
Introduction
Non-small cell lung cancer (NSCLC) accounts for 85% of all lung cancer cases [1]. Molecular characterization of NSCLC has identified dominant molecular pathways that drive tumorigenesis, expanding treatment options to include targeted therapies that have contributed to improved patient outcome [2-4]. The gene rearrangement between the anaplastic lymphoma kinase (ALK) gene and echinoderm microtubule-associated protein-like 4 (EML4) gene resulting in the EML4-ALK gene fusion is the most common ALK fusion in lung cancer and are present in approximately 3-8% of NSCLC tumors, resulting in a constitutively active protein kinase that is essential for transformation [5, 6]. EML4-ALK transgenic mouse models have shown that ALK is required for growth and proliferation in cells and inhibition of ALK leads to growth inhibition and apoptosis [7]. In this regard, targeted inhibitors of ALK have been developed and used as an effective therapeutic strategy. Crizotinib, a kinase inhibitor with specificity for ALK, ROS1, and MET, was the first ALK inhibitor evaluated for clinical use and received accelerated FDA approval for treatment of ALK rearranged NSCLC based on an overall response rate of 57% in these patients [2, 8]. Despite this initial success, durable clinical response to crizotinib is limited by inevitable development of drug resistance through various known and unknown mechanisms of resistance. These include secondary mutations in the kinase domain, ALK gene copy number gain or gene amplification, and activation of alternative signaling pathways [9, 10]. Although more potent ALK inhibitors have been FDA approved, such as ceritinib, alectinib, and brigatinib, drug resistance still ultimately develops following treatment [11-14].
One approach to minimize drug resistance is to identify additional critical cellular pathways beyond the dominant oncogene and develop strategies targeting these vulnerabilities. Since there are numerous potential signaling mechanisms that may cooperate with the dominant oncogene, the use of functional genetic screens provides a powerful tool to explore mechanisms of drug resistance in preclinical cancer models. We describe here a genome-wide short hairpin RNA (shRNA) loss-of-function genetic screen to identify genes whose suppression can confer sensitivity to crizotinib in ALK+ NSCLC lines. We identified c-MYC binding protein (MYCBP) as a determinant of crizotinib sensitivity in ALK+ cell lines. This is of significance since MYCBP has been shown to interact with and regulate oncogenic c-MYC transcriptional activity through promoting enhanced E-box recognition and transcription of c-MYC target genes [15]. In this report, we identified the MYC signaling network as critical for crizotinib sensitivity and that MYC activity is regulated in an ALK-dependent manner in ALK rearranged NSCLC.
Results
RNAi-based screen identifies MYCBP as synthetic lethal with crizotinib in ALK+ NSCLC
In order to identify genes and pathways whose inhibition synergizes with crizotinib to induce cell death in ALK rearranged non-small cell lung cancer, we performed a genome-wide RNAi-based synthetic lethal screen in two ALK+ NSCLC adenocarcinoma cell lines, H2228 and H3122 (Figure 1A). The screen used a lentiviral shRNA library carrying 3-5 target sequences for approximately 50,000 human gene transcripts. This library was transduced into H2228 and H3122 cells. After a selection period, transduced cells were divided into two groups and treated with either vehicle or crizotinib for 72h hours. shRNA sequences were amplified, sequenced, mapped to gene transcripts and analyzed for statistically significant changes between crizotinib and vehicle treated groups as described in Material and Methods.
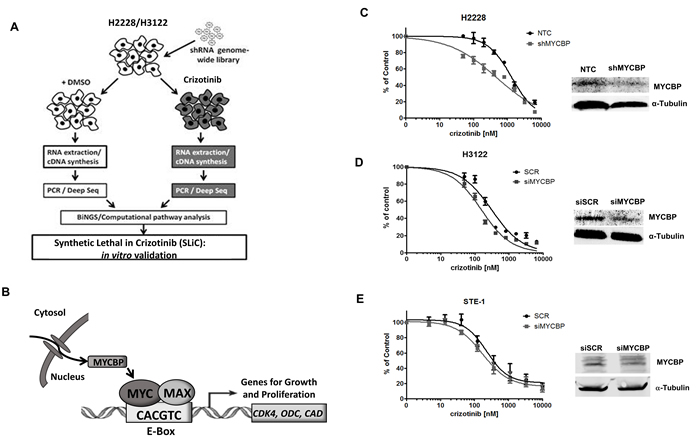
Figure 1: RNAi-based screen identifies MYCBP as synthetic lethal in ALK+ NSCLC cell lines. A. Schematic overview of genetic screen and data analysis workflow. B. Schematic representation of c-MYC and MYCBP interaction. MYC encodes the transcription factor c-MYC which heterodimerizes with Max and promotes transcription of genes involved in growth and proliferation. The mechanisms through which MYC mediates its broad range of biological effects are thought to include co-factors that can interact with c-MYC to regulate its transcriptional activity. MYCBP is one such co-factor that interacts with MYC through binding to its N-terminal domain to promote E-box dependent transcription of c-MYC target genes. C. H2228 cells expressing either non-gene targeting control (NTC) or shRNA targeting MYCBP (shMYCBP) were treated with crizotinib for 72 hours and proliferation was measured by MTS assay. Western blot demonstrates knockdown efficiency of MYCBP. H3122 D. or STE-1 E. cells expressing either scrambled siRNA control (siSCR) or SmartPool siRNA targeting MYCBP (siMYCBP) were treated with crizotinib for 72 hours and proliferation was measured by MTS assay. Knockdown efficiency was determined by Western blot.
Synthetic lethality was determined by identifying shRNAs that are underrepresented in the crizotinib treatment group as target genes whose silencing sensitized cells to crizotinib. To reduce false positives and to identify those genes that were indeed synthetic lethal hits, we considered genes with Z-transformed p-values (P(wZP)) of <0.05 as synthetic lethal hits (325 and 226 genes for H2228 and H3122; Supplementary Table 1A and 1B, respectively) as previously described [16]. Based on this criteria, a total of 12 genes were significant hits in both H2228 and H3122 (Table 1). We have termed these genes as Synthetic Lethal in Crizotinib (SLiC) hits.
