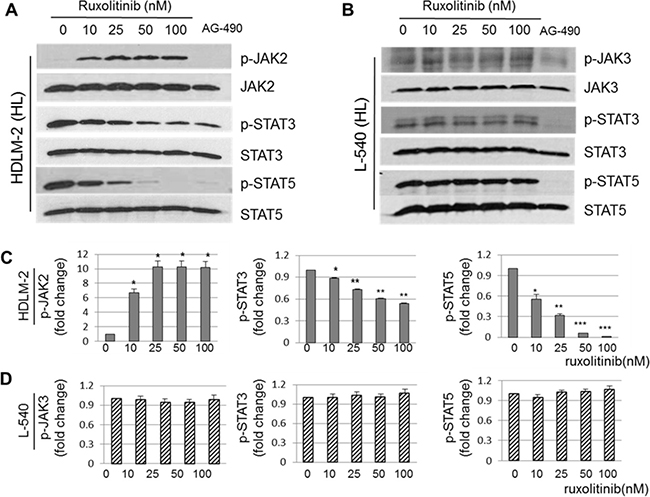
Figure 1: Selective activity of ruxolitinib on the JAK2/STAT signaling pathway in HL cells. Ruxolitinib (0, 10, 25, 50, and 100 nM) treated HDLM-2 and L-540 HL cells were collected and the lysates were separated in SDS-PAGE gels (A and B, respectively). The pan-JAK inhibitor, AG490 was used as a negative control treatment in each cell. Significant differences of p-JAK2, p-STAT3 and p-STAT5 with the quantification on the intensity of the bands by western blotting in HDLM-2 (C) (n = 3) and L-540 (D) (n = 3) cells. Data are represented as the mean ± SD. *p < 0.005; **p < 0.0005; ***p < 0.0001.
Effect of ruxolitinib on phosphorylation of STAT-3, -5 and -6 in HL and PMBL cells
To investigate the effects of ruxolitinib on the JAK2/STAT pathway in HL and PMBL cells, L-428 and HDLM-2 HL cells, and Karpas-1106P PMBL cells, each were treated with vehicle (dimethyl sulfoxide [DMSO]) alone or ruxolitinib (0, 25, 100, 400 and 1000 nM) for 48 hours and western blots were performed. Ruxolitinib significantly downregulated phosphorylation of STAT3, STAT5 and STAT6 in L-428 (Figure 2A and Figure 2D), HDLM-2 (Figure 2B and Figure 2E) following 25 nM ruxolitinib treatment compared to DMSO control whereas the expression level of total STAT3, STAT5 and STAT6 did not significantly change. Similarly, we observed significant inhibition of phosphorylation of STAT3 and STAT6 in 25 nM ruxolitinib treated Karpas-1106P cells (Figure 2C and Figure 2F). However, there was no significant inhibition of phosphorylation of STAT5 in Karpas-1106P following ruxolitinib treatment.
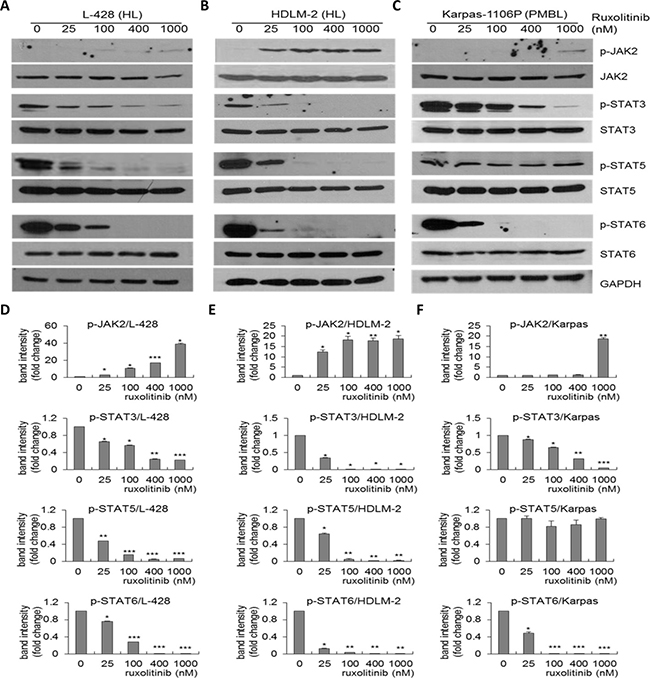
Figure 2: Downregulation of p-STAT3, p-STAT5 and p-STAT6 expression in HL and PMBL by ruxolitinib. Total cell lysates were collected treated L-428 (A) and HDLM-2 (B) HL, and Karpas-1106P PMBL cells (C) following ruxolitinib treatment (0, 25, 100, 400, and 1000 nM), and western blotting of protein expression of p-JAK2, JAK2, p-STAT3, STAT3, p-STAT5, STAT5, p-STAT6 and STAT6 were performed. (n = 3). Band intensity was quantified by western blot in ruxolitinib treated L-428 (D), *p < 0.0005;**p < 0.00005;***p < 0.00001) and HDLM-2 (E) HL, and Karpas-1106P PMBL cells (F). Data are represented as the mean ± SD of triplicates (paired t test). *p < 0.001;**p < 0.0001;***p < 0.00001.
Significant anti-proliferative in vitro effects of ruxolitinib in HL and PMBL cells
We examined the effect of ruxolitinib on HL and PMBL in vitro cell proliferation. Cells were treated with vehicle (DMSO) alone or ruxolitinib at various concentrations (0, 1, 10, and 100 uM) for 48 hours and cell proliferation was determined using MTS assay. There was a significant decrease in cell proliferation in the ruxolitinib treated L-428, HDLM-2 and Karpas-1106P vs control cells with DMSO treatment (20 - 79% reduction, p-value between p < 0.05 and p < 0.01) (Figure 3A). Specifically, there were significant decreases in cell proliferation with 20% reduction (p < 0.01) and 64% reduction (p < 0.01) with 10 and 100 uM of ruxolitinib treatment, respectively, in ruxolitinib treated L-428 cells. We also observed significant inhibition of cell proliferation with 38% (p < 0.05), 49% (p < 0.01) and 79% (p < 0.01) reduction in 1, 10 and 100 uM ruxolitinib treated HDLM-2 cells, respectively, compared to DMSO treated control cells. Ruxolitinib treated Karpas-1106P cells also showed significant inhibition with 20% (p < 0.05), 44% (p < 0.05) and 59% (0 < 0.05) reduction in 1, 10 and 100 uM ruxolitinib treated Karpas-1106P cells, respectively, compared to control cells following DMSO treatment.
