
INTRODUCTION
Philadelphia chromosome–like B-cell lymphoblastic leukemia (Ph-like B-ALL) is a B-ALL subgroup of patients whose transcriptional profile resembles BCR-ABL1-positive B-ALL but does not contain an actual BCR-ABL1 fusion [2]. This subgroup comprises 10-20% of children with high-risk pediatric B-ALL and nearly 30% of young adults with B-ALL [3]. Patients with Ph-like B-ALL experience high rate of relapse and have poor overall survival [4]. Novel therapeutic strategies are urgently needed to improve outcomes.
The vast majority of patients with Ph-like B-ALL harbor kinase-activating alterations, including chromosomal rearrangement, structural variations, and sequence mutations [3, 5], resulting in cytokine-independent cell proliferation and activation of multiple cell signaling pathways. Rearrangement of cytokine receptor-like factor 2 (CRLF2-re), resulting in CRLF2 overexpression, is the most frequent alteration, occurring in about 50% of Ph-like B-ALL cases [3, 4]. The rearrangement links the full-length CRLF2 coding region to transcription control elements driving elevated expression. Rearrangements include translocations to immunoglobulin heavy chain (IGH) locus enhancer elements or to the P2RY8 promoter through intra-chromosomal deletion [6]. Approximately half of CRLF2-re cases harbor focal deletions or sequence mutations in JAK pathway–associated genes, including IL7R, FLT3, SH2B3, JAK1, and most frequently JAK2 [7]. Activation of the JAK/STAT signaling pathway promotes cell survival, proliferation, and migration.
Constitutive activity of the JAK/STAT pathway may also facilitate upregulation of the downstream PI3K/AKT/mTOR pathway contributing towards ALL pathogenesis. The PI3K/AKT/mTOR signaling network is one of the most frequently activated pathways in human cancer and is subject to complex cross-talk and feedback. mTOR has two different kinase complexes: mTOR complex 1 (mTORC1, with RAPTOR) and mTORC2 (with RICTOR) [8]. The mTORC1 kinase complex binds RAPTOR to phosphorylate downstream proteins S6 kinase (S6K) and 4E-BP1, in turn regulating the cap-dependent mRNA translation of proteins that are critical for cell cycle progression from G1 to S phase [9]. The mTORC2 kinase complex binds RICTOR to phosphorylate AKT on Ser473, regulating cell survival [8].
Type I JAK2 inhibitors such as ruxolitinib bind within the ATP-binding pocket of the active conformation of JAK2, to compete with ATP, thereby inhibiting the phosphorylation of STAT5 and p-STAT5 signaling within tumor cells. However, such effects have limited effect on survival of ALL cell lines and in patient-derived xenograft (PDX) models [10, 11]. Recent work has already proposed favorable combination of JAK2 inhibition and PI3K/mTOR inhibition [11]. However, first generation mTOR inhibitors such as rapamycin only partially inhibit mTORC1, thus only transiently inhibiting 4E-BP1 phosphorylation [12] and do not affect mTORC2/AKT. On the contrary, inhibition of S6K can activate p-AKT through a feedback mechanism which promotes cell survival [13].
This led to our hypothesis that combined inhibition of both JAK/STAT and mTOR pathways with next generation kinase inhibitors may be beneficial in patients with CRLF2-re or JAK2 driven Ph-like B-ALL. To test this hypothesis, we utilized BBT594, a dihydroindole pharmacophore type II inhibitor originally developed as an inhibitor of the T315I BCR-ABL mutant, which also inhibited other isoforms of BCR-ABL and receptor tyrosine kinase such as JAK2 [1] and RET [14]. As a type II inhibitor, upon binding BBT594 extends to a hydrophobic site adjacent to the ATP-binding site of JAK2 thereby stabilizing the kinase in an inactive conformation, preventing phosphorylation of the activation loop [15]. Transphosphorylation of JAK2 by JAK1 or TYK2 does not confer resistance to BBT594 [16], and CHZ868, a benzimidazole analogue JAK2 type II inhibitor, was recently shown to have superior activity in blocking JAK2 signaling in Ph-like cell line model BaF/3 expressing Ph-like related genes and murine transgenic Ph-like ALL model co-expressing CRLF2 and mutant JAK2 R683G [15]. We further utilized second generation mTOR inhibitor AZD2014 that targets both mTORC1 and mTORC2. In this study, we examined combined efficacy of these agents in human and murine engineered cell lines harboring rearranged CRLF2 and/or JAK2 mutation and in patient-derived Ph-like xenografts in vitro and in vivo.
RESULTS
Combined inhibition of JAK2 and mTOR induced synergistic inhibition of cell growth in cell lines with CRLF2 rearrangement and JAK2 mutation
We first evaluated the effects of JAK2 and mTOR inhibitors in two JAK2-driven human Ph-like B-ALL cell lines: MHH-CALL-4, which harbors IGH-CRLF2 translocation and the JAK2 I682F mutation, and MUTZ-5 with IGH-CRLF2 translocation and the JAK2 R683G mutation. Cell line REH, which harbors ETV6-RUNX1 translocation and is wild-type for CRLF2 and JAK2, served as a negative control. In the proliferation assay, ruxolitinib used at a concentration range between 0.025μM and 0.4μM had minimal or no effect on all tested cell lines (Supplementary Figure 1A-1B). In contrast, the same concentration range of type II JAK2 inhibitor BBT594 potently inhibited growth of JAK2-driven cell lines with an IC50 of 0.1 ± 0.005 μM in MHH-CALL-4 and 0.1± 0.008 μM in MUTZ-5 (Figure 1A-1B). In non–JAK2-driven REH cells, only high concentrations exceeding 1μM of BB T594 showed minimal growth-inhibitory effects, possible due to off-target activity (Supplementary Figure 1C). Rapamycin alone or when combined with ruxolitinib had very modest growth-inhibitory effects in MHH-CALL-4 and MUTZ-5 cells (Supplementary Figure 1A-1B). In contrast, AZD2014 caused more pronounced growth inhibition in all three cell lines, and the combination of ruxolitinib and AZD2014 was synergistic in JAK2-mutated cells (CIs 0.7±0.3 and 0.3±0.1, respectively) but not in REH cells (Figure 1A-1B, Supplementary Figure 1A-1C). BBT594 showed additive/moderate synergy when combined with rapamycin (CIs 0.4± 0.03 and 0.9± 0.02) (Supplementary Figure 1A-1B), and strong synergy upon combined use with AZD2014 (CIs 0.5±0.02 and 0.7± 0.11) (Figure 1A-1B) (summarized in Table 1).