Table 1: Common synthetic lethal in crizotinib (SLiC) hits in H2228 and H3122
GENE SYMBOL |
GENE NAME |
CCDC59 |
Coiled-coil domain containing 59 |
SLC29A3 |
Solute carrier family 29 (nucleoside transporters), member 3 |
MCFD2 |
Multiple coagulation factor deficiency 2 |
RPL28 |
Ribosomal protein 28 |
PSMD1 |
Proteasome 26S subunit, non-ATPase 1 |
MYCBP |
c-MYC binding protein |
IKBKB |
Inhibitor of nuclear factor kappa B kinase subunit Beta |
RRM1 |
Ribonucleotide reductase subunit M1 |
GNB1 |
Guanine nucleotide binding protein subunit Beta 1 |
SSX2IP |
Synovial sarcoma X breakpoint 2 interacting protein |
PHC1 |
Polyhomeotic homolog 1 |
M6PR |
Mannose 6-phophate receptor |
Among the genes identified as SLiCs in both H2228 and H3122 ALK+ cell lines was c-MYC binding protein (MYCBP), demonstrating post-analysis statistical significance determined by a weighted Z-transformed p-value and E-value. MYCBP encodes a small binding protein that functions as a c-MYC co-factor through binding to the N-terminal domain of c-MYC and stimulating E-box dependent transcriptional activation (Figure 1B) [15, 17]. Thus, identification of MYCBP in our synthetic lethal genetic screen suggests that the MYC signaling axis may be critical in ALK+ NSCLC.
To validate MYCBP as synthetic lethal gene in our ALK+ NSCLC cell lines, we used shRNAs targeting MYCBP whose shRNA sequences are distinct from those used in the initial screen. Vector shRNA targeting MYCBP and non-gene targeting negative control (NTC) were introduced into the EML4-ALK cell line H2228 to generate stable gene knockdown and negative control lines, respectively (Figure 1C). Knockdown of MYCBP in H2228 demonstrated a 3-fold increase in sensitivity to crizotinib. Stable knockdown of MYCBP was unsuccessful in the H3122 cell line, therefore validation of synthetic lethal inhibition was demonstrated in H3122 cells using a transient siRNA approach to silencing MYCBP (Figure 1D). MYCBP gene silencing decreased cell proliferation by 2-fold in response to crizotinib as compared to scrambled siRNA control (SCR). MYCBP gene silencing was performed in an additional ALK+ cell line, STE-1 (Figure 1E). Despite minimal knockdown efficiency, we still observe a modest increase in crizotinib sensitivity in this cell line. Finally, to demonstrate these effects were specific in ALK-activated NSCLC, MYCBP was effectively silenced in the EGFR mutant cell line HCC827 and treated with crizotinib (Supplementary Figure 1). Crizotinib had no effect on proliferation in this cell line and this remained unchanged upon MYCBP knockdown. Overall these results validate MYCBP as a critical gene whose inhibition increases sensitivity to crizotinib in ALK+ NSCLC cell lines.
MYC is regulated in an ALK-dependent manner in ALK+ NSCLC cells
MYCBP can regulate the oncogenic transcriptional activity of MYC, which is frequently dysregulated in many human malignancies [18-20]. Furthermore, several studies have shown that aberrant ALK signaling in neuroblastoma cells regulates MYC and MYCN transcriptional initiation and protein stability [21, 22]. To determine if differential expression of MYC or MYCBP was present in ALK+ NSCLC compared to other mutant-driver lung cancer subtypes, we measured basal mRNA and protein expression of MYC and MYCBP in a panel of cell lines that included three ALK+ NSCLC cell lines (H3122, H2228, and STE-1), a ROS1 gene fusion cell line (HCC78), two mutant EGFR cell lines (H1650 and HCC827) and two KRAS cell lines (A549 and H460) and NRAS (H1299) NSCLC cell lines (Figure 2A, 2B and Supplementary Figure 2). MYC mRNA expression at baseline is variable across all cell lines, reflecting the ubiquitous nature of MYC, and suggesting different mechanisms of regulation in a resting state. Analysis of MYCBP expression in this cell line panel showed that the ALK+ cell lines showed a trend towards greater transcript levels of MYCBP compared with the other genomic subtypes.
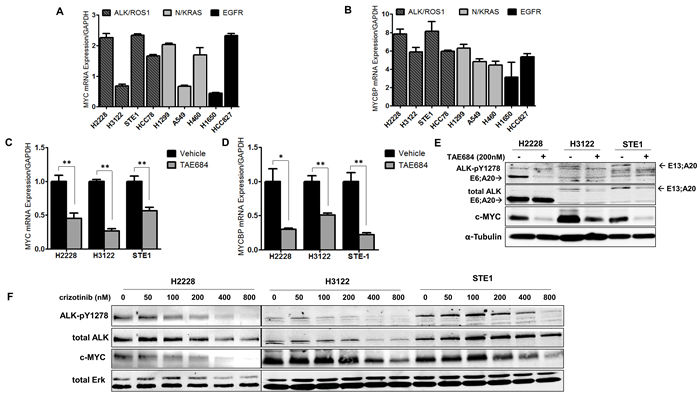
Figure 2: Regulation of MYC and MYCBP expression is ALK-dependent in NSCLC cells. A. and B. Quantitative real-time PCR (qRT-PCR) analysis of MYC or MYCBP in NSCLC cells. NSCLC cell line panel includes ALK+ cell lines (H2228, H3122, STE-1), a ROS1 fusion line (HCC78), two KRAS cell lines (A549; G12S, H460; Q61H) and NRAS (H1299; codon 61), two mutant EGFR cell lines (H1650, HCC827; del 19). C. and D. Quantitative real-time PCR analysis of MYC or MYCBP in ALK+ cell lines in response to 24 hour treatment with 200nM TAE684. (**P < 0.01, *P < 0.05). E. Western blot analysis of H2228, H3122 and STE-1 cells after 24 hour treatment with 200nM TAE684. F. Western blot analysis of ALK+ cells treated for 24 hours with the indicated dose of crizotinib.
To further investigate MYC regulation in ALK+ NSCLC, we examined the expression of MYC and MYCBP in response to pharmacologic ALK inhibition in ALK+ NSCLC cell lines. To eliminate any potential off-target effects, we used TAE684, a potent, highly specific ALK inhibitor [23], as a complementary inhibitor tool for these studies. Treatment with TAE684 reduced MYC transcript levels in each of the ALK+ cell lines tested (Figure 2C). We also examined expression of MYCBP in response to ALK inhibition, as it has been demonstrated that MYCBP expression can be regulated indirectly through MYC [24, 25]. Similar to that of MYC, we observed decreased transcript levels of MYCBP in response to TAE684 (Figure 2D). We observe similar results in response to crizotinib (Supplementary Figure 3). Immunoblot analysis revealed loss of c-MYC protein expression in the three ALK+ cell lines tested in response to TAE684 treatment (Figure 2E). To verify inhibition of the auto-activated ALK kinase domain, an antibody recognizing phosphorylated tyrosines 1278/1282/1283 was employed. Analysis of several NSCLC ROS1 fusion cell lines also demonstrated loss of c-MYC protein in response to TAE684 (Supplementary Figure 4). This is important since ALK and ROS1 are evolutionary conserved kinases and share 77% amino acid identity within the ATP-binding sites, permitting parallel therapeutic approaches against the two kinases [26]. Additionally, we demonstrate reduction of c-MYC protein in response to a dose range of crizotinib (Figure 2F). Furthermore, the observed loss of MYC protein in response to ALK inhibitor is ALK-specific, as we do not see concomitant reduction in MYC protein levels in KRAS or EGFR mutant cell lines treated with an ALK inhibitor (Supplementary Figure 5).