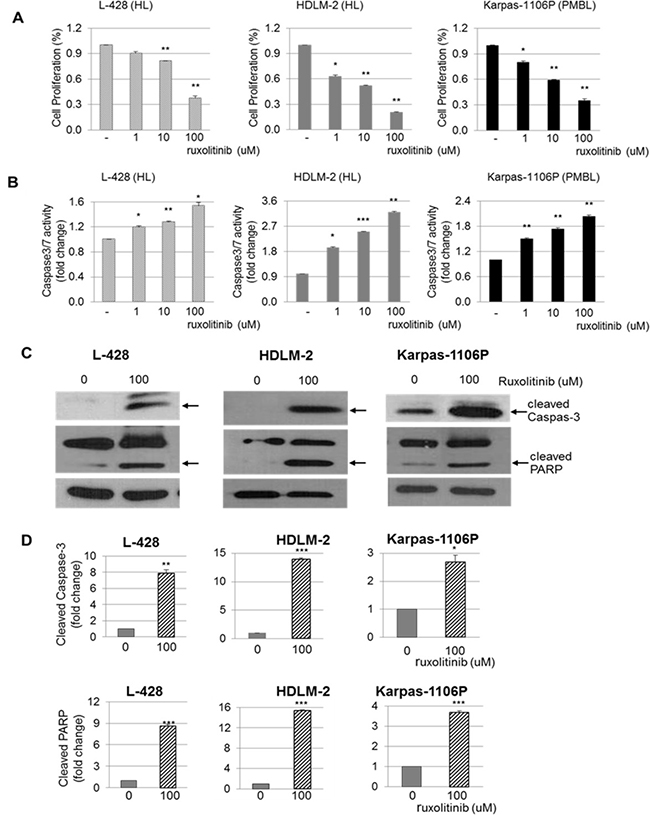
Figure 3: Cell proliferation, Caspase 3/7-dependent apoptosis and cleaved Caspas-3 and PARP in HL and PMBL cells. Cells were plated (1x105) into 48 well plates and cell growth was measured every 48 hours via MTS assay following ruxolitinib treatment (0, 1, 10 and 100 uM) in L-428 (left) and HDLM-2 (middle) HL cells, and Karpas-1106P PMBL cells (right) (A) (n = 3). Caspase 3/7 activity was measured using the Clarity Luminescence microplate reader (Biotek) in L-428 (left) and HDLM-2 (middle) HL cells, and Karpas-1106P PMBL cells (right) (B) (n = 3). Data are represented as the mean ± SD . *p < 0.05; **p < 0.01; ***p < 0.001. Cleaved Caspas-3 and PARP protein expression were measured by western blotting in L-428 (left) and HDLM-2 (middle) HL cells, and Karpas-1106P PMBL cells (right), and the arrow indicates cleaved Caspas-3 and PARP (C) (n = 3) and the intensity of the bands on gels by western blot was quantified in cells (D) (n = 3). Data are represented as the mean ± SD. *p < 0.005; **p < 0.001; ***p < 0.0005.
Significant increase in ruxolitinib-induced caspase-3/7 activity
To further investigate the mechanisms of the anti-proliferative activity of ruxolitinib, we determined the caspase-3/7 activity in ruxolitinib treated HL and PMBL cells. HL and PMBL cells were evaluated for fold change of caspase 3/7 activity over 48 hours with escalating doses of ruxolitinib (0, 1, 10 and 100 uM) (Figure 3B). There was a significant increase on caspase 3/7 activity at day 2 with >1.2 fold (p < 0.05), >1.3 fold (p = 0.01), and >1.5 fold (p < 0.05) increase at 1, 10, and 100 uM of ruxolitinib treatment, respectively, in L-428 cells compared to DMSO treated control cells. We also observed significant increases in caspase 3/7 activity in ruxolitinib treated HDLM-2 cells (>1.9 fold, p < 0.05; >2.5, p < 0.001; >3.2 fold, p < 0.01 in 1, 10 and 100 uM ruxolitinib, respectively). In addition, ruxolitinib significantly induced Caspase-3/7 activity in ruxolitinib treated Karpas-1106P cells (<1.5 fold, p < 0.01; >1.8 fold, p < 0.01; >2.0 fold, p < 0.01 in 1, 10 and 100 uM ruxolitinib, respectively) compared to control cells following DMSO treatment in Karpas-1106P cells. The cleavage of PARP (Figure 3C) and caspase-3 (Figure 3D), another hallmark of apoptosis, was significantly increased in ruxolitinib treated L-428 (p < 0.001 and p < 0.0005 at cleaved Caspase-3 and PARP, respectively), HDLM-2 (p < 0.0005 at both cleaved proteins) and Karpas-1106P cells (p < 0.005 and p < 0.0005 at cleaved caspase-3 and PARP, respectively) in the same dose-dependent manner following 100 uM of ruxolitinib treatment.
Significant changes in the expression of pro- and anti-apoptotic protein and gene expression in ruxolitinib treated HL and PMBL cells
We also examined the effects of ruxolitinib on the expression of pro- and anti-apoptotic proteins to enhance our understanding of the effects of ruxolitinib on apoptosis in HL and PMBL cells. Cells were treated with 100 uM ruxolitinib for 48 hours and the expression of anti-apoptotic proteins such as Bcl-2, Bcl-xL and Mcl-1 were analyzed. There was significant decrease of Bcl-2 (p < 0.005), Bcl-xL (p < 0.0005) and Mcl-1 (p < 0.0001) in 100uM ruxolitinib treated HDLM-2 cells compared to DMSO treated cells (Figure 4A). Additionally, we observed significant inhibition of the protein expression of Bcl-2 (p < 0.0001), Bcl-xL and Mcl-1 (p < 0.0005) following 100 uM ruxolitinib exposure to Karpas-1106P cells (Figure 4B). We also observed significant inhibition of expression of Bcl-2 mRNA in 100 uM ruxolitinib treated HDLM-2 (p < 0.0005) and Karpas-1106P cells (p < 0.0005) (Figure 4C). Interestingly, ruxolitinib also significantly inhibited the expression of Bcl-xL mRNA in 100 uM ruxolitinib treated HDLM-2 (p < 0.005) and Karpas-1106P cells (p < 0.001) (Figure 4D). These results suggest that ruxolitinib decreases HL and PMBL survival by in part inducing programmed cell death via down-regulating the expression of pro- and anti-apoptotic genes in HL and PMBL cells.
