
Introduction
Approximately 70% of primary breast tumors express estrogen receptor α (ER) and/or progesterone receptor (PR). Patients with such hormone receptor-positive breast cancers are typically treated with adjuvant anti-estrogen therapies such as tamoxifen or aromatase inhibitors (AIs) for 5-10 yrs after surgical removal of the primary tumor [1]. While adjuvant anti-estrogen therapies have been used to successfully treat hormone-dependent breast cancer, approximately one-third of patients develop recurrent advanced/metastatic disease that is rarely cured and frequently fatal [2, 3].
Mechanistic target of rapamycin (mTOR) complex I (mTORC1) is a critical signaling hub that regulates cellular and organismal homeostasis by balancing anabolic and catabolic processes with nutrient, energy, oxygen availability, and growth factor input signaling [4]. mTORC1 inhibitors continue to be investigated as therapeutics for many cancer subtypes. Hyperactivation of the phosphatidylinositol 3-kinase (PI3K)/AKT/mTORC1 pathway is associated with anti-estrogen resistance in ER+ breast cancer [5]. In patients with recurrent/metastatic ER+ breast cancer resistant to a non-steroidal AI (e.g., letrozole, anastrozole), treatment with the steroidal AI exemestane plus the mTORC1 inhibitor everolimus (RAD001; afinitor) significantly improved median progression-free survival (but not overall survival) compared to exemestane/placebo (11 vs. 4.1 months) in the BOLERO-2 study [6-8]. The TAMRAD study similarly evaluated the use of tamoxifen +/- everolimus for the treatment of advanced, anti-estrogen-resistant ER+ breast cancer; patients in the combination arm experienced a prolonged time-to-progression compared to tamoxifen/placebo (8.6 vs. 4.5 months), and a 55% reduction in risk of death [9]. Such findings supported the U.S. FDA approval of everolimus for use in combination with exemestane for patients with ER+ breast cancer resistant to a non-steroidal AI. However, the benefit of continued anti-estrogen therapy (with exemestane) in the context of mTORC1 inhibition (with everolimus) remains unproven (being tested in the ongoing BOLERO-6 trial [10]); indeed, single-agent everolimus elicited clinical benefit in 6/22 (27%) patients with advanced ER+ breast cancer [11]. Positive findings with exemestane/everolimus in the setting of advanced disease prompted the ongoing clinical testing of adjuvant everolimus in combination with anti-estrogen therapy for patients with high-risk early-stage ER+/HER2- breast cancer [12]; many such cases will have anti-estrogen-sensitive micrometastatic disease.
Preclinical data show that combined therapeutic targeting of ER and mTORC1 is more effective than single agents to inhibit growth and induce apoptosis of breast cancer cells and xenografts [13, 14]. However, the synergistic mechanism of anti-cancer action of such drug combinations is incompletely understood. These signaling pathways exhibit crosstalk at several levels. mTORC1 and its effector ribosomal protein S6 kinase β-1 (p70S6K) catalyze serine phosphorylation of insulin receptor substrate-1 (IRS-1), inducing IRS-1 downregulation [15]. ER drives the transcription of genes encoding growth factor receptor tyrosine kinases (RTKs), ligands, and signaling adaptors, including IRS-1 and its activator, insulin-like growth factor-1 receptor (IGF-1R). Estrogen can also stimulate non-genomic ER-mediated activation of IGF-1R/insulin receptor (InsR) complexes, epidermal growth factor receptor (EGFR), Src, PI3K, and mitogen-activated protein kinase kinase (MEK) [16]. Estrogen stimulation induces ER binding to the mTORC1 component regulatory-associated protein of mTOR (Raptor), which drives nuclear translocation of Raptor. mTORC1 also phosphorylates ER on S104/106 to promote transcriptional activity [17]. We and others have shown that inhibition of mTORC1 relieves negative feedback on upstream activators, including IRS-1, IGF-1R, human epidermal growth factor receptor 3 (HER3), and PI3K, which may subsequently promote cell survival and antagonize the anti-cancer effects of mTORC1 inhibitors [18-21]. Herein, we demonstrate a role for ER in such feedback activation of survival pathways in breast cancer cell lines and human tumors, providing supportive rationale for using anti-estrogens to enhance the effects of mTORC1 inhibitors in ER+ breast cancer.