ALK regulates MYC protein stabilization and transcription initiation in ALK+ NSCLC
Regulation of c-MYC can occur through post-translational modifications at MYC-homology Box 1 (MB1) within the N-terminal domain [27]. Phosphorylation events at serine 62 (S62) and threonine 58 (T58) can regulate c-MYC protein stability, where phosphorylation at S62 and T58 together promotes MYC protein stability, but phosphorylation of T58 alone leads to ubiquitination and degradation [28]. To explore c-MYC protein stability in ALK+ NSCLC we analyzed the phosphorylation of these sites in our three ALK+ cell lines in response to TAE684 (Figure 3A). Overall, we observe sustained S62 phosphorylation, indicating c-MYC protein stability is not significantly altered through loss of ALK kinase signaling. However, we observe increased phosphorylation at T58 in H2228 cells in response to TAE684, indicating c-MYC protein stability may be regulated by ALK in this cell line. The levels of S62 (which promotes c-MYC protein stability) remain unchanged suggesting balanced c-MYC protein levels. Minor changes in T58 and S62 phosphorylation are observed in H3122 and STE-1 cells, suggesting ALK may in part play a role in mediating c-MYC protein stability in these cell lines, but does not fully explain significant reduction in total c-MYC protein levels.
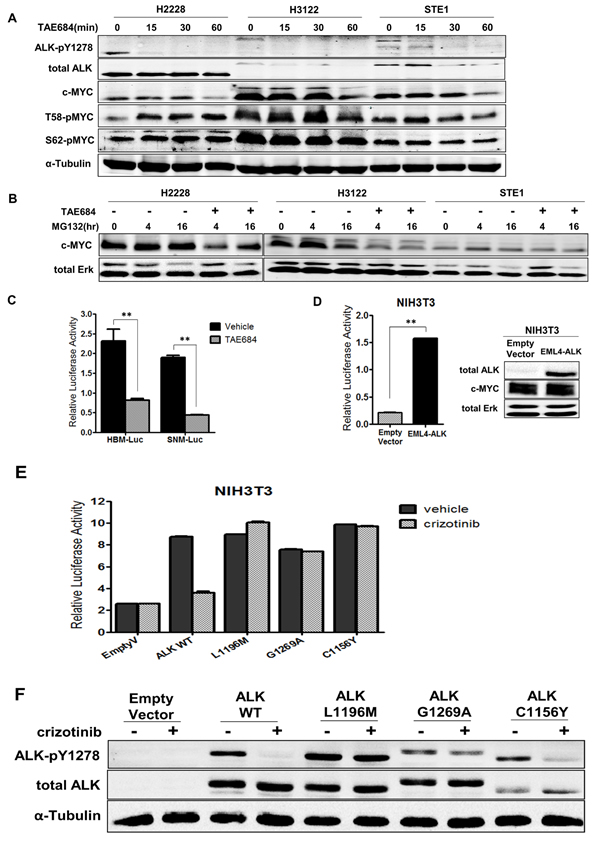
Figure 3: ALK regulates the MYC promoter and minimally regulates c-MYC protein stabilization in ALK+ NSCLC cells. A. Western blot analysis of c-MYC phosphorylation sites in ALK+ cell lines treated with 200nM TAE684 for up to 60 minutes. B. Western blot analysis of ALK+ cell lines treated with proteasome inhibitor MG132 alone or in combination with TAE684 for 0, 4, or 16 hours. C. Luciferase activity measuring MYC promoter activity in H3122 cells expressing HBM-Luc or SNM-Luc promoter construct treated with 200nM TAE684. (**P < 0.01). D. Luciferase activity measuring MYC promoter activity in NIH3T3 cells co-transfected with EML4-ALK or pCDH-Empty Vector and HBM-Luc constructs. Western blot showing expression of ALK in EML4-ALK-expressing NIH3T3 cells. (**P < 0.01). E. Luciferase activity measuring MYC promoter activity in NIH3T3 cells co-transfected with pCDH-empty vector or EML4-ALK wild-type (WT) or the cDNA encoding L1196M, G1269A or C1156Y mutations and the HBM-Luc MYC reporter construct. F. Western blot analysis of NIH3T3 lysates expressing the indicated cDNA vector treated with crizotinib.
To further investigate regulation of c-MYC protein levels in ALK+ cells, we examined c-MYC protein turnover by treating cells with crizotinib and the proteasome inhibitor MG132 (Figure 3B). Overall, we observe increased c-MYC protein levels within 4 hours of MG132 treatment alone, indicating protein is accumulating and not undergoing degradation. In H3122 and STE-1 cells, pre-treatment with ALK inhibitor TAE684 demonstrates reduced c-MYC expression that is not rescued by addition of the proteasome inhibitor, indicating ALK-mediated regulation of MYC is not at the protein level. In the H2228 cell line, we observed some recovery of c-MYC protein after 16 hour treatment with TAE684 in presence of MG132. Since H2228 also demonstrated increased T58 phosphorylation in response to ALK inhibition, this indicates that ALK may be involved in regulating c-MYC protein levels in this cell line.
We next investigated if ALK is involved in transcriptional regulation of MYC, as demonstrated in other ALK-activated tumor types [22, 29, 30]. In order to investigate the effect of ALK activity on initiation of transcription of MYC, we employed promoter-reporter luciferase technology. We used two MYC reporter constructs, SNM-Luc and HBM-Luc, which contain the required P1 and P2 MYC transcription start sites and engineered with a luciferase reporter (Supplementary Figure 6). HBM-Luc is the full-length promoter sequence where SNM-Luc is a truncated reporter, created by terminal deletion of a 502-bp fragment from the 5’ end of the HBM-Luc construct while still containing both P1 and P2 transcription start sites [31, 32]. We transiently transfected these constructs into H3122 cells and measured the luciferase activity after treatment with TAE684 (Figure 3C). As demonstrated, both the HBM-Luc and SNM-Luc promoter constructs were responsive to ALK inhibition indicating that only the minimal sequences for correct transcription of MYC are required for ALK to regulate MYC promoter activity. Similarly, we performed these experiments with a dose range of crizotinib and observe reduced promoter activity within the known IC50 range of H3122 (~150nM) (Supplementary Figure 7). Additionally, in the ALK+ NSCLC cell line STE-1, we observe reduced MYC-promoter activity in response to TAE684, in both reporter assays (Supplementary Figure 8).