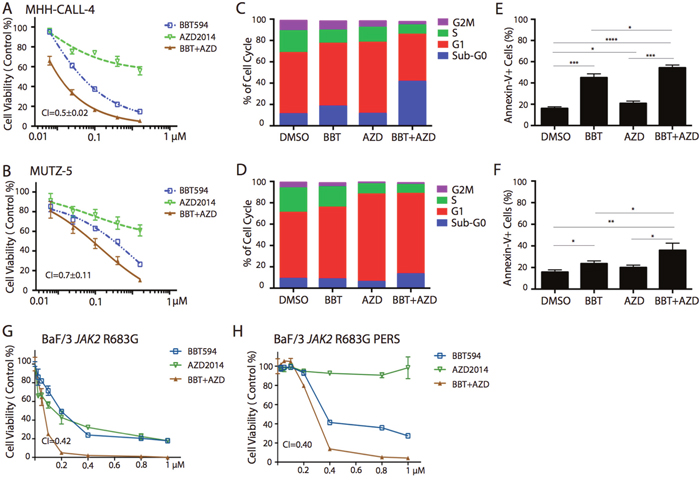
Figure 1: In vitro antileukemia efficacy of dual JAK2 and mTOR inhibition in Ph-like B-ALL cell lines. MHH-CALL4 and MUTZ-5 cells were treated with 0.25-0.8μM BBT594 (BBT), AZD2014 (AZD), or combinations for 72 h, then the numbers of viable cells were determined by CTG assay. The cell inhibition curves were plotted with the live cell number normalized to those of DMSO-treated controls: (A) MHH-CALL4 cells, (B) MUTZ-5 cells. Treated cells were fixed in 90% methanol and then stained with propidium iodine to determine the effects of the treatments on the cell cycle by flow cytometry: (C) MHH-CALL4 cells, (D) MUTZ-5 cells. The cells were stained with annexin V/DAPI to quantify cell apoptosis by flow cytometry: (E) MHH-CALL4 cells, (F) MUTZ-5 cells. * p<0.05, ** p<0.005, *** p<0.0005 as determined by unpaired Student t-test. (G, H) BaF/3 cells expressing the Ph-like B-ALL-associated JAK2 R683G parental (G) and ruxolitinib-“persistent” cells (H) were treated with 0.25-1.0μM BBT594, AZD2014 or the combination for 72 h, and cell viability was analyzed by CTG assay. The viability of cells treated was selected inhibitors was to those of DMSO-treated controls, and expressed as % viable cells.
Table 1: Combination Index (CI) of the compound combinations in MHH-CALL-4 and MUTZ-5 cell lines
CI |
MHH-CALL-4 |
MUTZ-5 |
||
|---|---|---|---|---|
Rapamycin |
AZD2014 |
Rapamycin |
AZD2014 |
|
Ruxolitinib |
148.6±166.3 |
0.7±0.3 |
0.6±0.6 |
0.3±0.1 |
BBT594 |
0.4±0.03 |
0.5±0.02 |
0.9±0.02 |
0.7±0.11 |
Compared to the control REH cell line, in which only mTOR inhibitors affected cell cycle distribution, the combinations of a JAK2 inhibitor and an mTOR inhibitor arrested the MHH-CALL-4 and MUTZ-5 cells in the G1/sub-G0 phase with concomitant reductions in the percentages of cells in G2M/S phase (Figure 1C-1D, Supplementary Figure 1D-1F). While ruxolitinib alone or in combination with mTOR inhibitors failed to or induced only minimal cell death, BBT594 used as a single agent and more so in combination with AZD2014 induced modest apoptosis in MHH-CALL-4 and MUTZ-5 cells (Figure 1E-1F, Supplementary Figure 1G-1I).
Combined inhibition of JAK2 and mTOR induced synergistic inhibition in BaF/3 cell lines with Ph-like B-ALL–associated JAK mutations
We next compared efficacy of ruxolitinib or BBT594 in combination with AZD2014 in parental or ruxolitinib-“persistent” BaF/3 cells expressing the Ph-like B-ALL–associated JAK1 V658F mutation, the JAK2 R683G mutation, or co-expressing wild-type CRLF2 and the JAK2 R683S mutation, described previously [15, 17]. In these cells generated by chronic exposure to ruxolitinib, JAK2 is stabilized and reactivated secondary to heterodimeric association of JAK2 with JAK1/TYK2 and JAK2 transactivation [16]. Three parental BaF/3 cells engineered to express JAK1 V658F and JAK2 R683G mutations with or without CRLF2 were sensitive to inhibitors, and combined inhibition of JAK and mTOR synergistically inhibited cell growth in all except JAK2 R683G cells (Figure 1G and Supplementary Figure 2A-2C). As reported, ruxolitinib-“persistent” cells were resistant to up to 8μM ruxolitinib, while the median inhibitory concentration (IC50) was 0.2 to 0.8 μM in the parental cells. In turn, BaF/3 JAK1 V658F and BaF/3 CRLF2/JAK2 R683S (but not JAK2 R683G) “persistent” cells remained sensitive to mTOR C1/2 inhibitor AZD2014, which in combination with ruxolitinib induced higher apoptosis in these cells (p=0.01 and p=0.007) (Figure 1H and Supplementary Figure 2A-2C).
Furthermore, even though variably, ruxolitinib-“persistent” cells responded to BBT594, and there was synergistic cell inhibition when exposed to BBT594/AZD2014 combination (Figure 1H and Supplementary Figure 2A-2C). These data suggest that combinations of JAK2 and mTOR inhibitors induce synergistic antileukemia activity in Ph-like ALL cells, and that blockade of mTOR signaling can partially reverse resistance to ruxolitinib, further enhancing sensitivity to BBT594.
JAK2 and mTOR inhibitors reduce TSLP-stimulated signaling in Ph-like ALL cells
We next examined the effects of these inhibitors on components of JAK/STAT and PI3K/mTOR pathways using phospho-flow cytometry. In these experiments, we utilized a recombinant ligand of CRLF2, thymic stromal lymphopoietin (TSLP), to simulate conditions of the BM microenvironment [18]. TSLP robustly stimulated the phosphorylated (p)-rS6, p-ERK, and p-JAK2 signals and moderately induced the p-AKT, p-STAT5, and p-4E-BP1 signals in MHH-CALL-4 or MUTZ-5 cells (Figure 2A). Ruxolitinib inhibited TSLP-induced but not constitutive (baseline) p-JAK2 and reduced p-STAT5 below baseline levels. In turn, BBT594 fully inhibited both, constitutive and TSLP-induced p-JAK2 and inhibited p-STAT5 to the baseline level. Rapamycin inhibited p-rS6 and p-4E-BP1, but increased levels of p-AKT, while AZD2014 fully inhibited all three phospho-proteins. Neither rapamycin nor AZD2014 fully inhibited p-ERK, while both JAK2 inhibitors blocked TSLP-induced p-ERK activation. The combinations of ruxolitinib+rapamycin and ruxolitinib+AZD2014 fully inhibited all the measured signals except for p-JAK2. In comparison, the combinations of BBT594+ rapamycin and BBT594+ AZD2014 fully inhibited p-JAK2 and other signaling, albeit less potently reducing p-STAT5 level. In turn, rapamycin alone or in combination failed to fully inhibit p-4E-BP1, while this was successfully achieved by AZD2014 in combinations with either JAK2 inhibitor.