Results
Loss of ER activity abrogates mTORC1 inhibitor-induced feedback activation of PI3K/AKT
Prior reports showed that ER+ breast cancer cells are growth-suppressed by treatment with anti-estrogens, estrogen deprivation, or mTORC1 inhibition [13, 14]. In line with these findings, we observed that treatment of 4/4 ER+ breast cancer cell lines with fulvestrant (fulv) or RAD001 suppressed growth (Supplementary Figure 1). Combined targeting of ER and mTORC1 with the combination of fulv/RAD001 synergistically inhibited growth (CI≤1 in 4/4 cell lines; Figure 1A and Supplementary Figure 1).
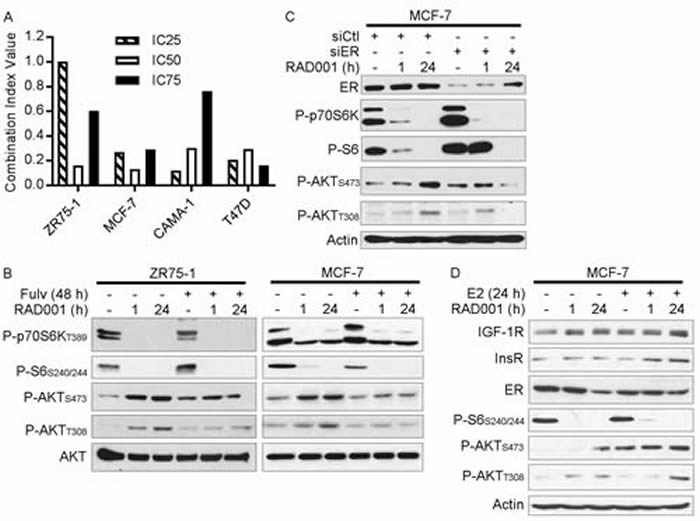
Figure 1: Loss of ER activity abrogates mTORC1 inhibitor-induced feedback activation of PI3K/AKT. A. ER+ breast cancer cells were treated with dose ranges of RAD001 and/or fulv for 5 d. Relative numbers of viable cells were measured by SRB assay (full growth curves are shown in Supplementary Figure 1). Combination Index Values (CI) at the IC25, IC50, and IC75 for the drug combination were determined by the Median Effect method. B-D) Cell lysates were analyzed by immunoblot using the indicated antibodies. In B., cells were pretreated +/- 1 µM fulv for 24 h, then treated +/- 20 nM RAD001 for an additional 1 h or 24 h (total fulv treatment time = 48 h). In C., siRNA was used to knock down ER. Two days later, cells were treated +/- 20 nM RAD001 for 1 h or 24 h. In D., cells were pretreated with hormone-depleted medium (10% DCC-FBS) for 3 d, then treated +/- 1 nM E2 for 24 h, 20 nM RAD001 for 1 h or 24 h, or combinations (total E2/RAD001 treatment time = 24 h).
To explore the mechanism underlying the synergistic effects of anti-estrogens and RAD001 on growth, we pre-treated ER+ breast cancer cells +/- fulv for 24 h (to allow time for ER transcriptional effects to subside), and then co-treated +/- RAD001 for 1 h or 24 h. Treatment with RAD001 inhibited mTORC1 activity, as indicated by decreased levels of phospho-p70S6K (an mTORC1 substrate) and phospho-S6 (a p70S6K substrate) (Figure 1B). As we and others observed previously [19, 22], short-term (1 h) and longer-term (24 h) mTORC1 inhibition induced increased phosphorylation of AKT at T308 (substrate of PDK1; marker of PI3K activity) and S473 (substrate of mTORC2), implying increased activation of the PI3K/AKT pathway. Co-treatment with fulv mitigated the RAD001-induced increases in P-AKTT308 and P-AKTS473, suggesting that ER is required for mTORC1 inhibitor-induced PI3K/AKT activation in endocrine-sensitive ER+ breast cancer cells (Figure 1B and Supplementary Figure 2A/2B). The ER-specific effects of fulv were confirmed by RNAi-mediated knockdown of ER, which similarly suppressed RAD001-induced AKT phosphorylation (Figure 1C). In contrast, fulv less effectively mitigated RAD001-induced AKT activation in long-term estrogen-deprived MCF-7/LTED cells, and RAD001 increased AKT activation in fulv-resistant MCF-7/FR and T47D/FR cells despite ER inhibition with fulv (Supplementary Figure 3). Thus, combined anti-estrogen/mTORC1 inhibitor therapy may be most effective in the setting of endocrine-sensitive disease.