To determine if the activation of the MYC promoter is ALK-specific, we co-transfected NIH3T3 cells with EML4-ALK cDNA and HBM-Luc MYC reporter construct and measured luciferase activity compared to cells co-transfected with pCDH-empty vector cDNA and HBM-Luc MYC reporter construct (Figure 3D). The MYC promoter activity in the cells co-transfected with the EML4-ALK vector was 5-fold higher compared to empty vector indicating that ALK is indeed activating the promoter activity of MYC.
To confirm ALK activity increases MYC promoter activity and the observed decrease in luciferase activity with ALK inhibitor is not an off-target (ALK non-specific) pharmacological effect, we utilized EML4-ALK cDNA vectors encoding mutations in the ALK kinase domain that have previously been determined to confer resistance to crizotinib [9]. We co-transfected NIH3T3 cells with cDNA vectors containing pCDH-empty vector or wild-type EML4-ALK, or the same cDNA encoding L1196M, G1269A or C1156Y mutations plus the HBM-Luc MYC reporter construct and measured luciferase activity in response to crizotinib (Figure 3E and 3F). The NIH3T3 cells expressing the EML4-ALK resistant mutations abrogated the impact of crizotinib treatment on MYC promoter activity as compared to those containing wild type EML4-ALK, conferring resistance in the context of MYC expression. Overall, these results implicate ALK in the transcriptional regulation of MYC in ALK rearranged NSCLC.
ALK regulates transcriptional activity of c-MYC in ALK+ NSCLC
We sought to determine if transcriptional activity of c-MYC was mediated in an ALK-dependent manner. We used a luciferase promoter-reporter construct, MBS-Luc, containing four c-MYC E-box consensus sequence binding sites (MBS) upstream of a promoter of the known c-MYC target gene, CDK4 (Supplementary Figure 9) [33]. We transfected H3122 cells with the MBS-Luc reporter construct and measured luciferase activity after treatment with 150nM of crizotinib (Figure 4A). In response to crizotinib, we observe over a 5-fold reduction in luciferase activity, indicating ALK is critical in mediating c-MYC transcriptional activation. Next, we tested whether ALK could induce c-MYC transcriptional activity in a non-tumor cell line ectopically expressing ALK. We employed NIH3T3 cells and co-transfected them with wild-type EML4-ALK or pCDH empty vector in addition to the MBS-Luc reporter construct (Figure 4B). Here we observe a nearly 8-fold induction of c-MYC transcriptional activity in ALK-expressing cells demonstrating that ALK induces c-MYC transcriptional activity. To further assess ALK-dependent regulation of c-MYC activity, we examined mRNA expression of known c-MYC target genes in response to ALK inhibitor TAE684 (Figure 4C). The c-MYC target genes analyzed include cyclin-dependent kinase (CDK4), ornithine decarboxylase (ODC), and carbamoyl phosphate synthase-aspartate transcarbamylase-dihydroorotase (CAD), all of which are implicated in oncogenic signaling. After treatment with TAE684 we observe decreased expression of each of the c-MYC target genes in all three ALK+ cell lines tested. To test the ability of ALK to induce c-MYC target gene expression, we transfected NIH3T3 cells with wild-type EML4-ALK cDNA or empty vector and demonstrate that expression of ALK in the NIH3T3 cells induces expression of these oncogenic c-MYC target genes (Figure 4D). Overall, these results demonstrate that ALK mediates regulation of c-MYC transcriptional activities and induces expression of c-MYC target genes, therefore establishing ALK as a key activator of oncogenic MYC signaling in ALK+ NSCLC.
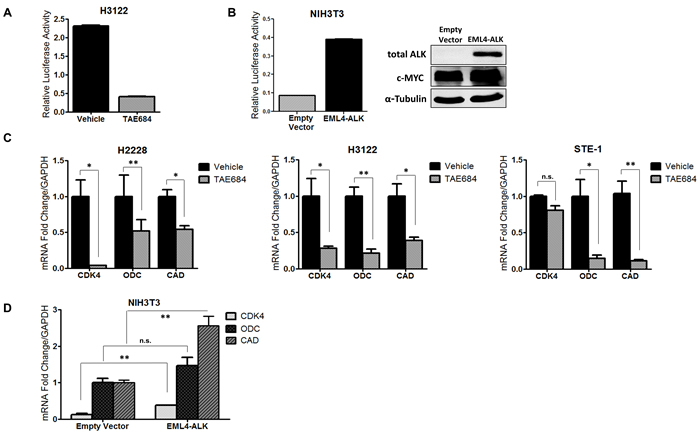
Figure 4: ALK regulates transcriptional activity of c-MYC in ALK+ NSCLC cells. A. Luciferase activity measuring c-MYC target gene promoter activity in H3122 cells expressing the MBS-Luc reporter construct upon treatment with 200nM TAE684. B. Luciferase activity measuring c-MYC target gene promoter activity in NIH3T3 cells co-transfected with EML4-ALK or pCDH-Empty Vector and MBS-Luc constructs. C. Quantitative real-time PCR analysis of c-MYC target genes cyclin-dependent kinase (CDK4), ornithine decarboxylase (ODC), carbamoyl phosphate synthase-aspartate transcarbamylase-dihydroorotase (CAD) in ALK+ cell lines treated with 200nM TAE684. D. Quantitative real-time PCR analysis of c-MYC target genes CDK4, ODC, and CAD in NIH3T3 cells co-transfected with EML4-ALK or pCDH-Empty Vector. (**P < 0.01, *P < 0.05).
Inhibition of MYC as a potential treatment strategy in ALK+ NSCLC
Aberrant expression of MYC is a common feature in many human malignancies and may be critical in ALK+ NSCLC. MYC therefore represents an attractive therapeutic target. To observe the effects of MYC loss in ALK+ NSCLC, we employed RNAi technology to silence MYC in ALK+ cell lines and measured the proliferation in response to crizotinib (Figure 5A). We observed increased crizotinib sensitivity with MYC silencing (siMYC). Interestingly, this appears to be dose-dependent with the level of knockdown observed across the cell lines tested (Figure 5B). In order to correlate the observed effects of RNA silencing to a pharmacologically relevant target, we used TMPyP4, a small molecule capable of stabilizing a specific G-quadruplex within the promoter of MYC and thus inhibiting its expression [34]. We demonstrate reduced MYC mRNA expression and loss of c-MYC protein expression upon treatment with TMPyP4 in the ALK+ NSCLC cells (Figure 5C and 5D). In order to see the effect of combined ALK inhibition with TMPyP4, we treated ALK+ cell lines with increasing dose of crizotinib in the presence of 5μM of the TMPyP4 compound (Figure 5E). We observe increased sensitivity to crizotinib in the presence of the TMPyP4 MYC inhibitor, similar to the MYC RNAi analysis. Taken together, these results demonstrate reduction of MYC increases sensitivity to ALK inhibition in ALK+ NSCLC and provides rationale for dual inhibition in the therapeutic setting.