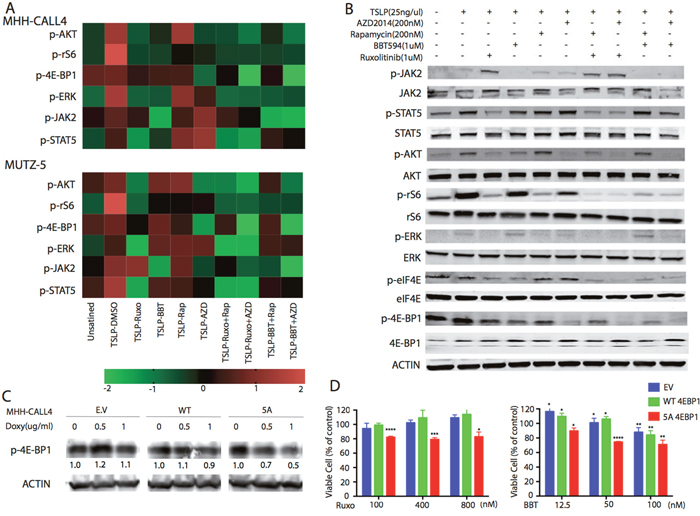
Figure 2: Inhibitory effects of dual JAK2 and mTOR inhibition on the PI3K/mTOR pathway. (A) Phospho-flow cytometry analysis demonstrated increased phosphorylation of JAK-2(Tyr1008), STAT5(Ty694)), ERK(T202/Y204) and AKT/pS6 [AKT(Ser473)],4E-BP1(T37/46), and rS6(S240/244) after stimulation with TSLP(25ng/mL) for 30 min. MHH-CALL4 and MUTZ-5 cells were treated with indicated compounds for 1 h, then the phosphorylation signals of PI3K/mTOR pathway proteins were determined by phospho-flow cytometry. Heat map data depict the changes in phosphoprotein levels by median fluorescence intensity. Color scale represents normalization (Z-score) of each condition for each signal. (B) MHH-CALL4 cells were stimulated with TSLP for 30min followed by treatment with ruxolitinib (Ruxo), BBT594 (BBT), rapamycin (Rap), or AZD2014 (AZD) or one of their combinations at the indicated doses for 1 h, after which the expression of PI3K/mTOR pathway proteins were probed by Western blot. (C) MHH-CALL4 cells were infected with empty vector (EV), with a wild-type (WT) or with mutant (5A) 4E-BP1 constructs, then treated with indicated concentrations of doxycycline (Doxy) for 72 h to induce p-4E-BP1 expression. Levels of p-4E-BP1 were determined by Western blot. (D) MHH-CALL4 cells transfected with 4E-BP1 constructs were treated with doxycycline (1μg/mL) for 72 h to induce 4E-BP1 and then with ruxolitinib (Ruxo) or BBT594 (BBT) for 72 h. Cell viability was analyzed by CTG assay. * p<0.05, ** p<0.005, *** p<0.0005, **** p<0.0001 as determined by unpaired Student t-test.
The flow cytometry results were validated by immunoblotting in MHH-CALL-4 cells (Figure 2B). TSLP stimulation increased phosphorylation of all tested phospho-proteins. BBT594 completely inhibited p-JAK2, and AZD2014 inhibited p-AKT and p-4E-BP1 to a greater extent than rapamycin.
Since more efficient blockade of mTORC1, in particular 4E-BP1, has been implicated in the higher activity of next generation TORC inhibitors, we engineered MHH-CALL-4 cells to express doxycycline-inducible mutant 4E-BP1 5A [19]. Phosphorylation of 4E-BP1 is known to disrupt the complex between 4E-BP1 and the translation factor eIF4E, releasing free active eIF4E [20]. Phosphorylation-defective mutant 4E-BP1 5A was reported to tightly associate with eIF4E [19], inhibiting its translation activation and signaling functions downstream of 4E-BP1, mimicking effects of AZD2014 (as shown by decreased p-4E-BP1 level after doxycycline induction in Figure 2C). While there was no toxicity of 0.1ug/ml of doxycycline when 5A 4EBP1 was induced up to 72h (not shown), growth of cells induced to express a mutant non-phosphorylatable 4E-BP1 5A was further inhibited by BBT594 and by ruxolitinib compared to cells transfected with empty vector or wild-type plasmid constructs (Figure 2D).
To explore additional mechanisms underlying the synergy, we performed the reverse phase protein array (RPPA) in MHH-CALL-4 and MUTZ-5 cells (Figure 3). In addition to anticipated changes in p70-S6K-Thr389, p-S6, and phospho-rictor (on T1135, an S6K1 site), ruxolitinib and BBT594 combined with AZD2014 decreased c-myc expression and phospho-Bad (Bad_pS112), both of which are targets of mTOR and JAK/STAT networks [21]. TSLP stimulated samples showed high level of tyrosine-protein phosphatase non-receptor type 11 (PTPN11, SHP2), human epidermal growth factor receptor 2 (HER2), which were in turn inhibited by JAK inhibitor and further reduced by combinations. On the other hand, TSLP stimulation decreased the level of CD49, stem cell factor receptor kinase (c-KIT), and dual specificity phosphatase 4 (DUSP4), which were strongly elevated in samples treated with combination of inhibitors. These findings indicate cross-talk with other pathways which could conceivably contribute to the synergy effect, to be explored in future studies.
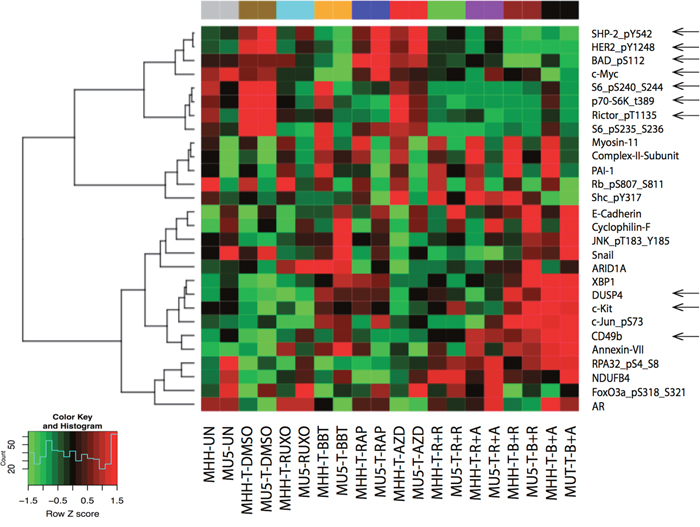
Figure 3: Inhibitory effects of dual JAK2 and mTOR inhibition by Reverse Phase Protein Array (RPPA) analysis. MHH-CALL4 and MUTZ-5 cells were stimulated with TSLP for 30 min, treated with indicated compounds for 1 h, after which the cell lysates were prepared for RPPA analysis. The heatmap showed the differentially expressed proteins in at least on treatment group with adjust ANOVA p (FDR) < 0.1. Pearson distance metric and Ward’s minimum variance was used to cluster the samples and proteins.
Combination of JAK type II inhibitor BBT594 with an mTOR inhibitor induced synergistic cell apoptosis in Ph-like B-ALL PDX samples ex vivo
To extend the cell killing and cell signaling in vitro findings to samples from patients with Ph-like B-ALL, we developed multiple PDX models of Ph-like B-ALL (Table 2). Notably, a subset of CRLF2-re/JAK2 wild-type PDX cells (four of the seven tested) failed to grow ex vivo and were excluded from subsequent analysis. All PDX samples harboring CRLF2-re and JAK2 mutation (four with P2RY8-CRLF2 and three with IGH-CRLF2) and one sample with wild-type CRLF2 and a JAK2 fusion maintained viability ex vivo and were tested with the JAK2/mTOR inhibitors.
Table 2: Genomic information of Ph-like ALL PDX samples
PDX |
Ph-like ALL Genomic Lesions |
|
|---|---|---|
CRLF2 |
JAK2 |
|
1 |
IGH-CRLF2 |
WT |
2 |
P2RY8-CRLF2 |
WT |
3 |
P2RY8-CRLF2 |
WT |
4 |
IGH-CRLF2 |
WT |
5 |
IGH-CRLF2 |
WT |
6 |
IGH-CRLF2 |
WT |
7 |
IGH-CRLF2 |
WT |
8 |
P2RY8-CRLF2 |
T875N |
9 |
IGH-CRLF2 |
R683G |
10 |
IGH-CRLF2 |
R683S |
11 |
P2RY8-CRLF2 |
R683G |
12 |
IGH-CRLF2 |
R683S |
13 |
P2RY8-CRLF2 |
R683S |
14 |
P2RY8-CRLF2 |
R683G |
15 |
N/A |
PAX5-JAK2 |
16 |
N/A |
ATF7IP-JAK2 |
PDX 13, 14 were confirmed strong Ph-like signature by microarray and prediction analysis of microarray, which reported previously as SJBALL021755 and SJBALL191 [3].