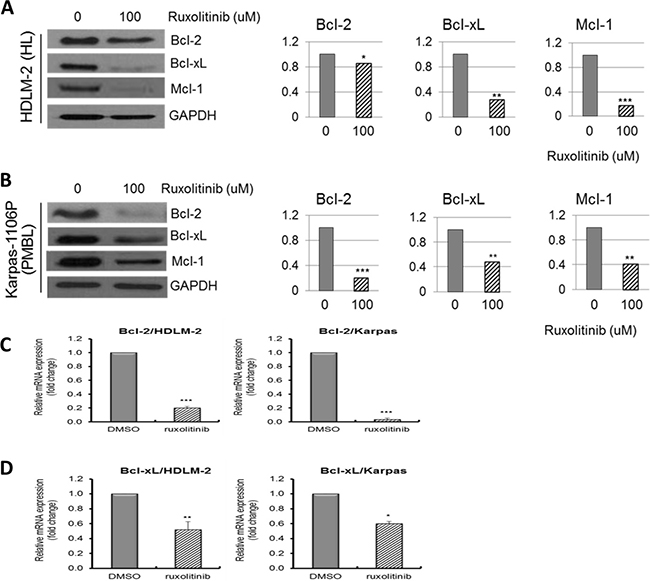
Figure 4: Downregulation of pro- and anti-apoptotic proteins and gene expression in HL and PMBL cells. Ruxolitinib significantly induced apoptosis through down-regulation of anti-apoptotic proteins, Bcl-2, Bcl-xL and Mcl-1 in ruxolitinib HDLM-2 (A) (n = 3) HL and Karpas-1106P PMBL cells (B) (n = 3). The intensity of the bands on gels by western blot was quantified in cells. Data are represented as the mean ± SD. *p < 0.005; **p < 0.0005; ***p < 0.0001. Real time qRT-PCR expression levels of Bcl-2 mRNA (C) (n = 3) and Bcl-xL mRNA (D) (n = 3) in ruxolitinib treated HDLM-2 HL cells and Karpas-1106P PMBL cells are shown. GAPDH was used as endogenous control for qRT-PCR normalization. *p < 0.01;**p < 0.005; ***p < 0.0005.
Effect of ruxolitinib on survival in PMBL and HL xenografted NSG mice
We further examined the efficacy of ruxolitinib in L-428 HL cells xenografted NSG mice and in Karpas-1106P PMBL cells xenografted NSG mice. Most importantly, ruxolitinib (45.0 mg/kg) treated L-428 (p < 0.03) and Karpas-1106P (p < 0.002) xenograft NSG mice had a significantly prolonged survival compared to control mice. Finally, we observed that ruxolitinib (45.0 mg/kg) treated L-428 xenografted NSG mice (n = 16) had significantly prolonged survival time with a median of 51.5 days compared to control mice (n = 15) (21 days, p = 0.0001) (Figure 5A), along with significant prolonged survival in ruxolitinib treated Karpas-1106P xenografted NSG mice (n = 16, median of 41.5 days) compared to control mice (n = 15, median of 20 days) (p < 0.0001) (Figure 5B). Furthermore, we demonstrated a significant decrease in L-428 HL and Karpas-1106 PMBL in bio-luminescence intensity (tumor burden) following ruxolitinib treatment in HL xenografted NSG mice (45.0 mg/kg, p < 0.05) at day 21 and 28 (Figure 5C), and Karpas-1106P PMBL cells xenografted NSG mice (45.0 mg/kg) (p < 0.05) at day 21 and 28 compared to control mice (Figure 5D).
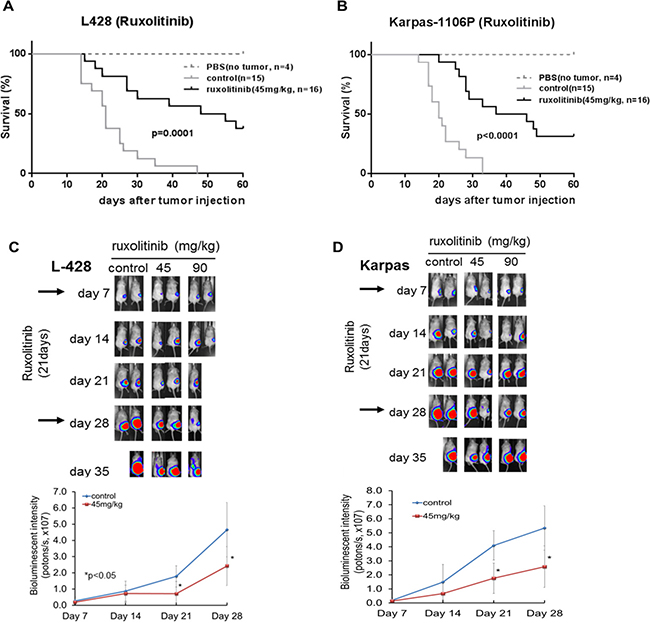
Figure 5: Survival in ruxolitinib treated HL and PMBL xenografted NSG mice. Survival rates were analyzed following ruxolitinib treatment (45 mg/kg) by Kaplan-Meier method and differences evaluated by log-rank test. A significant difference in ruxolitinib treated L428 HL cells xenografted NSG mice (n = 16) compared to control (n = 15, p = 0.0001) (A). Ruxolitinib treated Karpas-1106P xenografted NSG mice (n = 16) had significantly extended survival time compared to the control mice (n = 15, p < 0.0001) (B). A significant decrease in tumor luminescence intensity following ruxolitinib treated HL 28 (45 mg/kg, p < 0.05) (C) and PMBL (45 mg/kg) (p < 0.05) xenografted NSG mice (D) at day 21 and compared to control.*p < 0.05.