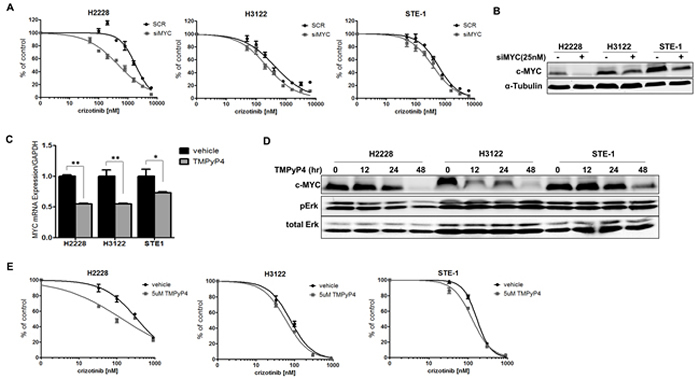
Figure 5: Inhibition of MYC sensitizes ALK+ NSCLC cells to crizotinib. A. H2228, H3122 or STE-1 cells expressing either scrambled siRNA control (siSCR) or SmartPool siRNA targeting MYC (siMYC) were treated with crizotinib for 72 hours and proliferation was measured by MTS assay. B. Western blot demonstrates knockdown efficiency of MYC. C. Quantitative real-time PCR analysis of MYC in ALK+ cells after treatment with 5μM TMPyP4 for 48 hours. (**P < 0.01, *P < 0.05). D. Western blot analysis demonstrating c-MYC protein expression after treatment with 5μM TMPyP4 for up to 48 hours. E. ALK+ cells were treated with crizotinib in combination with either vehicle or 5μM TMPyP4 for 72 hours and proliferation was measured by MTS assay.
Discussion
The profound initial responses observed with the use of targeted therapies, such as ALK inhibitors, in the treatment of oncogene-driven lung adenocarcinoma has demonstrated the value of identifying the dominant signaling pathways driving the cancer. However, complete responses are not durable and resistance inevitably emerges. Furthermore, some ALK positive lung cancer patients do not exhibit significant tumor shrinkage with ALK-inhibitor treatment, suggesting refractory residual disease may result from compensatory signaling pathways [11, 35]. Recent work by our lab demonstrated a critical role for EGFR signaling in ALK+ cancer cells [36]. By understanding the additional signaling requirements of these oncogene-positive cancer cells, we can begin to identify compensatory survival pathways that mediate the intrinsic and acquired resistance and improve treatments strategies.
Alternative or effector signaling pathways are emerging as mechanisms of resistance in the era of oncogene-targeted therapy. As reported recently, ALK+ lung cancer cells demonstrate a dependence on MAPK signaling for growth and survival and as a mechanism of ALK inhibitor resistance [37]. In order to identify genes (and pathways) whose suppression can confer sensitivity to crizotinib, we used a loss-of-function genetic screen in ALK rearranged lung cancer cell lines. The results from the screen uncovered MYCBP as a target in sensitizing ALK+ NSCLC to crizotinib, implicating the MYC signaling axis as critical in this tumor type. MYC encodes the oncogenic transcription factor c-MYC, which heterodimerizes with Max and promotes transcription of genes required for growth and proliferation. The mechanisms through which c-MYC mediates its broad range of biological effects are thought to include co-factors that can interact with c-MYC to regulate its transcriptional activity. MYCBP is one such co-factor that interacts with c-MYC through binding to the N-terminal transactivation domain and promotes E-box dependent transcription of c-MYC target genes [15]. This co-factor regulation of c-MYC may be critical in ALK+ NSCLC survival as we demonstrate that RNAi-mediated loss of MYCBP increases sensitivity to ALK-inhibitor crizotinib in ALK+ NSCLC cell lines, therefore validating our findings from the synthetic lethal gene screen. It is notable that another hit in this synthetic lethal screen, EGFR, has recently been validated by our lab [36].
The association between activating ALK mutations and MYC or MYCN is observed in other malignancies including neuroblastoma and anaplastic large cell lymphoma (ALCL). A connection between ALK and MYC has been observed in neuroblastoma, where ALK gain-of-function mutations are more frequent in MYCN amplified tumors and activation of full length ALK increases mRNA, protein expression, and transformation potential of MYCN in neuroblastoma cell lines [21, 22]. In ALCL, activation of ALK through a conditionally dimerized construct results in increased c-MYC protein and MYC mRNA expression [38]. Furthermore, it was demonstrated that NPM-ALK-induced expression of MYC is mediated through interferon regulatory factor (IRF4), a transcription factor shown to bind the MYC promoter and induce MYC expression [29]. In neuroblastoma, ALK activity has been shown to promote transcription of MYCN through phosphorylation of ERK5, a member of the MAPK family, in a PI3K-dependant manner thus integrating ALK effector signaling and MYC regulation [22, 30].
In this present study, we observe decreased abundance of MYC mRNA and c-MYC protein expression in response to ALK inhibition implicating ALK in regulation of MYC in NSCLC. The loss of c-MYC protein expression was also observed in several ROS1 fusion cell lines in response to TAE684, consistent with highly homologous kinase domains between ALK and ROS1. Furthermore, we demonstrate regulation of MYC promoter activity by ALK in ALK+ cell lines and non-tumor NIH3T3 ectopically expressing EML4-ALK. Additional analysis using mutant ALK kinase domain expression vectors with known crizotinib resistance activity in NIH3T3 cells, demonstrates abrogated responses to crizotinib in the ALK-mutants thus diminishing the effect of ALK inhibitor on c-Myc transcriptional activity. This finding not only demonstrates that ALK is necessary for MYC transcription in this model, but also points to a possible mechanism of oncogene co-operation in the resistance setting. Previous studies have demonstrated enhanced MYCN protein stability by constitutive activation of ALK [21]. In our study, we do not observe definitive regulation at the protein level as ALK inhibition fails to alter c-MYC phosphorylation status in H3122 and STE-1 cells and furthermore, cells pre-treated with ALK inhibitor and then exposed to proteasome inhibition exhibit loss of c-MYC expression indicating possible regulation at the level of transcription. However, based on the observation of modest increase in phosphorylation at T58 upon crizotinib treatment in H2228 cells, we cannot completely rule out protein stability as a mechanism of MYC regulation by ALK. Additionally, it is thought that regulation of protein stability by ALK effector signaling and ALK-mediated regulation of MYC transcription are not mutually exclusive [21, 30, 39]. MYC is a potent activator of oncogenic transcription programs and demonstrated to be required for tumor growth in multiple tumor types. In a retrospective analysis in lung adenocarcinomas, c-MYC copy number gain was an independent poor-prognostic factor for disease-free and overall survival, with a possible association with EGFR mutation status [40]. We examined the role of ALK in c-MYC transactivation of known target genes and find that c-MYC transactivation can be regulated by ALK. The mechanism through which ALK induces activation of c-MYC target genes is unknown. Our group recently performed analysis of circulating tumor DNA from ALK+ patients collected prior to initiating treatment and at time of progression on ALK inhibitor. MYC amplification was observed in 2/42 ALK-inhibitor resistant patient samples suggesting MYC overexpression or amplification as a potential mechanism of resistance to ALK inhibitors [41].