In three CRLF2-re/JAK2 wild-type samples, ruxolitinib had no effect on cell death, while BBT594 alone or combined with rapamycin/AZD2014 induced modest cell apoptosis in 2 P2RY8-CRLF2 samples and significant cell death in IGH-CRLF2 sample ex vivo without notable synergistic effects with mTOR inhibitors (Figure 4A).
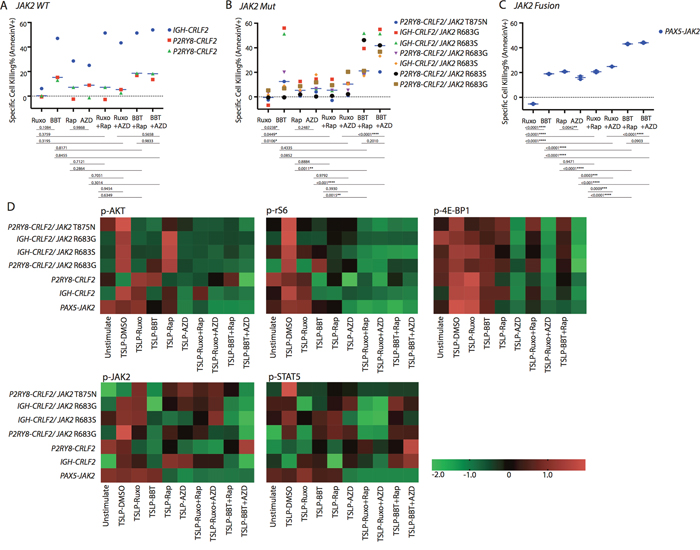
Figure 4: Antileukemia efficacy of dual JAK2 and mTOR inhibition ex vivo in Ph-like B-ALL patient-derived xenograft (PDX) models. Cells collected from Ph-like B-ALL PDX models were treated with DMSO, ruxolitinib (Ruxo; 1 μM), BBT594 (BBT; 1 μM), rapamycin (Rap; 0.2 μM), AZD2014 (AZD; 0.2 μM), or one of their combinations for 48 h. The cells were then analyzed for (A-C) apoptosis by annexin V/DAPI or (D) the mTOR and JAK2/STAT phosphoproteins gated on CD19+ cells. (D) Heat map data depict the changes in phosphoprotein levels by fluorescence median intensity. Color scale represents normalization (Z-score) of each condition for each signal.
In the CRLF2-re/JAK2-mutated samples, BBT594 achieved significantly greater cell apoptosis than any other compound. The combinations of BBT594 with either mTOR inhibitor caused additional cell killing in the majority of samples tested (Figure 4B). In turn, ruxolitinib alone had no effect, while ruxolitinib+ AZD2014 showed additive induction of cell death (p=0.010). Similar results were seen in a single sample with a JAK2 fusion (Figure 4C).
Combined inhibition of JAK2 and mTOR achieved greater cell signaling inhibition in Ph-like B-ALL PDX models ex vivo
Phospho-signaling studies in samples with CRLF2-re with or without JAK2 mutation showed findings consistent with cell lines studies, i.e. greater inhibition of AKT/mTOR outputs upon JAK2/mTOR inhibitors combinations, whereby only AZD2014-based combinations were capable of inhibiting p-4E-BP1 (Figure 4D). BBT594 fully inhibited constitutive or TSLP-induced p-JAK2 in all CRLF2-re samples, while ruxolitinib incompletely reduced p-JAK2 in 3 PDX and failed to do so in 3 additional PDX; co-treatment of BBT594 and AZD2014 had most profound inhibition of p-JAK2 in all samples tested. The combinations with ruxolitinib produced greater inhibition of p-STAT5, which was consistent with cell line data. In PDX with PAX5-JAK2 fusion, ruxolitinib or BBT594 alone failed to inhibit either AKT/mTOR outputs or p-JAK2 but this was accomplished in combinations with either rapamycin or AZD2014, which on their own suppressed p-JAK2.
Combination treatment in PDX models in vivo
Based on the in vitro and ex vivo data, we hypothesized that the combinations of a type II JAK2 inhibitor and an mTOR inhibitor would achieve better anti-leukemia control in the in vivo Ph-like B-ALL PDX models. For these studies, we selected three Ph-like B-ALL PDX models, two CRLF2-re (P2RY8-CRLF2/JAK2 T875N, P2RY8-CRLF2/JAK2 R683S), and one with ATF7IP-JAK2 fusion. Mice engrafted with ALL PDX were dosed for a total of 7 days with ruxolitinib, BBT594, AZD2014 or combinations as indicated in Figure 5A, after which engraftment in spleen, PB or BM was compared and spleen weight recorded. All mice tolerated therapy without significant weight loss (not shown). In both CRLF2-re PDX, spleen weights were significantly reduced by ruxolitinib alone or when combined with mTOR inhibitors, and most pronounced in the BBT594/AZD2014 combination group, indicating significant reduction of total tumor burden (Figure 5B, 5F). In contrast, the relative fraction (%) of ALL cells was minimally affected by all treatments with exception of decrease in PB ALL cells in the AZD2014/BBT594 combination group (Figure 5D, 5H). BBT594 at selected doses/duration did not significantly affect engraftment and spleen weights in CRLF2-re PDX. AZD2014 reduced spleen weights but did not affect engraftment. In stark contrast, in the ATF7IP-JAK2 fusion model all inhibitors drastically reduced spleen weights and also significantly decreased fractions of leukemic cells in PB, BM and spleen, with BBT594/AZD2014 exhibiting most profound effect (Figure 6A-6D). In turn, ruxolitinib/AZD2014 combination failed to show better efficacy than ruxolitinib alone in any of the three models tested.