DISCUSSION
We investigated the efficacy of ruxolitinib in HL and PMBL cells and found significant in vitro anti-proliferative and proapoptotic effects of ruxolitinib treated L-428 and HDLM-2 HL cells and Karpas-1106P PMBL cells. Additionally, we observed significant downregulation in the phosphorylation of STAT3, STAT5 and STAT6 in HL, and p-STAT3 and STAT6 in PMBL following ruxolitinib treatment. Most importantly, ruxolitinib significantly decreased tumor burden and prolonged survival in HL and PMBL xenografted NSG mice.
HL and PMBL are highly curable diseases in first complete remission; however, patients with relapsed or refractory HL and PMBL have typically been associated with high failure rates despite aggressive chemo-radiotherapeutic approaches and/or autologous stem cell marrow transplantation [6, 33–35]. Furthermore, late effects in both HL and PMBL are significant, which affect both morbidity and mortality. Thus, novel salvage regimens are needed and less toxic new approaches in newly diagnosed patients are also warranted. Patients with the myelofibrosis (MF) carrying activating mutant allele of JAK2 exhibited rapid, significant reduction in disease burden and durable clinical benefit in patients following ruxolitinib [31, 36].
Ruxolitinib is an orally available pyrrolo [2,3-d] pyrimidine analog that exhibits subnanomolar affinity for JAK2 and JAK1 but exhibits lower activity against JAK3 [30]. Our observations in this study are consistent with these findings that ruxolitinib has selective activity of JAK2 against JAK3 protein, as demonstrated by inhibition of phosphorylation of the downstream STAT3 and STAT5 proteins at 10-100 nM ruxolitinib in HDLM-2 JAK2/STAT signaling vs L-540 JAK3/STAT signaling. In agreement with earlier studies, increased levels of JAK2 phosphorylation were observed despite inhibition of downstream STA3, STAT5 and STAT6 signaling in HL and PMBL cells. Similar paradoxical phosphorylation has been previously reported after incubation with JAK2 inhibitor Gö6976 and SB1518 in primary acute myeloid leukemia cells HEL92.1.7, [37, 38] and JAK inhibitor AZD 1480 in HL cells [39]. These findings could be related to the induction of negative-feed-back loops involving activating cytokines. Suppressor of cytokine signaling 1(SOCS1) works as the key effector of a classic negative feedback loop in JAK/STAT signaling. SOCS1 has been identified as a recurrently mutated tumor suppressor gene in HL and Karpas-1106P PMBL cells [40, 41]. Further, mutated SOCS1 may have contributed to the hyper-phosphorylation of JAK2 in HL and PMBL. Finally, ruxolitinib has also been demonstrated to inhibit the phosphorylation of ERK1/2, especially downstream to RAS-RAF-MEK-ERF signaling pathway [30, 42].
In this study, we demonstrated that ruxolitinib significantly inhibits HL and PMBL JAK/STAT signaling pathways. Furthermore, ruxolitinib significantly suppresses cell proliferation in HL and PMBL cells. This is consistent with inhibition of cell proliferation reported by Ju et al. in 4 HL cell lines [43]. The half maximal inhibitory concentration (IC50) is shown for L-428 (IC50 = 74.9 uM), HDLM-2 (IC50 = 15.7 uM) and Karpas-1106P (IC50 = 43.8 uM). Compared to the abrogation of downstream STAT phosphorylation by ruxolitinib at sub-micromolar and micromolar concentrations, relatively less sensitive anti-proliferative effects were observed. The relatively high IC50 of HL and PMBL cells imply that SOCS1 negative-feedback loop may activate secondary signaling pathways other than JAK2/STAT in HL and PMBL.
The maximum tolerated dose (MTD) for ruxolitinib in MF was established at 25 mg twice daily or 100 mg once daily [31, 44]. When investigated in children with relapsed/refractory malignancies in a phase 1 consortium study by the Children’s Oncology Group at five dose levels, ruxolitinib did not exceed the MTD; dose limiting toxicity (DLT) was observed in ≤1 of 6 patients per cohort [45]. In a phase I/II study of ruxolitinib in patients >14 years old with relapsed/refractory acute myeloid leukemia (AML), ruxolitinb was reasonably well tolerated in doses ranging from 50 mg twice daily to 200 mg twice daily [46]. In the present study, while we saw some effects at the lower concentration, the effect was augmented with higher dosing. These findings, with the relatively high IC50 in HL and PMBL cells in our study, suggest that higher dosing of ruxolitinib may be necessary for optimum activity, and may be well tolerated.
A kinetic analysis of caspase 3/7 activity following ruxolitinib therapy in HL and PMBL cells revealed that ruxolitinib significantly activates caspase 3/7 and induces apoptosis. This is in part presumably secondly to down- regulating the expression of anti-apoptotic factors, such as Bcl-2 and Bcl-xL in HL and PMBL cells. Our results suggest that therapy with ruxolitinib significantly decreased tumor burden and prolonged survival in HL and PMBL xenografted NSG mice. This is consistent with in vitro and in vivo JAK2 inhibition in HL and MLBL using fedratinib [26, 29]. Kim et al. demonstrated in a phase I study the safety and efficacy of ruxolitinib in adult patients with HL and PMBL, with disease control achieved in 54% (7/13) patients with heavily pretreated relapsed or refractory HL [47]. Most recently, Neste et al. demonstrated in a phase II study, a best ORR (1 CR/5 PR) of 19% (6/32) of ruxolitinib in adults with relapsed/refractory HL [48]. These results are consistent with our preclinical studies of mild to modest activity.
In summary, we have demonstrated that ruxolitinib, a JAK1/JAK2 inhibitor, significantly blocks in vitro STATs signaling, induces apoptosis and inhibits cell proliferation in vitro against HL and PMBL cell lines and in vivo prolonged survival in HL and PMBL xenografted NSG mice. Thus ruxolitinib has the potential to be an alternative therapeutic strategy in patients with high risk HL and PMBL. In PMBL, in addition to gains of chromosome 9p (including JAK2, JMJD2C, PDL1, PDL2), gain of chromosome 2p (including REL, BCL11A) has been consistently reported in up to 50% of patients [49, 50]. Gains of the same regions are also detected in HL [19, 51]. These genetic features suggest that there may be options to improve the therapeutic effect of ruxolitinib by combining it with other targeted therapies such as PD-1 or PDL-1 blockades or brentuximab-vedotin, in cHL [26, 43].