Since MYC and ALK could be cooperative in oncogenic signaling and therapy resistance, we examined the effect of combination inhibition in ALK+ NSCLC cells. RNAi-mediated silencing of MYC increased sensitivity to ALK inhibition. Additionally, use of a G-quadruplex inhibitor that inhibits transcription of MYC also resulted in increased sensitivity to ALK inhibition. These results indicate that the MYC axis is critical and has the potential to be therapeutically exploited in ALK+ NSCLC. In summary, through a genome-wide RNAi screen we identified MYCBP as a potential target in sensitization to ALK kinase inhibition. We demonstrate that the MYC signaling axis is critical in ALK+ NSCLC and is positively regulated by ALK providing a potential therapeutic opportunity for combination therapy.
Materials and Methods
Cell lines and reagents
H2228, H3122, and HCC78 were a kind gift from Dr. John D. Minna (The University of Texas, Southwestern Medical Center, Dallas, TX) and STE-1 were a kind gift from Dr. Christine M. Lovly (Vanderbilt University School of Medicine, Nashville, TN). ROS1 fusion cell lines CUTO2 (SDC4-ROS1), CUTO23 and CUTO27 (CD74-ROS1), and CUTO28 (TPM3-ROS1) were generated at the University of Colorado from patient tumor samples following informed consent under an IRB-approved protocol. ALK and ROS1 fusion cell lines were grown in RPMI supplemented with 10% FBS. H1299, A549, H460, H1650, HCC827 were obtained from the University of Colorado Cancer Center Tissue Culture Core and cultured in RPMI supplemented with 5% FBS. NIH3T3 cells were obtained from American Type Culture collection and grown in Dulbecco’s Modified Eagle’s Media with 5% FBS. Crizotinib (PF-02341066) was obtained from Pfizer, Inc. NVP-TAE684 and MG132 were purchased from Selleck Chemicals. Antibodies used were ALK pY1278/1282/1283 (#3983), total ALK (#3791), ERK pT202/Y204 (#9191), total ERK (#9107), c-MYC (#5605), c-MYC pS62 (#13748) were purchased from Cell Signaling Technology. MYCBP (ab172444) c-MYC pT58 (28842) was purchased from Abcam, and alpha-tubulin (sc-8035), was purchased from Santa Cruz biotechnology.
Proliferation assays
Proliferation was measured using the MTS CellTiter 96 Aqueous Proliferation Assay from Promega according to the manufacturer’s instructions. Briefly, cells were seeded into 96-well plates at a density to permit exponential growth throughout the length of the assay 24 hours before drug treatment, and proliferation was measured 72 hours after treatment. The absorbance at 490 nm was measured in 96-well plates using a Microplate Reader from Molecular Devices. The IC50 values were calculated using Prism v5.02 from GraphPad Software.
Immunoblotting
Immunoblotting was conducted as previously described with minor modifications [9]. Briefly, cells were lysed in modified RIPA buffer supplemented with Halt Protease and Phosphatase Inhibitor Cocktail purchased from Thermo Scientific. Total protein was separated by SDS-PAGE, transferred to nitrocellulose, and stained with the indicated primary antibodies. Protein detection was achieved by imaging with an Odyssey Imager and Odyssey Version 3.0 image analysis software from LI-COR Biotechnology.
Quantitative real-time PCR (qRT-PCR)
Total RNA (1ug) was reverse transcribed using High Capacity cDNA Reverse Transcription Kit from Life Technologies. 1uL of cDNA was PCR amplified in a 20uL reaction including TaqMan 2X Universal Master Mix and TaqMan gene expression probe/primer set for MYC (Hs00153408), MYCBP (Hs01894873), and GAPDH (Hs02758991) as an internal control for normalized gene expression. Samples were run in triplicate for a total of 3 separate experiments.
Luciferase reporter assays
H3122, STE-1 or NIH3T3 cells were transfected with 5ug of promoter-driven firefly luciferase plasmid and 5ug TK-Renilla using TransIT-2020 reagent (Mirus, Madison, WI). After treatment incubation, cells were lysed and assayed following Dual-Luciferase Reporter Assay System instructions (Promega, Madison, WI). The HBM-Luc, SNM-Luc, and MBS-Luc promoter luciferase constructs were obtained from Addgene (Cambridge, MA). Lentivirus preparation
Lentivirus production was performed by transfecting viral packaging vectors pCMV-VSV-G and pΔ8.9 into 293T cells using TransIT-293 reagent (Mirus) as previously described [42]. For the genome-wide screen GeneNet Lentiviral Human 50K library was used (pS1H1-H1 Puro; Systems Biosciences). shRNA vectors used for validation were sourced from TRC (pLKO.1).
shRNA synthetic lethal gene screen
The screen used a lentiviral shRNA library carrying 3-5 target sequences for approximately 50,000 human gene transcripts (SBI, Mountain View, CA). This library was transduced into H2228 and H3122 cells at a low MOI to allow for one gene knockdown per cell and selected in puromycin for two weeks to eliminate untransduced cells. This period of growth also allowed for elimination of shRNAs that target essential genes. The population of transduced cells was then divided into two groups for treatment with vehicle control or the ALK inhibitor crizotinib at an IC80 for both cell lines (H2228: 300nM, H3122: 150nM). Total RNA was isolated and reverse-transcribed, cDNA was amplified by nested PCR with addition of adapter sequences. Sequencing was performed on an Illumina Genome Analyzer and shRNAs were identified and the number of clusters for each shRNA sequence was quantified.
Bioinformatics analysis
The Bioinformatics for Next Generation Sequencing! (BiNGS!) workflow was used to analyze and interpret the genome-wide synthetic lethal data [16]. A pre-processing step removed low quality and erroneous reads, typically non-barcoded sequences. Reads were then mapped to the shRNA reference library using Bowtie [43]. Additional filtering removed shRNAs mapping to sequences of unannotated genes, shRNAs where the median raw count was greater in the control than the maximum raw count in the treatment group if the shRNA is enriched in the treatment group, and vice versa (medC > maxT). A negative binomial was used to model the read count distribution in the data using edgeR [44]. Post-analysis determined the q-value of false discovery rate for multiple comparisons of the shRNAs and collapsed shRNAs to genes by combining q-values for all the shRNAs representing the same gene using weighted Z-transformation [45]. The associated weighted Z-transformed p-value (P(wZP)) is used to sort the gene list demonstrating differentially represented shRNAs, identifying synthetic lethal genes.