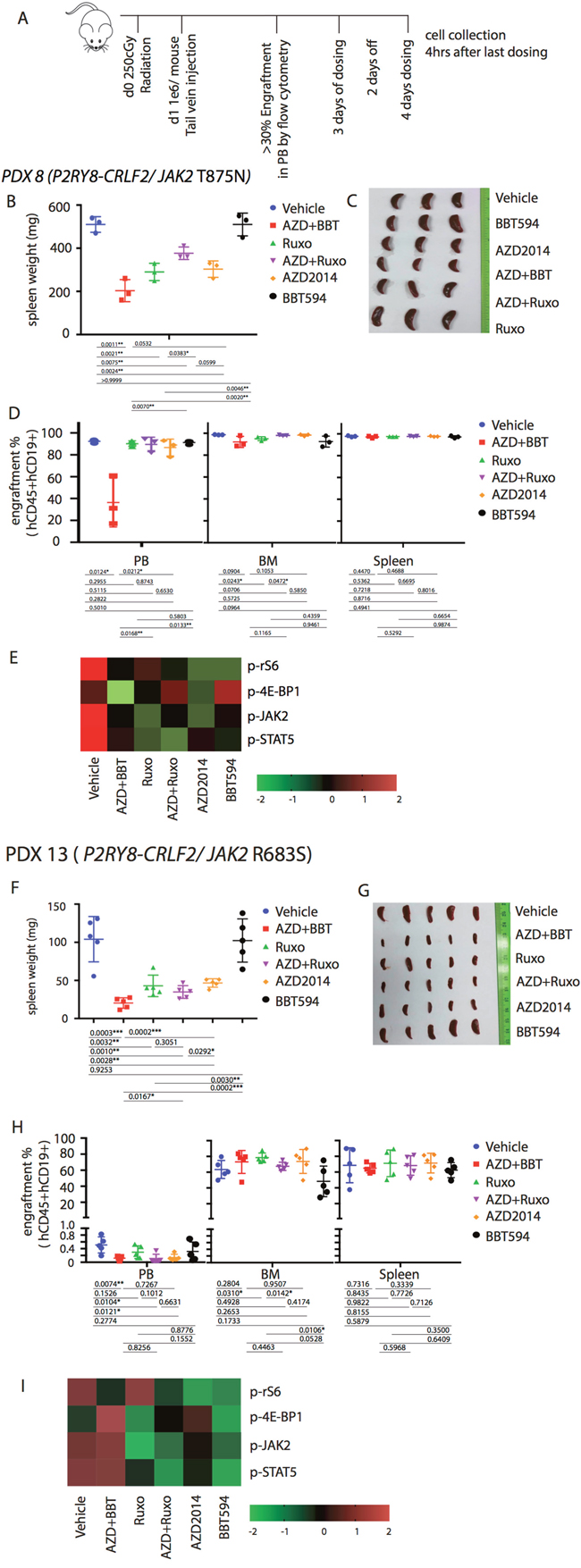
Figure 5: Anti-leukemia efficacy of dual JAK2 and mTOR inhibition in vivo in the P2RY8-CRLF2/JAK2 mutation PDX. (A) Treatment schema for in vivo experiment with PDX 8 (P2RY8-CRLF2/JAK2 T875N) and PDX 13 (R2RY8-CRLF2/JAK2 R683S). Mice were treated with vehicle, ruxolitinib (Ruxo, 2gm/mouse/day) BBT594 (BBT; 100mg/kg/d), AZD2014 (AZD; 30 mg/kg/d), or the combinations when higher than 30% leukemia cells engraftment was detected in peripheral blood (PB) for a total of 7 days (3 days on, 2 days off, 4 days on). 4 h after the last dosing was completed, mice were sacrificed for tumor burden assessment. Human leukemia cell percentages in the peripheral blood, bone marrow (BM), and spleen were measured by quantitative flow cytometry. Tumor burden was represented by spleen weight and morphology (B, F, C and G) and fraction of engraftment (D, H). Heat map data depict the changes in phosphoprotein levels by fluorescence median intensity (E, I).
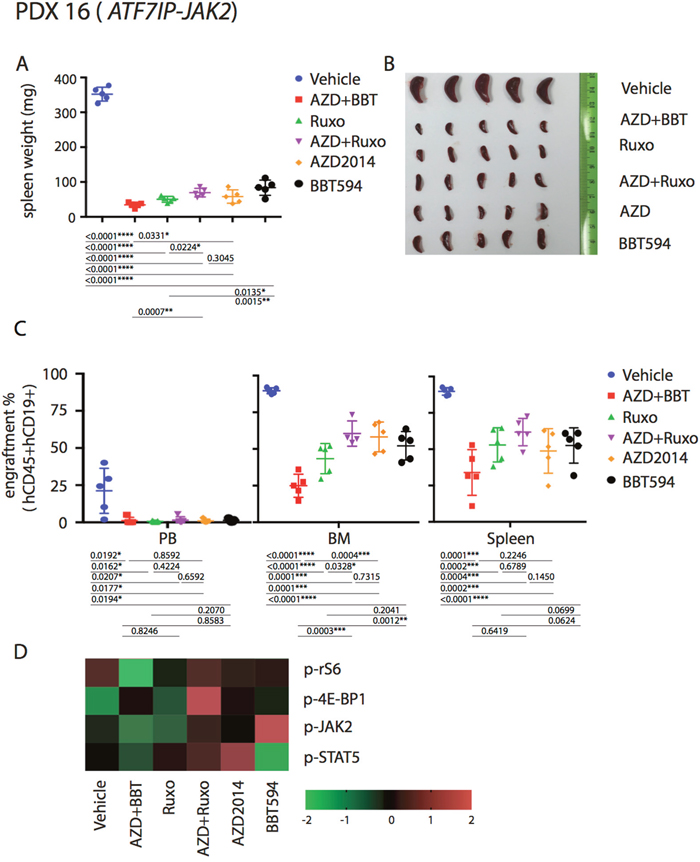
Figure 6: Anti-leukemia efficacy of dual JAK2 and mTOR inhibition in vivo in the ATF7IP-JAK2 fusion PDX. Mice were injected with PDX 16 (ATF7IP-JAK2) and treated as indicated in Figure 5A. Tumor burden was represented by spleen weight and morphology (A, B), engraftment (C) and signaling (D).
We next analyzed the signaling perturbations by JAK2 and mTOR inhibitors in the leukemia cells (gated on hCD19 by flow cytometry) collected from the murine spleens (Figure 5E, 5I, Figure 6D). Ex vivo signaling results exhibited differences compared to in vitro findings, possibly due to the dynamic signaling changes in vivo that cannot be fully reflected by single time point measurements; these inconsistences were noted in prior studies [11, 18]. For example, in P2RY8-CRLF2/JAK2 R683S model BBT594/AZD2014, and in ATF7IP-JAK2 model ruxolitinib/AZD2014 combination increased rather than decreased p-4E-BP1. In P2RY8-CRLF2/JAK2 T875N model, both JAK2 inhibitors and additionally AZD2014 reduced p-JAK2, consistent with in vitro results. Ruxolitinib had stronger impact on p-STAT5 compared with BBT594. In ATF7IP-JAK2 fusion model, BBT594 failed to inhibit p-JAK2, while AZD2014 alone and more so when combined with BBT594 or ruxolitinib, fully blocked p-JAK2. AZD2014 inhibited p-4E-BP1 and p-S6 (Figure 5E, 5I). In turn, greater combination effects of BBT594/AZD2014 were observed on p-S6 and p-STAT5 phospho-signals in the ATF7IP-JAK2 fusion model (Figure 6D).
DISCUSSION
Several prior studies have highlighted abnormal activation of PI3K/AKT/mTOR and JAK/STAT pathways in childhood Ph-like ALL models [18] and have demonstrated improved anti-leukemia efficacy upon combining type I JAK1/2 inhibitor ruxolitinib and PI3K/mTOR inhibitors [11, 18, 22]. While allosteric mTOR inhibitors such as rapamycin or its analogues have limited efficacy in various cancers in part due to activation of AKT signaling [23–26], second generation mTOR inhibitors can avoid the compensatory activation and elicit deeper inhibition of both mTOR complexes and downstream signaling [13, 27–29]. Further, recent studies in MPN and Ph-like ALL models have demonstrated superior activity of type II JAK2 inhibitors that fully inhibit p-JAK2 activity, and showed collateral activation of p-JAK2 as a mechanism of acquired resistance to ruxolitinib [15, 30].