Future studies will be required to determine how best to incorporate this therapeutic agent in both newly diagnosed high risk and in relapsed/refractory patients with both HL and PMBL.
MATERIALS AND METHODS
Cell lines
The HL cell lines HDLM-2, L-428, L-540 and PMBL cell line Karpas-1106P cells were obtained from the German Collection of Microorganisms and Cell Cultures (DSMZ, Germany). All of these 4 cell lines were previously demonstrated to have a significant 9p24.1 and JAK2 amplification [25]. The L-428 cells were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (GIBCO BRL, Gaithersburg, MD, USA) 1% glutamine and penicillin-streptomycin in a humid environment of 5% CO2 at 37° C. The HDLM-2, L-540 and Karpas-1106P cell lines were cultured in RPMI 1640 medium supplemented with 20% heat-inactivated fetal bovine serum.
Cell proliferation assay
Cell growth was determined by the Cell titer 96 Aqueous One solution cell proliferation assay (MTS) (Promega, Madison, WI). At 24, 48 and 72 hours after treatment with INC424 or DMSO, MTS reagent was added and cells were incubated for 1 hour at 37° C. Cell proliferation was measured by OD 490 nm using Multilabel Counter (Perkin Elmer, Massachusetts, USA).
Caspase 3/7 assay
Caspase 3/7 activity was directly measured at 24, 48 and 72 hours after treatment using Caspase-Glo 3/7 Activity kit (Promega) as per the manufacturer’s protocol. Briefly, the cells (5 × 105/ml) were seeded into 24-well plated and treated with vehicle (DMSO) alone or ruxolitinib at various concentrations for 48 and 72 hours, Caspase-Glo reagent was added and cells were incubated for 1 hour at room temperature in the dark. Relative light intensity was measured in each well using Clarity Luminescence microplate reader (BioTek, Vermont, USA).
Reagents and antibodies
Ruxolitinib (INC424) was generously provided by Incyte Corporation (Wilmington, DE, USA. AG-490). Antibodies specific for phospho-JAK3, JAK3, STAT3, STAT5, were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies specific for phospho-STAT3, phospho-STAT5, phospho-STAT6, STAT6, phospho-JAK2, JAK2, Bcl-xL, Bcl-2, Mcl-1, Caspas-3, poly adenosine diphosphate ribose polymerase (PARP) were purchased from Cell Signaling Technology (Cambridge, MA, USA).
Western blot analysis
Cell pellets were suspended in a lysis buffer containing 50 mmol/l Tris–HCl, pH 7.4, 350 mmol/l NaCl, 1% TritonX-100, 0.5% Nonidet P-40, 10% glycerol, 0.1% sodium dodecyl sulfate (SDS), 1 mmol/l EDTA, 1 mmol/l EGTA, 1 mmol/l Na3VO4, 1 mmol/l phenylmethylsulphonyl fluoride (PMSF) and phosphatase inhibitor cocktails on ice. Whole-cell extracts were resolved on SDS-PAGE, transferred to nitrocellulose membrane, and probed with appropriate antibodies. Membranes were blocked in 5% non-fat dried milk in Tris-buffered saline (pH 7.4) containing 0.1% Tween 20 (TBST) for 1 hour and subsequently incubated with primary antibodies diluted in TBST at 4° C overnight. Membranes were then probed with horseradish Peroxidase-conjugated secondary antibodies and developed using enhanced chemiluminescence (ECL) reagent (GE Healthcare Bio-Sciences, Piscataway, NJ, USA). Band intensities on SDS-PAGE gel were measured using ImageJ software program [52].
Quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR)
Total RNA was prepared using Trizol reagent (Invitrogen) according to the manufacturer’s directions, and cDNA was synthesized using qScript™ cDNA Synthesis Kit (Quantas) using 1ug of RNA. The qRT-PCR was performed using CFX96 Real-time system (Bio-rad) and SsoFast™ EvaGreen® Supermix (Bio-rad) as PCR reagents. Relative quantification (ddCt) of mRNA expression of genes was determined by normalizing to the housekeeping gene (GAPDH). Primers used in qRT-PCR are previously described [53].
In vivo xenograft mouse models
The experimental animal protocols, procedures and care were approved by the Institutional Animal Care and Use Committee at New York Medical College (NYMC), Valhalla, NY, USA. Kapas-1106P PMBL cell line and L-428 HL cell line were stably transfected with a firefly luciferase expression plasmid (ffluc-zeo), kindly provided by Laurence Cooper MD, PhD, and then stable clones were selected under zeocin (Invitrogen) selection. The mice were γ-irradiated (2.5 Gy) 1 day before tumor cell transplantation. Six- to eight- week-old female NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice (The Jackson laboratory, Bar Harbor, ME) were subcutaneously injected with 1 × 106 ffluc-zeo Karpas-1106P or L-428 cells. The tumor burden was verified by bioluminescence imaging (BLI) system using the Xenogen IVIS-200 (Caliper Life Sciences) for up to 50 days as we have previously described [54]. Mice were orally gavaged with either 0.5% methyl cellulose vehicle or ruxolitinib (45.0 mg/kg) for 21 days. Tumor progression was monitored at day 7 and once every week by BLI. The survival rate was analyzed using humane endpoints and all mice were sacrificed when tumor size was greater than 2.0 cm3 ([length × width2]/2]) or with signs of ulceration, or if mice were moribund.
Statistical analysis
Data obtained from independent experiments are represented as means ± standard deviation (SD) and significant differences between two groups were determined by using Student’s t-test. Statistical analysis was performed using a two-tailed Student’s t test. P values less than 0.05 were considered significant. In vivo study, survival rates were analyzed by the Kaplan-Meier method and differences evaluated by log-rank test using the Prism Version 6.0 software.