Statistical analysis
Statistical significance was assessed by Student’s t-Test (two-tailed distribution, two-sample, unequal variance) and considered statistically significant with P-value <0.05, (**P < 0.01, *P < 0.05).
Acknowledgments
We thank Dr. Christine Lovly for the generous gift of the STE-1 cell line. Dr. James DeGregori’s contribution to development of the synthetic lethal gene screen was supported by R01CA157850.
Financial Support
This work was supported by a Career Development Award from the University of Colorado Lung Cancer SPORE (funded by the National Cancer Institute (NCI) of the National Institutes of Health (NIH) grant P50CA058187), a research grant from Pfizer, and funds from the Boettcher Foundation’s Webb-Waring Biomedical Research Program to RCD.
Conflicts of Interest
RCD has received honorarium, consulting fees and a research grant from Pfizer and consulting fees from OxOnx.
References
1. Ramalingam SS, Owonikoko TK, Khuri FR. Lung cancer: New biological insights and recent therapeutic advances. CA Cancer J Clin. 2011; 61:91-112.
2. Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, Ou SH, Dezube BJ, Janne PA, Costa DB, Varella-Garcia M, Kim WH, Lynch TJ, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010; 363:1693-1703.
3. Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004; 350:2129-2139.
4. Pao W, Miller VA, Kris MG. ‘Targeting’ the epidermal growth factor receptor tyrosine kinase with gefitinib (Iressa) in non-small cell lung cancer (NSCLC). Semin Cancer Biol. 2004; 14:33-40.
5. Choi YL, Takeuchi K, Soda M, Inamura K, Togashi Y, Hatano S, Enomoto M, Hamada T, Haruta H, Watanabe H, Kurashina K, Hatanaka H, Ueno T, et al. Identification of novel isoforms of the EML4-ALK transforming gene in non-small cell lung cancer. Cancer research. 2008; 68:4971-4976.
6. Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H, Bando M, Ohno S, Ishikawa Y, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007; 448:561-566.
7. Soda M, Takada S, Takeuchi K, Choi YL, Enomoto M, Ueno T, Haruta H, Hamada T, Yamashita Y, Ishikawa Y, Sugiyama Y, Mano H. A mouse model for EML4-ALK-positive lung cancer. Proc Natl Acad Sci U S A. 2008; 105:19893-19897.
8. Shaw AT, Yeap BY, Solomon BJ, Riely GJ, Gainor J, Engelman JA, Shapiro GI, Costa DB, Ou SH, Butaney M, Salgia R, Maki RG, Varella-Garcia M, et al. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. Lancet Oncol. 2011; 12:1004-1012.
9. Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, Kondo KL, Linderman DJ, Heasley LE, Franklin WA, Varella-Garcia M, Camidge DR. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012; 18:1472-1482.
10. Zhang S, Wang F, Keats J, Zhu X, Ning Y, Wardwell SD, Moran L, Mohemmad QK, Anjum R, Wang Y, Narasimhan NI, Dalgarno D, Shakespeare WC, et al. Crizotinib-resistant mutants of EML4-ALK identified through an accelerated mutagenesis screen. Chem Biol Drug Des. 2011; 78:999-1005.
11. Shaw AT, Engelman JA. Ceritinib in ALK-rearranged non-small-cell lung cancer. N Engl J Med. 2014; 370:2537-2539.
12. Gadgeel SM, Gandhi L, Riely GJ, Chiappori AA, West HL, Azada MC, Morcos PN, Lee RM, Garcia L, Yu L, Boisserie F, Di Laurenzio L, Golding S, et al. Safety and activity of alectinib against systemic disease and brain metastases in patients with crizotinib-resistant ALK-rearranged non-small-cell lung cancer (AF-002JG): results from the dose-finding portion of a phase 1/2 study. Lancet Oncol. 2014; 15:1119-1128.
13. Larkins E, Blumenthal GM, Chen H, He K, Agarwal R, Gieser G, Stephens O, Zahalka E, Ringgold K, Helms W, Shord S, Yu J, Zhao H, et al. FDA Approval: Alectinib for the Treatment of Metastatic, ALK-Positive Non-Small Cell Lung Cancer Following Crizotinib. Clin Cancer Res. 2016; 22:5171-5176.
14. Kim DW, Tiseo M, Ahn MJ, Reckamp KL, Hansen KH, Kim SW, Huber RM, West HL, Groen HJM, Hochmair MJ, Leighl NB, Gettinger SN, Langer CJ, et al. Brigatinib in Patients With Crizotinib-Refractory Anaplastic Lymphoma Kinase-Positive Non-Small-Cell Lung Cancer: A Randomized, Multicenter Phase II Trial. J Clin Oncol. 2017;35:2490-2498.
15. Taira T, Maeda J, Onishi T, Kitaura H, Yoshida S, Kato H, Ikeda M, Tamai K, Iguchi-Ariga SM, Ariga H. AMY-1, a novel C-MYC binding protein that stimulates transcription activity of C-MYC. Genes Cells. 1998; 3:549-565.
16. Kim J, Tan AC. BiNGS!SL-seq: a bioinformatics pipeline for the analysis and interpretation of deep sequencing genome-wide synthetic lethal screen. Methods in molecular biology. 2012; 802:389-398.
17. Sakamuro D, Prendergast GC. New Myc-interacting proteins: a second Myc network emerges. Oncogene. 1999; 18:2942-2954.
18. Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nature reviews Cancer. 2008; 8:976-990.
19. Nesbit CE, Tersak JM, Prochownik EV. MYC oncogenes and human neoplastic disease. Oncogene. 1999; 18:3004-3016.
20. Zhou ZQ, Hurlin PJ. The interplay between Mad and Myc in proliferation and differentiation. Trends Cell Biol. 2001; 11:S10-14.
21. Berry T, Luther W, Bhatnagar N, Jamin Y, Poon E, Sanda T, Pei D, Sharma B, Vetharoy WR, Hallsworth A, Ahmad Z, Barker K, Moreau L, et al. The ALK(F1174L) mutation potentiates the oncogenic activity of MYCN in neuroblastoma. Cancer Cell. 2012; 22:117-130.