We then assessed whether ER activation primes the PI3K/AKT pathway for activation in response to mTORC1 inhibition. MCF-7 cells were pretreated with hormone-depleted medium (DCC-FBS) for 3 d, and then treated +/- E2 for 24 h, RAD001 for 1 h or 24 h, or combinations. Although RAD001 modestly induced P-AKT in hormone-depleted cells, the addition of E2 enhanced RAD001-induced AKT phosphorylation (Figure 1D). Thus, ligand-dependent ER signaling promotes PI3K/AKT activation in response to mTORC1 inhibition.
mTORC1 inhibitor-induced activation of PI3K/AKT requires IGF-1R/InsR signaling
ER promotes the transcription of genes encoding RTKs (e.g., IGF-1R), ligands, and signaling adaptors (e.g., IRS-1) that activate PI3K [16]. Since fulv decreased IGF-1R levels in MCF-7 and ZR75-1 cells (Supplementary Figure 2A), we considered whether this effect blunted PI3K/AKT activation in response to mTORC1 inhibition. ZR75-1 and MCF-7 cells were pretreated +/- the IGF-1R/InsR dual kinase inhibitor OSI-906 for 2 h, then treated +/- RAD001 or OSI-906 for an additional hour. OSI-906 blocked the RAD001-induced increase in P-AKT (Figure 2A). However, OSI-906 treatment partially downregulated phospho-p70S6K, suggesting partial mTORC1 inhibition that may have triggered feedback activation of AKT. To confirm the effects of OSI-906 on IGF-1R/InsR, we used RNAi to knock down IGF-1R and InsR. Dual knockdown of both RTKs blocked RAD001-induced P-AKT, while single-RTK knockdown elicited variable effects (Figure 2B), possibly due to compensation between IGF-1R and InsR [23].
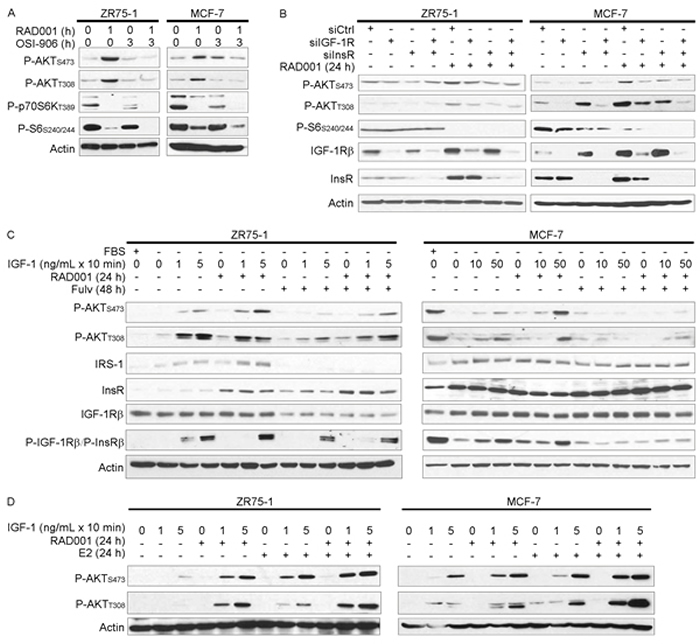
Figure 2: mTORC1 inhibitor-induced feedback activation of PI3K/AKT requires IGF-1R/InsR kinase activity. Cell lysates were analyzed by immunoblot using the indicated antibodies. A. Cells were pretreated +/- 4 µM OSI-906 for 2 h, then treated +/- 20 nM RAD001 for an additional hour (total OSI-906 treatment time = 3 h). B. siRNA against IGF-1R, InsR, or non-silencing control was used to knock down expression. Two days post-transfection, cells were treated +/- 20 nM RAD001 for 24 h. C. Cells were pretreated +/- fulv (48 h) or RAD001 (24 h), with the final 24 h of pretreatment in serum-free medium as indicated. Cells were then stimulated +/- IGF-1 for 10 min. D. Cells were pretreated with hormone-depleted medium (DMEM/10% DCC-FBS) for 3 d, then treated +/- 20 nM RAD001 or 1 nM E2 for 24 h in serum-free medium. Cells were then stimulated with IGF-1 for 10 min.