Abbreviations
AML: acute myeloid leukemia; AYA: adolescents and young adults; BID: bis in die (twice daily); BLI: bioluminescence imaging; DLBCL: diffuse large B-cell lymphoma; DLT: dose limiting toxicity; DMSO: dimethyl sulfoxide; ECL: enhanced chemiluminescence; EFS: event-free survival; Ffluc: firefly luciferase; HL: Hodgkin lymphoma; JAK: Janus Kinase; MF: myelofibrosis; MLBL: mediastinal large B-cell lymphoma; MPNs: myeloproliferative neoplasms; MTD: maximum tolerated dose; NFKB: nuclear factor kappa B; NSG : non-obese diabetic severe combined immunodeficiency gamma; NYMC: New York Medical College; PDL-1: programmed death ligand-1; PMBL: primary mediastinal large B-cell lymphoma; qRT-PCR: Quantitative reverse-transcriptase polymerase chain reaction; SD: Standard deviation; SOCS1: Suppressor of cytokine signaling 1; STAT: signal transducer and activator of transcription; TBST: Tris-buffered saline Tween.
Author contributions
SL organized, designed and performed the experiments, analyzed the data and wrote the manuscript. CY performed experiments, analyzed the data and wrote the manuscript. TS, JH, EM, JA, and CV reviewed and revised the manuscript. MSC designed the study, analyzed the data and wrote the manuscript.
ACKNOWLEDGMENTS
The authors would like to thank Virginia Moore for her expert assistance in the preparation of this manuscript.
CONFLICTS OF INTEREST
All authors declare no conflicts of interest.
FUNDING
Funding was provided by grants from the Pediatric Cancer Research Foundation, Children’s Cancer Fund, Marisa Fund and St. Baldrick Foundation (MSC).
REFERENCES
1. Abramson JS, Shipp MA. Advances in the biology and therapy of diffuse large B-cell lymphoma: moving toward a molecularly targeted approach. Blood. 2005; 106:1164–74.
2. Rosenwald A, Wright G, Leroy K, Yu X, Gaulard P, Gascoyne RD, Chan WC, Zhao T, Haioun C, Greiner TC, Weisenburger DD, Lynch JC, Vose J, et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med. 2003; 198:851–62.
3. Glaser SL, Jarrett RF. The epidemiology of Hodgkin’s disease. Baillieres Clin Haematol. 1996; 9:401–16.
4. Hochberg J, Waxman IM, Kelly KM, Morris E, Cairo MS. Adolescent non-Hodgkin lymphoma and Hodgkin lymphoma: state of the science. Br J Haematol. 2009; 144:24–40.
5. Brenner H, Gondos A, Pulte D. Ongoing improvement in long-term survival of patients with Hodgkin disease at all ages and recent catch-up of older patients. Blood. 2008; 111:2977–83.
6. Harris RE, Termuhlen AM, Smith LM, Lynch J, Henry MM, Perkins SL, Gross TG, Warkentin P, Vlachos A, Harrison L, Cairo MS. Autologous peripheral blood stem cell transplantation in children with refractory or relapsed lymphoma: results of Children’s Oncology Group study A5962. Biol Blood Marrow Transplant. 2011; 17:249–58.
7. Satwani P, Jin Z, Martin PL, Bhatia M, Garvin JH, George D, Chaudhury S, Talano J, Morris E, Harrison L, Sosna J, Peterson M, Militano O, et al. Sequential myeloablative autologous stem cell transplantation and reduced intensity allogeneic hematopoietic cell transplantation is safe and feasible in children, adolescents and young adults with poor-risk refractory or recurrent Hodgkin and non-Hodgkin lymphoma. Leukemia. 2015; 29:448–55.
8. Place AE, Frederick NN, Sallan SE. Therapeutic approaches to haematological malignancies in adolescents and young adults. Br J Haematol. 2014; 164:3–14.
9. Wood WA, Lee SJ. Malignant hematologic diseases in adolescents and young adults. Blood. 2011; 117:5803–15.
10. Swerdlow S, Campo E, Harris NL. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissue. Geneva, Switzerland: World Health Organization. International Agency for Research on Cancer. 2008: 1-439.
11. Jaffe ES, Harris NL, Stein H, Vardiman JW. Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. WHO Classification of Tumours. International Agency for Research on Cancer; 2001:3.
12. Bhatt VR, Mourya R, Shrestha R, Armitage JO. Primary mediastinal large B-cell lymphoma. Cancer Treat Rev. 2015; 41:476–85.
13. Grant C, Dunleavy K, Eberle FC, Pittaluga S, Wilson WH, Jaffe ES. Primary mediastinal large B-cell lymphoma, classic Hodgkin lymphoma presenting in the mediastinum, and mediastinal gray zone lymphoma: what is the oncologist to do? Curr Hematol Malig Rep. 2011; 6:157–63.
14. Lones MA, Perkins SL, Sposto R, Kadin ME, Kjeldsberg CR, Wilson JF, Cairo MS. Large-cell lymphoma arising in the mediastinum in children and adolescents is associated with an excellent outcome: a Children’s Cancer Group report. J Clin Oncol. 2000; 18:3845–53.
15. Burkhardt B, Zimmermann M, Oschlies I, Niggli F, Mann G, Parwaresch R, Riehm H, Schrappe M, Reiter A, and BFM Group. The impact of age and gender on biology, clinical features and treatment outcome of non-Hodgkin lymphoma in childhood and adolescence. Br J Haematol. 2005; 131:39–49.
16. Gerrard M, Waxman IM, Sposto R, Auperin A, Perkins SL, Goldman S, Harrison L, Pinkerton R, McCarthy K, Raphael M, Patte C, Cairo MS, and French-American-British/Lymphome Malins de Burkitt 96 (FAB/LMB 96) International Study Committee. Outcome and pathologic classification of children and adolescents with mediastinal large B-cell lymphoma treated with FAB/LMB96 mature B-NHL therapy. Blood. 2013; 121:278–85.
17. Cairo MS, Sposto R, Gerrard M, Auperin A, Goldman SC, Harrison L, Pinkerton R, Raphael M, McCarthy K, Perkins SL, Patte C. Advanced stage, increased lactate dehydrogenase, and primary site, but not adolescent age (>/= 15 years), are associated with an increased risk of treatment failure in children and adolescents with mature B-cell non-Hodgkin’s lymphoma: results of the FAB LMB 96 study. J Clin Oncol. 2012; 30:387–93.