22. Schonherr C, Ruuth K, Kamaraj S, Wang CL, Yang HL, Combaret V, Djos A, Martinsson T, Christensen JG, Palmer RH, Hallberg B. Anaplastic Lymphoma Kinase (ALK) regulates initiation of transcription of MYCN in neuroblastoma cells. Oncogene. 2012; 31:5193-5200.
23. Galkin AV, Melnick JS, Kim S, Hood TL, Li N, Li L, Xia G, Steensma R, Chopiuk G, Jiang J, Wan Y, Ding P, Liu Y, et al. Identification of NVP-TAE684, a potent, selective, and efficacious inhibitor of NPM-ALK. Proc Natl Acad Sci U S A. 2007; 104:270-275.
24. Sibbesen NA, Kopp KL, Litvinov IV, Jonson L, Willerslev-Olsen A, Fredholm S, Petersen DL, Nastasi C, Krejsgaard T, Lindahl LM, Gniadecki R, Mongan NP, Sasseville D, et al. Jak3, STAT3, and STAT5 inhibit expression of miR-22, a novel tumor suppressor microRNA, in cutaneous T-Cell lymphoma. Oncotarget. 2015; 6:20555-20569. https://doi.org/10.18632/oncotarget.4111.
25. Xiong J, Du Q, Liang Z. Tumor-suppressive microRNA-22 inhibits the transcription of E-box-containing c-Myc target genes by silencing c-Myc binding protein. Oncogene. 2010; 29:4980-4988.
26. Shaw AT, Ou SH, Bang YJ, Camidge DR, Solomon BJ, Salgia R, Riely GJ, Varella-Garcia M, Shapiro GI, Costa DB, Doebele RC, Le LP, Zheng Z, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med. 2014; 371:1963-1971.
27. Gustafson WC, Weiss WA. Myc proteins as therapeutic targets. Oncogene. 2010; 29:1249-1259.
28. Marshall GM, Liu PY, Gherardi S, Scarlett CJ, Bedalov A, Xu N, Iraci N, Valli E, Ling D, Thomas W, van Bekkum M, Sekyere E, Jankowski K, et al. SIRT1 promotes N-Myc oncogenesis through a positive feedback loop involving the effects of MKP3 and ERK on N-Myc protein stability. PLoS Genet. 2011; 7:e1002135.
29. Weilemann A, Grau M, Erdmann T, Merkel O, Sobhiafshar U, Anagnostopoulos I, Hummel M, Siegert A, Hayford C, Madle H, Wollert-Wulf B, Fichtner I, Dorken B, et al. Essential role of IRF4 and MYC signaling for survival of anaplastic large cell lymphoma. Blood. 2015; 125:124-132.
30. Umapathy G, El Wakil A, Witek B, Chesler L, Danielson L, Deng X, Gray NS, Johansson M, Kvarnbrink S, Ruuth K, Schonherr C, Palmer RH, Hallberg B. The kinase ALK stimulates the kinase ERK5 to promote the expression of the oncogene MYCN in neuroblastoma. Sci Signal. 2014; 7:ra102.
31. Facchini LM, Chen S, Marhin WW, Lear JN, Penn LZ. The Myc negative autoregulation mechanism requires Myc-Max association and involves the c-myc P2 minimal promoter. Mol Cell Biol. 1997; 17:100-114.
32. Han Y, San-Marina S, Liu J, Minden MD. Transcriptional activation of c-myc proto-oncogene by WT1 protein. Oncogene. 2004; 23:6933-6941.
33. Hermeking H, Rago C, Schuhmacher M, Li Q, Barrett JF, Obaya AJ, O’Connell BC, Mateyak MK, Tam W, Kohlhuber F, Dang CV, Sedivy JM, Eick D, et al. Identification of CDK4 as a target of c-MYC. Proc Natl Acad Sci U S A. 2000; 97:2229-2234.
34. Gonzalez V, Guo K, Hurley L, Sun D. Identification and characterization of nucleolin as a c-myc G-quadruplex-binding protein. The Journal of biological chemistry. 2009; 284:23622-23635.
35. Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, De Pas T, Besse B, Solomon BJ, Blackhall F, Wu YL, Thomas M, O’Byrne KJ, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013; 368:2385-2394.
36. Vaishnavi A, Schubert L, Rix U, Marek LA, Le AT, Keysar SB, Glogowska MJ, Smith MA, Kako S, Sumi NJ, Davies KD, Ware KE, Varella-Garcia M, et al. EGFR Mediates Responses to Small-Molecule Drugs Targeting Oncogenic Fusion Kinases. Cancer research. 2017; 77:3551-3563.
37. Hrustanovic G, Olivas V, Pazarentzos E, Tulpule A, Asthana S, Blakely CM, Okimoto RA, Lin L, Neel DS, Sabnis A, Flanagan J, Chan E, Varella-Garcia M, et al. RAS-MAPK dependence underlies a rational polytherapy strategy in EML4-ALK-positive lung cancer. Nature medicine. 2015; 21:1038-1047.
38. Raetz EA, Perkins SL, Carlson MA, Schooler KP, Carroll WL, Virshup DM. The nucleophosmin-anaplastic lymphoma kinase fusion protein induces c-Myc expression in pediatric anaplastic large cell lymphomas. Am J Pathol. 2002; 161:875-883.
39. Bisikirska BC, Adam SJ, Alvarez MJ, Rajbhandari P, Cox R, Lefebvre C, Wang K, Rieckhof GE, Felsher DW, Califano A. STK38 is a critical upstream regulator of MYC’s oncogenic activity in human B-cell lymphoma. Oncogene. 2013; 32:5283-5291.
40. Seo AN, Yang JM, Kim H, Jheon S, Kim K, Lee CT, Jin Y, Yun S, Chung JH, Paik JH. Clinicopathologic and prognostic significance of c-MYC copy number gain in lung adenocarcinomas. British journal of cancer. 2014; 110:2688-2699.
41. Doebele R, Banks K, Ihuegbu N, Diaz J, Lanman R, Blakely C. P1.02-057 Clinical Utility of ctDNA for Detecting ALK Fusions and Resistance Events in NSCLC: Analysis of a Laboratory Cohort. Journal of Thoracic Oncology. 2016; 12:S522.
42. Doebele RC, Schulze-Hoepfner FT, Hong J, Chlenski A, Zeitlin BD, Goel K, Gomes S, Liu Y, Abe MK, Nor JE, Lingen MW, Rosner MR. A novel interplay between Epac/Rap1 and mitogen-activated protein kinase kinase 5/extracellular signal-regulated kinase 5 (MEK5/ERK5) regulates thrombospondin to control angiogenesis. Blood. 2009; 114:4592-4600.
43. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009; 10:R25.
44. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139-140.
45. Whitlock MC. Combining probability from independent tests: the weighted Z-method is superior to Fisher’s approach. J Evol Biol. 2005; 18:1368-1373.