We then tested the effects of mTORC1 and ER inhibition on sensitivity to IGF-1 ligand in the absence of other exogenous growth factors. Serum-deprived cells were pretreated +/- fulv or RAD001, then stimulated +/- IGF-1 for 10 min. mTORC1 inhibition with RAD001 sensitized cells to IGF-1-induced activation of AKT without appreciably changing ligand-induced phospho-IGF-1Rβ/P-InsRβ levels (Figure 2C and Supplementary Figure 4). Fulv treatment mitigated RAD001-induced AKT phosphorylation, which may have occurred by different mechanisms in different cell lines as suggested by differences between T308 and S473 phosphorylation patterns. Fulv hindered IGF-1-induced IGF-1R/InsR phosphorylation in MCF-7 cells, but had only modest effects in ZR75-1 and HCC-1428 cells (Figure 2C and Supplementary Figure 4). Serum deprivation increased total IGF-1R, InsR, and IRS-1 levels in MCF-7 cells (Figure 2C), which may reflect a loss of negative feedback regulation of expression due to decreased AKT activation [24]. In contrast, fulv decreased IRS-1 and IGF-1R levels in serum-deprived ZR75-1 cells, and IRS-1 in HCC-1428 cells (Figure 2C and Supplementary Figure 4). Thus, we postulate that fulv hindered RAD001-induced sensitization to IGF-1 in ZR75-1 and HCC-1428 cells by decreasing expression of signaling components (IRS-1, IGF-1R), and in MCF-7 cells by decreasing activation of IGF-1R/InsR; the mechanism underlying the latter is unclear.
To determine whether ER activation primes cells for response to IGF-1, cells were cultured in hormone-depleted conditions for 3 d, serum-deprived for 1 d in the presence or absence of RAD001 or E2, and then stimulated +/- IGF-1 for 10 min. Inhibition of mTORC1 or treatment with E2 each enhanced AKT activation in response to IGF-1, and combined RAD001/E2 was most effective (Figure 2D). Taken together, these data implicate ER-modulated IGF-1R/InsR signaling in mTORC1 inhibitor-induced feedback activation of PI3K/AKT.
Requirement for IRS-1/2 in mTORC1 inhibitor-induced feedback activation of PI3K/AKT
IGF-1R and InsR activate PI3K by phosphorylating adaptor proteins such as IRS-1 and -2, and Tyr phosphorylated IRS-1/2 engage the p85 regulatory subunit of Class IA PI3K to promote PI3K activity [15, 25]. IRS-1/2 levels increased upon RAD001 treatment in MCF-7 cells under growth conditions (Figure 3A), which is likely due to loss of p70S6K phosphorylation that targets IRS-1/2 for proteasomal degradation [19, 26]. Using RNAi against IRS-1 and -2, we observed that dual knockdown decreased AKT activation induced by RAD001 (Figure 3A). RAD001 also increased association between the p85 regulatory subunit of PI3K and IRS-1 that was partly mitigated by fulv, possibly due to the fulv-induced decrease in IRS-1 expression (Figure 3B and Supplementary Figure 5). Thus, ER-regulated IRS-1/2 expression and interaction with PI3K are implicated in feedback induced by mTORC1 inhibition.
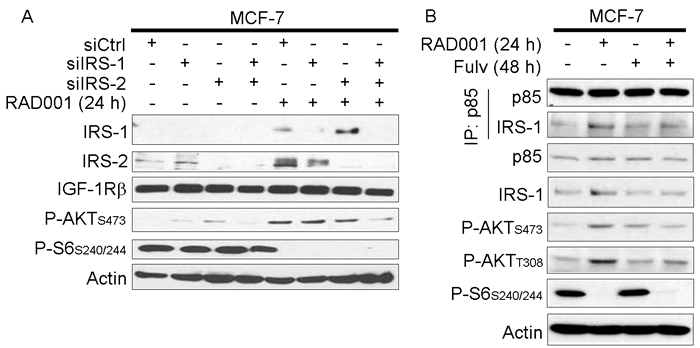
Figure 3: mTORC1 inhibitor-induced feedback activation of PI3K/AKT requires IRS-1/2. A. siRNA was used to knock down IRS-1, IRS-2, or non-silencing control. Two days later, cells were treated +/- 20 nM RAD001 for 24 h. Cell lysates were analyzed by immunoblot using the indicated antibodies. B. Cells were treated +/- fulv (48 h) or RAD001 (24 h), then lysates were used for immunoprecipitation of p85. Lysates and immunoprecipitates were analyzed as in (A).