18. Barth TF, Bentz M, Leithauser F, Stilgenbauer S, Siebert R, Schlotter M, Schlenk RF, Dohner H, Moller P. Molecular-cytogenetic comparison of mucosa-associated marginal zone B-cell lymphoma and large B-cell lymphoma arising in the gastro-intestinal tract. Genes Chromosomes Cancer. 2001; 31:316–25.
19. Joos S, Granzow M, Holtgreve-Grez H, Siebert R, Harder L, Martin-Subero JI, Wolf J, Adamowicz M, Barth TF, Lichter P, Jauch A. Hodgkin’s lymphoma cell lines are characterized by frequent aberrations on chromosomes 2p and 9p including REL and JAK2. Int J Cancer. 2003; 103:489–95.
20. Joos S, Menz CK, Wrobel G, Siebert R, Gesk S, Ohl S, Mechtersheimer G, Trumper L, Moller P, Lichter P, Barth TF. Classical Hodgkin lymphoma is characterized by recurrent copy number gains of the short arm of chromosome 2. Blood. 2002; 99:1381–7.
21. MacLeod RA, Spitzer D, Bar-Am I, Sylvester JE, Kaufmann M, Wernich A, Drexler HG. Karyotypic dissection of Hodgkin’s disease cell lines reveals ectopic subtelomeres and ribosomal DNA at sites of multiple jumping translocations and genomic amplification. Leukemia. 2000; 14:1803–14.
22. Nedomova R, Papajik T, Prochazka V, Indrak K, Jarosova M. Cytogenetics and molecular cytogenetics in diffuse large B-cell lymphoma (DLBCL). Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2013; 157:239–47.
23. Guiter C, Dusanter-Fourt I, Copie-Bergman C, Boulland ML, Le Gouvello S, Gaulard P, Leroy K, Castellano F. Constitutive STAT6 activation in primary mediastinal large B-cell lymphoma. Blood. 2004; 104:543–9.
24. Gunawardana J, Chan FC, Telenius A, Woolcock B, Kridel R, Tan KL, Ben-Neriah S, Mottok A, Lim RS, Boyle M, Rogic S, Rimsza LM, Guiter C, et al. Recurrent somatic mutations of PTPN1 in primary mediastinal B cell lymphoma and Hodgkin lymphoma. Nat Genet. 2014; 46:329–35.
25. Green MR, Monti S, Rodig SJ, Juszczynski P, Currie T, O’Donnell E, Chapuy B, Takeyama K, Neuberg D, Golub TR, Kutok JL, Shipp MA. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood. 2010; 116:3268–77.
26. Hao Y, Chapuy B, Monti S, Sun HH, Rodig SJ, Shipp MA. Selective JAK2 inhibition specifically decreases Hodgkin lymphoma and mediastinal large B-cell lymphoma growth in vitro and in vivo. Clin Cancer Res. 2014; 20:2674–83.
27. Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, Catalano JV, Deininger MW, Miller CB, Silver RT, Talpaz M, Winton EF, Harvey JH Jr, et al, and COMFORT-I investigators. Efficacy, safety, and survival with ruxolitinib in patients with myelofibrosis: results of a median 3-year follow-up of COMFORT-I. Haematologica. 2015; 100:479–88.
28. Jamieson C, Hasserjian R, Gotlib J, Cortes J, Stone R, Talpaz M, Thiele J, Rodig S, Pozdnyakova O. Effect of treatment with a JAK2-selective inhibitor, fedratinib, on bone marrow fibrosis in patients with myelofibrosis. J Transl Med. 2015; 13: 294.
29. Pardanani A, Harrison C, Cortes JE, Cervantes F, Mesa RA, Milligan D, Masszi T, Mishchenko E, Jourdan E, Vannucchi AM, Drummond MW, Jurgutis M, Kuliczkowski K, et al. Safety and Efficacy of Fedratinib in Patients With Primary or Secondary Myelofibrosis: A Randomized Clinical Trial. JAMA Oncol. 2015; 1:643–51.
30. Quintas-Cardama A, Vaddi K, Liu P, Manshouri T, Li J, Scherle PA, Caulder E, Wen X, Li Y, Waeltz P, Rupar M, Burn T, Lo Y, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010; 115:3109–17.
31. Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, Estrov Z, Fridman JS, Bradley EC, Erickson-Viitanen S, Vaddi K, Levy R, Tefferi A. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010; 363:1117–27.
32. Mascarenhas J, Hoffman R. A comprehensive review and analysis of the effect of ruxolitinib therapy on the survival of patients with myelofibrosis. Blood. 2013; 121:4832–7.
33. Popat U, Przepiork D, Champlin R, Pugh W, Amin K, Mehra R, Rodriguez J, Giralt S, Romaguera J, Rodriguez A, Preti A, Andersson B, Khouri I, et al. High-dose chemotherapy for relapsed and refractory diffuse large B-cell lymphoma: mediastinal localization predicts for a favorable outcome. J Clin Oncol. 1998; 16:63–9.
34. Baker KS, Gordon BG, Gross TG, Abromowitch MA, Lyden ER, Lynch JC, Vose JM, Armitage JO, Coccia PF, Bierman PJ. Autologous hematopoietic stem-cell transplantation for relapsed or refractory Hodgkin’s disease in children and adolescents. J Clin Oncol. 1999; 17:825–31.
35. Bradley MB, Cairo MS. Stem cell transplantation for pediatric lymphoma: past, present and future. Bone Marrow Transplant. 2008; 41:149–58.
36. Pardanani A, Gotlib JR, Jamieson C, Cortes JE, Talpaz M, Stone RM, Silverman MH, Gilliland DG, Shorr J, Tefferi A. Safety and efficacy of TG101348, a selective JAK2 inhibitor, in myelofibrosis. J Clin Oncol. 2011; 29:789–96.
37. Grandage VL, Everington T, Linch DC, Khwaja A. Go6976 is a potent inhibitor of the JAK 2 and FLT3 tyrosine kinases with significant activity in primary acute myeloid leukaemia cells. Br J Haematol. 2006; 135:303–16.