In this study, we evaluated the combinatorial activity of the second generation mTOR inhibitor AZD2014 and of the type II JAK2 inhibitor BBT594 compared with allosteric mTOR inhibitor rapamycin and type I JAK2 inhibitor ruxolitinib in Ph-like ALL harboring CRLF2 rearrangement. Studies in CRLF2-re/JAK2-mutated ALL cell lines, engineered BaF/3 cells or primary ALL PDX cultured ex vivo showed very little activity of ruxolitinib at sub-micro molar concentrations used. While rapamycin-based combinations elicited no further increase in anti-leukemia efficacy, combinations of ruxolitinib and the mTORC1/2 inhibitor AZD2014 were synergistic in human ALL cell lines and in BaF/3 cells including those which acquired resistance to ruxolitinib (ruxolitinib-“persistent” cells). It is notable that the reduction of viability based on CTG assay appears more prominent than effects on cell cycle or apoptosis induction. In this study, CTG assay was used across multiple inhibitor concentrations, cell cycle and induction of cell death was only examined at selected doses. It is possible, however, that the inhibitors directly affect cellular metabolism, especially in light of mTOR inhibition known to directly affect glucose uptake and glycolysis [31].
As anticipated from prior studies, AZD2014 unlike rapamycin fully suppressed activity of both mTOR complexes, including reduction of mTORC1 outputs with diminished p-4E-BP1, and of mTORC2 with a decrease in p-AKT. Cells expressing the 4E-BP1 5A mutant that suppresses eIF4E-dependent translation became more responsive to JAK2 inhibition. Albeit the reduction in viability was statistically significant, the extent of reduction was relatively minor. These data support the contributory, but not the dominant, role of mTOR/4E-BP1 in the observed synergy between JAK2 inhibitors and mTORC1/2 inhibitor AZD2014. In primary ALL PDX cultured ex vivo ruxolitinib/AZD2014 showed mainly additive effects that were limited to samples harboring mutant JAK2 (Figure 4). It should be considered that the co-culture with stromal cells used in this study could have reversed cytotoxicity in the JAK WT cells and modified the outcomes of targeted therapies. Further, in vivo data in 2 PDX CRLF2-re/JAK2-mutated samples failed to show synergy of this combination, at least upon 7-day treatment regimen used (Figure 5), consistent with recently published data [11]. Another interesting finding is the anti-leukemia effect of ruxolitinib which was observed in vivo but not in vitro in PDX models, likely due to better proliferation of ALL PDX cells under nurturing conditions of murine microenvironment. In this study, we utilized 7-day treatment regimen and analyzed impact on tumor burden as end-point analysis, which prohibited survival assessments and remains a limitation of this pilot study. Curiously, we observed impressive reduction of spleen weights despite similar levels of engraftment. This could be related to the higher TSLP production in spleen reported by others [32], which triggers signaling in the CRLF2-re ALL cells, making them more vulnerable to the target inhibition. Overall, these findings indicate potential utility of second generation mTOR inhibitor/ruxolitinib combinations in CRLF2-re/JAK2-mutated Ph-like ALL, but additional in vivo studies using prolonged regimens of drug administration are needed to support this premise.
Our results strongly indicate superior activity of the type II JAK2 inhibitor BBT594, alone and more so when combined with AZD2014. This was demonstrated in both human and murine cell lines, including ruxolitinib-persistent BaF/3 cells which resistant up to 8μM ruxolitinib, and in JAK2-mutated ALL PDX samples cultured ex vivo. Analysis of intracellular signaling demonstrated superior ability of BBT594 to inhibit p-JAK2, while exhibiting less profound blockade of p-STAT5. Similar results were seen upon analysis of spleen cells from mice co-treated with BBT594 and AZD2014 (Figure 5). These findings indicate that other downstream JAK2 targets might be important for the observed anti-leukemia efficacy of BBT594. Deeper inhibition of JAK2 by BBT594 was also reported previously in Ph-like murine model [15] and in MPN murine models [1, 16]. Less likely, off-target potency against other kinases could contribute towards superior activity of BBT594, however none of the identified targeted kinases [15] have a known role in ALL survival. While these need further detailed studies, our preliminary RPPA studies demonstrated inhibition of c-myc and decreased levels of phosphorylated, i.e. inactivated, apoptotic protein Bad, both of which play an important role in cellular proliferation and survival. Induction of HER2 expression after TSLP stimulation in MHH-CALL4 and MUTZ-5 cells might indicate possible trastuzumab sensitivity which needs further study. Altogether, these results indicate superior activity of type II JAK2 and second generation mTOR inhibitors in CRLF2-re/JAK2-mutated Ph-like ALL, secondary to more profound inhibition of collateral JAK2 and 4E-BP1 signaling.
In our study, a single model with JAK2 fusion Ph-like ALL was evaluated. Consistent with previous studies [10], this PDX was more sensitive to ruxolitinib in vivo but not in vitro (Figure 4C and Figure 6A-6D). However, combination treatment with BBT594 and AZD2014 produced highest activity both in ex vivo and in vivo study, with 65%, 56% and 20% reduction in the fraction of ALL cells in BM, spleen, and PB, respectively (Figure 6C). Importantly, only combination of JAK2 inhibitors with mTOR inhibitors elicited full inhibition of p-JAK2, p-STAT5 and p-4E-BP1, consistent with previous findings that mTOR inhibitors could reduce activation of JAK/STAT pathway, ie. p-STAT5 [18, 22, 33]. Curiously, mTOR inhibitor reduced p-JAK2 in this model, invoking a possibility of mTOR pathway mediated phosphorylation of PAX-5 portion of the mutant fusion proteins, in turn controlling the dimerization and activation of PAX5-JAK2. Importantly, these fusions are observed more commonly in the adolescent and young adult (AYA) population and in adult Ph-like ALL and represent an important object for future studies [3, 34].
In summary, our findings indicate that the combined inhibition of the JAK/STAT and mTOR pathways by novel more potent inhibitors can effectively inhibit cell signaling and reduce leukemia burden in CRFL2-re Ph-like B-ALL cell lines and PDX models. Therefore, concurrent targeting of both pathways is a promising new therapeutic strategy for patients with CRFL2-re Ph-like B-ALL. While type II JAK2 inhibitors are not currently in clinical development, other strategies aimed at complete inhibition of JAK2 (such as HSP90 inhibitors) may produce greater anti-leukemia efficacy than ruxolitinib in CRLF2-re Ph-like ALL [35, 36].
MATERIALS AND METHODS
Information concerning cell origin, authentication, culture, reagents, proliferation and cytotoxicity assays, phospho-flow cytometry, Western blotting and reverse phase protein array (RPPA) are detailed in the Supplementary Materials.
PDX model establishment and ex vivo experiments
All samples were obtained with patient or parent/guardian provided informed consent under protocols approved by the Institutional Review Board at University of California-San Francisco Medical Center, Dana-Farber Cancer Institute, St. Jude Children’s Research Hospital and M.D. Anderson Cancer Center. PDX 13-16 were already published previously [3]. PDX 13 and 14 with P2RY8-CRLF2 expressed strong Ph-like signature by gene expression microarray, and were previously reported as SJBALL021755 and SJBALL191 [3].
All animal studies were conducted and animals handled in accordance with guidelines approved by the Institutional Animal Care and Use Committees at The University of Texas MD Anderson Cancer Center. Female NOD-scid IL2Rγnull-3/GM/SF (NSG-SGM3, NSGS) mice (Jackson Laboratory, Maine, USA, 6-8 weeks old) were injected intravenously with cells isolated from Ph-like B-ALL samples (1×106 cells/100μL/mouse). Four-twelve weeks after injection, engraftment of human CD45/CD19 cells was determined in peripheral blood samples by flow cytometry. Details of the samples are summarized in Table 2. Once tumor burden in peripheral blood was determined to be high (>90% CD45+ CD19+) or the mice presented tumor burden symptoms according to the Institutional Animal Care and Use Committee protocol, mice were humanely sacrificed and their spleens were harvested for ex vivo cell killing and cell signaling assays. The collected cells were seeded at 0.4×106 in 0.4ml medium in 48-well plates pre-coated with 200 MS-5 cells and treated with each inhibitor or one of their combinations for 72 h. The cytotoxicity was determined by annexin V/DAPI flow cytometry after gating on CD19+ cells. The specific cell killing was calculated by subtracting spontaneous cell death in DMSO–treated control cells from selected inhibitor-induced cell killing.
In vivo efficacy study
Mice were randomized into treatment groups based on the engraftment level determined by hCD45 flow cytometry. AZD2014 (30mg/kg/d by gavage) was formulated in 20% Captisol (Ligand Techonology, CA, USA). Ruxolitinib was formulated in chow, which was dosed at 2 g/mouse/day. BBT594 (100 mg/kg/d by gavage) was formulated in 0.5% methyl cellulose and 0.2% Tween 80. Upon high PB engraftment (>20%), mice were dosed with inhibitors for 7 days total, on a 3 days on - 2 days off - 4 days on schedule. Animals were humanely sacrificed after last dose, and BM and spleens collected for leukemia burden analysis by hCD45/CD19 flow cytometry.
Statistical analysis
All statistical analyses were performed using Prism software 7.0. Unless indicated, the results are expressed as the mean± standard deviation of at least three independent experiments. Differences with a p-value <0.05 were considered statistically significant.
Author contributions
Q.Z and C.S designed, directed, and performed research, analyzed data and wrote the manuscript; L.H, N.J, H.M, T.C, A.C, Y.T, R.J, H.M, Y.Z, J.W, Z.Z performed research, data analysis and interpretation; K.G.R, S.W, F.C, Z.J, Y.M, E.J, R.L, S.K.T, C.G.M, D.M.W, D.A.F and M.K contributed to research design and data interpretation; M.K edited the manuscript; and all authors critically reviewed the manuscript prior submission.
ACKNOWLEDGMENTS
We thank Dr. Thomas Radimerski and Paul Manley (Norvartis, Switzerland) for synthesizing and providing NVP-BBT594. We thank Dr. Markus Muschen (University of California-San Francisco, USA) for providing samples and instructions on research design.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
1 R21 HD081319-01 NIH/NCI’ “Combination strategies to enhance therapy for Ph-like B-ALL” to D.A.F. and M.K; Cancer Prevention & Research Institute of Texas (CPRIT) grant RP150006 “Defining and Treating Targetable Lesions in AYA Acute Lymphoblastic Leukemia” to M.K. and N.J; and K08 CA184418/CA/NCI NIH HHS to S.K.T.
REFERENCES
1. Andraos R, Qian Z, Bonenfant D, Rubert J, Vangrevelinghe E, Scheufler C, Marque F, Regnier CH, De Pover A, Ryckelynck H, Bhagwat N, Koppikar P, Goel A, et al. Modulation of activation-loop phosphorylation by JAK inhibitors is binding mode dependent. Cancer Discov. 2012; 2: 512-23. https://doi.org/10.1158/2159-8290.CD-11-0324.
2. Jain N, Roberts KG, Jabbour E, Patel K, Eterovic AK, Chen K, Zweidler-McKay P, Lu X, Fawcett G, Wang SA, Konoplev S, Harvey RC, Chen IM, et al. Ph-like acute lymphoblastic leukemia: a high-risk subtype in adults. Blood. 2017; 129: 572-81. https://doi.org/10.1182/blood-2016-07-726588.
3. Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, McCastlain K, Ding L, Lu C, Song G, Ma J, Becksfort J, Rusch M, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014; 371: 1005-15. https://doi.org/10.1056/NEJMoa1403088.
4. Den Boer ML, van Slegtenhorst M, De Menezes RX, Cheok MH, Buijs-Gladdines JG, Peters ST, Van Zutven LJ, Beverloo HB, Van der Spek PJ, Escherich G, Horstmann MA, Janka-Schaub GE, Kamps WA, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol. 2009; 10: 125-34. https://doi.org/10.1016/S1470-2045(08)70339-5.
5. Reshmi SC, Harvey RC, Roberts KG, Stonerock E, Smith A, Jenkins H, Chen IM, Valentine M, Liu Y, Li Y, Shao Y, Easton J, Payne-Turner D, et al. Targetable kinase gene fusions in high-risk B-ALL: a study from the Children's Oncology Group. Blood. 2017; 129: 3352-61. https://doi.org/10.1182/blood-2016-12-758979.
6. Mullighan CG, Collins-Underwood JR, Phillips LA, Loudin MG, Liu W, Zhang J, Ma J, Coustan-Smith E, Harvey RC, Willman CL, Mikhail FM, Meyer J, Carroll AJ, et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nat Genet. 2009; 41: 1243-6. https://doi.org/10.1038/ng.469.
7. Roll JD, Reuther GW. CRLF2 and JAK2 in B-progenitor acute lymphoblastic leukemia: a novel association in oncogenesis. Cancer Res. 2010; 70: 7347-52. https://doi.org/10.1158/0008-5472.CAN-10-1528.
8. Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, Gray NS, Sabatini DM. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009; 137: 873-86. https://doi.org/10.1016/j.cell.2009.03.046.
9. Dowling RJ, Topisirovic I, Fonseca BD, Sonenberg N. Dissecting the role of mTOR: lessons from mTOR inhibitors. Biochim Biophys Acta. 2010; 1804: 433-9. https://doi.org/10.1016/j.bbapap.2009.12.001.
10. Roberts KG, Morin RD, Zhang J, Hirst M, Zhao Y, Su X, Chen SC, Payne-Turner D, Churchman ML, Harvey RC, Chen X, Kasap C, Yan C, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. 2012; 22: 153-66. https://doi.org/10.1016/j.ccr.2012.06.005.
11. Tasian SK, Teachey DT, Li Y, Shen F, Harvey RC, Chen IM, Ryan T, Vincent TL, Willman CL, Perl AE, Hunger SP, Loh ML, Carroll M, et al. Potent efficacy of combined PI3K/mTOR and JAK or ABL inhibition in murine xenograft models of Ph-like acute lymphoblastic leukemia. Blood. 2017; 129: 177-87. https://doi.org/10.1182/blood-2016-05-707653.
12. Zhang Y, Zheng XF. mTOR-independent 4E-BP1 phosphorylation is associated with cancer resistance to mTOR kinase inhibitors. Cell Cycle. 2012; 11: 594-603. https://doi.org/10.4161/cc.11.3.19096.
13. Simioni C, Cani A, Martelli AM, Zauli G, Tabellini G, McCubrey J, Capitani S, Neri LM. Activity of the novel mTOR inhibitor Torin-2 in B-precursor acute lymphoblastic leukemia and its therapeutic potential to prevent Akt reactivation. Oncotarget. 2014; 5: 10034-47. https://doi.org/10.18632/oncotarget.2490.
14. Boulay A, Breuleux M, Stephan C, Fux C, Brisken C, Fiche M, Wartmann M, Stumm M, Lane HA, Hynes NE. The Ret receptor tyrosine kinase pathway functionally interacts with the ERalpha pathway in breast cancer. Cancer Res. 2008; 68: 3743-51. https://doi.org/10.1158/0008-5472.CAN-07-5100.