38. Hart S, Goh KC, Novotny-Diermayr V, Tan YC, Madan B, Amalini C, Ong LC, Kheng B, Cheong A, Zhou J, Chng WJ, Wood JM. Pacritinib (SB1518), a JAK2/FLT3 inhibitor for the treatment of acute myeloid leukemia. Blood Cancer J. 2011; 1:e44.
39. Derenzini E, Lemoine M, Buglio D, Katayama H, Ji Y, Davis RE, Sen S, Younes A. The JAK inhibitor AZD1480 regulates proliferation and immunity in Hodgkin lymphoma. Blood Cancer J. 2011; 1:e46.
40. Weniger MA, Melzner I, Menz CK, Wegener S, Bucur AJ, Dorsch K, Mattfeldt T, Barth TF, Moller P. Mutations of the tumor suppressor gene SOCS-1 in classical Hodgkin lymphoma are frequent and associated with nuclear phospho-STAT5 accumulation. Oncogene. 2006; 25:2679–84.
41. Melzner I, Weniger MA, Bucur AJ, Bruderlein S, Dorsch K, Hasel C, Leithauser F, Ritz O, Dyer MJ, Barth TF, Moller P. Biallelic deletion within 16p13.13 including SOCS-1 in Karpas1106P mediastinal B-cell lymphoma line is associated with delayed degradation of JAK2 protein. Int J Cancer. 2006; 118:1941–4.
42. Cante-Barrett K, Spijkers-Hagelstein JA, Buijs-Gladdines JG, Uitdehaag JC, Smits WK, van der Zwet J, Buijsman RC, Zaman GJ, Pieters R, Meijerink JP. MEK and PI3K-AKT inhibitors synergistically block activated IL7 receptor signaling in T-cell acute lymphoblastic leukemia. Leukemia. 2016; 30:1832–43.
43. Ju W, Zhang M, Wilson KM, Petrus MN, Bamford RN, Zhang X, Guha R, Ferrer M, Thomas CJ, Waldmann TA. Augmented efficacy of brentuximab vedotin combined with ruxolitinib and/or Navitoclax in a murine model of human Hodgkin’s lymphoma. Proc Natl Acad Sci U S A. 2016; 113:1624–9.
44. Shi JG, Chen X, McGee RF, Landman RR, Emm T, Lo Y, Scherle PA, Punwani NG, Williams WV, Yeleswaram S. The pharmacokinetics, pharmacodynamics, and safety of orally dosed INCB018424 phosphate in healthy volunteers. J Clin Pharmacol. 2011; 51:1644–54.
45. Loh ML, Tasian SK, Rabin KR, Brown P, Magoon D, Reid JM, Chen X, Ahern CH, Weigel BJ, Blaney SM. A phase 1 dosing study of ruxolitinib in children with relapsed or refractory solid tumors, leukemias, or myeloproliferative neoplasms: A Children’s Oncology Group phase 1 consortium study (ADVL1011). Pediatr Blood Cancer. 2015; 62:1717–24.
46. Pemmaraju N, Kantarjian H, Kadia T, Cortes J, Borthakur G, Newberry K, Garcia-Manero G, Ravandi F, Jabbour E, Dellasala S, Pierce S, Verstovsek S. A phase I/II study of the Janus kinase (JAK)1 and 2 inhibitor ruxolitinib in patients with relapsed or refractory acute myeloid leukemia. Clin Lymphoma Myeloma Leuk. 2015; 15:171–6.
47. Kim SJ, Kang HJ, Dong-Yeop S, Lee HS, Oh SY, Shin HJ, Yoon DH, Hong JY, Kong JH, Sakamoto K, Ko YH, Takeuchi K, Suh C, Kim WS. The Efficacy of JAK2 Inhibitor in Heavily Pretreated Classical Hodgkin Lymphoma: A Prospective Pilot Study of Ruxolitinib in Relapsed or Refractory Classical Hodgkin Lymphoma and Primary Mediastinal Large B-Cell Lymphoma. Blood. 2016; 128:1820.
48. Van Den Neste EW, Andre M, Gastinne T, Stamatoullas A, Haloun C, Belhabri A, Reman O, Casasnovas O, Chesquieres H, Verhoef G, Claessen MJ, Poirel HA, Chopin MC, et al. A Lysa phase II study of oral JAK1/2 inhibitor ruxolitinib in advanced relapsed/refractory (R/R) Hodgkin lymphoma (HL). Blood. 2016; 128:4160.
49. Weniger MA, Pulford K, Gesk S, Ehrlich S, Banham AH, Lyne L, Martin-Subero JI, Siebert R, Dyer MJ, Moller P, Barth TF. Gains of the proto-oncogene BCL11A and nuclear accumulation of BCL11A(XL) protein are frequent in primary mediastinal B-cell lymphoma. Leukemia. 2006; 20:1880–2.
50. Weniger MA, Gesk S, Ehrlich S, Martin-Subero JI, Dyer MJ, Siebert R, Moller P, Barth TF. Gains of REL in primary mediastinal B-cell lymphoma coincide with nuclear accumulation of REL protein. Genes Chromosomes Cancer. 2007; 46:406–15.
51. Martin-Subero JI, Gesk S, Harder L, Sonoki T, Tucker PW, Schlegelberger B, Grote W, Novo FJ, Calasanz MJ, Hansmann ML, Dyer MJ, Siebert R. Recurrent involvement of the REL and BCL11A loci in classical Hodgkin lymphoma. Blood. 2002; 99:1474–7.
52. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012; 9:671–5.
53. Lee S, Luo W, Shah T, Yin C, O’Connell T, Chung TH, Perkins SL, Miles RR, Ayello J, Morris E, Harrison L, van de Ven C, Cairo MS. The effects of DLEU1 gene expression in Burkitt lymphoma (BL): potential mechanism of chemoimmunotherapy resistance in BL. Oncotarget. 2017; 8:27839–53. https://doi.org/10.18632/oncotarget.15711.
54. Chu Y, Hochberg J, Yahr A, Ayello J, van de Ven C, Barth M, Czuczman M, Cairo MS. Targeting CD20+ Aggressive B-cell Non-Hodgkin Lymphoma by Anti-CD20 CAR mRNA-Modified Expanded Natural Killer Cells In Vitro and in NSG Mice. Cancer Immunol Res. 2015; 3:333–44.