15. Wu SC, Li LS, Kopp N, Montero J, Chapuy B, Yoda A, Christie AL, Liu H, Christodoulou A, van Bodegom D, van der Zwet J, Layer JV, Tivey T, et al. Activity of the type II JAK2 inhibitor CHZ868 in B cell acute lymphoblastic leukemia. Cancer Cell. 2015; 28: 29-41. https://doi.org/10.1016/j.ccell.2015.06.005.
16. Koppikar P, Bhagwat N, Kilpivaara O, Manshouri T, Adli M, Hricik T, Liu F, Saunders LM, Mullally A, Abdel-Wahab O, Leung L, Weinstein A, Marubayashi S, et al. Heterodimeric JAK-STAT activation as a mechanism of persistence to JAK2 inhibitor therapy. Nature. 2012; 489: 155-9. https://doi.org/10.1038/nature11303.
17. Weigert O, Lane AA, Bird L, Kopp N, Chapuy B, van Bodegom D, Toms AV, Marubayashi S, Christie AL, McKeown M, Paranal RM, Bradner JE, Yoda A, et al. Genetic resistance to JAK2 enzymatic inhibitors is overcome by HSP90 inhibition. J Exp Med. 2012; 209: 259-73. https://doi.org/10.1084/jem.20111694.
18. Tasian SK, Doral MY, Borowitz MJ, Wood BL, Chen IM, Harvey RC, Gastier-Foster JM, Willman CL, Hunger SP, Mullighan CG, Loh ML. Aberrant STAT5 and PI3K/mTOR pathway signaling occurs in human CRLF2-rearranged B-precursor acute lymphoblastic leukemia. Blood. 2012; 120: 833-42. https://doi.org/10.1182/blood-2011-12-389932.
19. Mallya S, Fitch BA, Lee JS, So L, Janes MR, Fruman DA. Resistance to mTOR kinase inhibitors in lymphoma cells lacking 4EBP1. PLoS One. 2014; 9: e88865. https://doi.org/10.1371/journal.pone.0088865.
20. Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009; 10: 307-18. https://doi.org/10.1038/nrm2672.
21. Yun S, Vincelette ND, Knorr KL, Almada LL, Schneider PA, Peterson KL, Flatten KS, Dai H, Pratz KW, Hess AD, Smith BD, Karp JE, Hendrickson AE, et al. 4EBP1/c-MYC/PUMA and NF-kappaB/EGR1/BIM pathways underlie cytotoxicity of mTOR dual inhibitors in malignant lymphoid cells. Blood. 2016; 127: 2711-22. https://doi.org/10.1182/blood-2015-02-629485.
22. Maude SL, Tasian SK, Vincent T, Hall JW, Sheen C, Roberts KG, Seif AE, Barrett DM, Chen IM, Collins JR, Mullighan CG, Hunger SP, Harvey RC, et al. Targeting JAK1/2 and mTOR in murine xenograft models of Ph-like acute lymphoblastic leukemia. Blood. 2012; 120: 3510-8. https://doi.org/10.1182/blood-2012-03-415448.
23. Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, Egia A, Sasaki AT, Thomas G, Kozma SC, Papa A, Nardella C, Cantley LC, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008; 118: 3065-74. https://doi.org/10.1172/JCI34739.
24. O'Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, Baselga J, Rosen N. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006; 66: 1500-8. https://doi.org/10.1158/0008-5472.CAN-05-2925.
25. Britschgi A, Andraos R, Brinkhaus H, Klebba I, Romanet V, Muller U, Murakami M, Radimerski T, Bentires-Alj M. JAK2/STAT5 inhibition circumvents resistance to PI3K/mTOR blockade: a rationale for cotargeting these pathways in metastatic breast cancer. Cancer Cell. 2012; 22: 796-811. https://doi.org/10.1016/j.ccr.2012.10.023.
26. Blachly JS, Baiocchi RA. Targeting PI3-kinase (PI3K), AKT and mTOR axis in lymphoma. Br J Haematol. 2014; 167: 19-32. https://doi.org/10.1111/bjh.13065.
27. Fransecky L, Mochmann LH, Baldus CD. Outlook on PI3K/AKT/mTOR inhibition in acute leukemia. Mol Cell Ther. 2015; 3: 2. https://doi.org/10.1186/s40591-015-0040-8.
28. Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014; 13: 140-56. https://doi.org/10.1038/nrd4204.
29. Mallon R, Feldberg LR, Lucas J, Chaudhary I, Dehnhardt C, Santos ED, Chen Z, dos Santos O, Ayral-Kaloustian S, Venkatesan A, Hollander I. Antitumor efficacy of PKI-587, a highly potent dual PI3K/mTOR kinase inhibitor. Clin Cancer Res. 2011; 17: 3193-203. https://doi.org/10.1158/1078-0432.CCR-10-1694.
30. Bhagwat N, Koppikar P, Keller M, Marubayashi S, Shank K, Rampal R, Qi J, Kleppe M, Patel HJ, Shah SK, Taldone T, Bradner JE, Chiosis G, et al. Improved targeting of JAK2 leads to increased therapeutic efficacy in myeloproliferative neoplasms. Blood. 2014; 123: 2075-83. https://doi.org/10.1182/blood-2014-01-547760.
31. Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, Vander Heiden MG, MacKeigan JP, Finan PM, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010; 39: 171-83. https://doi.org/10.1016/j.molcel.2010.06.022.
32. Fagerberg L, Hallstrom BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, Habuka M, Tahmasebpoor S, Danielsson A, Edlund K, Asplund A, Sjostedt E, Lundberg E, et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics. 2014; 13: 397-406. https://doi.org/10.1074/mcp.M113.035600.
33. Bartalucci N, Tozzi L, Bogani C, Martinelli S, Rotunno G, Villeval JL, Vannucchi AM. Co-targeting the PI3K/mTOR and JAK2 signalling pathways produces synergistic activity against myeloproliferative neoplasms. J Cell Mol Med. 2013; 17: 1385-96. https://doi.org/10.1111/jcmm.12162.
34. Roberts KG, Gu Z, Payne-Turner D, McCastlain K, Harvey RC, Chen IM, Pei D, Iacobucci I, Valentine M, Pounds SB, Shi L, Li Y, Zhang J, et al. High frequency and poor outcome of Philadelphia chromosome-like acute lymphoblastic leukemia in adults. J Clin Oncol. 2017; 35: 394-401. https://doi.org/10.1200/JCO.2016.69.0073.
35. Marubayashi S, Koppikar P, Taldone T, Abdel-Wahab O, West N, Bhagwat N, Caldas-Lopes E, Ross KN, Gonen M, Gozman A, Ahn JH, Rodina A, Ouerfelli O, et al. HSP90 is a therapeutic target in JAK2-dependent myeloproliferative neoplasms in mice and humans. J Clin Invest. 2010; 120: 3578-93. https://doi.org/10.1172/JCI42442.
36. Fiskus W, Verstovsek S, Manshouri T, Rao R, Balusu R, Venkannagari S, Rao NN, Ha K, Smith JE, Hembruff SL, Abhyankar S, McGuirk J, Bhalla KN. Heat shock protein 90 inhibitor is synergistic with JAK2 inhibitor and overcomes resistance to JAK2-TKI in human myeloproliferative neoplasm cells. Clin Cancer Res. 2011; 17: 7347-58. https://doi.org/10.1158/1078-0432.CCR-11-1541.