Estrogen deprivation in patients with ER+ breast cancer prevents tumor AKT activation in response to mTORC1 inhibition ex vivo
Based on our preclinical findings, we hypothesized that inhibition of ER decreases IGF-1R/InsR/PI3K/AKT signaling induced by mTORC1 inhibition in ER+ breast cancer. To test this hypothesis in human tumors without exposing patients to an mTORC1 inhibitor, we conducted a presurgical study in which patients with Stage I-III ER+/HER2- breast cancer were treated +/- the AI letrozole for 10-21 d to induce estrogen deprivation; the 10-21-day period allows time for maximal estrogen suppression and downstream changes in the levels of ER-regulated transcripts [27]. At the time of surgical resection of the tumor, viable tumor fragments were acquired, adapted to serum-free medium ex vivo for 5 h, then treated +/- RAD001 or OSI-906 for 1 h. Tumor lysates were analyzed by immunoblot to assess AKT phosphorylation.
We first evaluated tumors from 10 patents in Arm A who did not receive presurgical anti-cancer treatment. Ex vivo treatment of tumor fragments with RAD001 increased the levels of AKT phosphorylation on both T308 and S473 (Figure 4A). When quantifying (phospho-)protein levels by densitometry (control-treated tumors were set at “1”), we observed a 45% to 2,400% increase in P-AKTT308 compared to control (mean % change +/- SD = 388% +/- 623%), and up to a 431% increase in P-AKTS473 (mean % change +/- SD = 157% +/- 35%). Treatment with OSI-906 alone or in combination with RAD001 significantly decreased P-AKT levels compared to single-agent RAD001 (Figure 4A), confirming our in vitro findings (Figure 2A). These results suggest that mTORC1 inhibition induces PI3K/AKT activation in an IGF-1R/InsR kinase-dependent manner in human ER+ breast tumors.
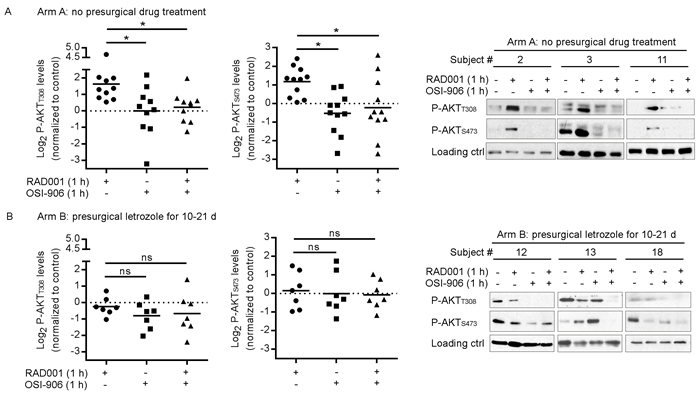
Figure 4: Presurgical estrogen deprivation in patients with ER+ breast cancer prevents RAD001-induced PI3K/AKT activation in tumors ex vivo. Patients with early-stage ER+/HER2- breast cancer received either A. no presurgical treatment (Arm A, n = 10), or B. presurgical treatment with letrozole for 10-21 d (Arm B, n = 7). Within 1 h after surgical resection, 1-mm3 punch cores were taken from primary tumors ex vivo. Tumor cores were put in serum-free medium for 5 h, then treated +/- 20 nM RAD001, 4 µM OSI-906, or the combination for 1 h. Tumor lysates were analyzed by immunoblot using the indicated antibodies. Immunoblot film signals were quantified by densitometry. Signal values from control-treated tumors were set at “1.” Data are shown as log2 P-AKT of inhibitor-treated sample relative to control. Horizontal bars indicate mean values. *p < 0.05 by Bonferroni multiple comparison-adjusted post-hoc test. C. Representative results are shown at right from 3 patients’ tumors from Arms A and B; actin or vinculin was assessed to confirm equal loading.
We then analyzed tumors from 7 patents in Arm B who received 10-21 d of letrozole treatment prior to surgery. Ex vivo treatment of Arm B tumors with RAD001 did not significantly increase P-AKT levels: P-AKTT308 ranged from -51% to 163% compared to control (mean % change +/- SD = -10% +/- 38%), and P-AKTS473 measured -51% to 281% compared to control (mean +/- SD = 135% +/- 91%). Accordingly, OSI-906/RAD001 co-treatment did not significantly alter P-AKT levels compared to RAD001 alone (Figure 4B). These data suggest that estrogen-induced ER activation is required for mTORC1 inhibitor-induced activation of PI3K/AKT in human ER+ breast tumors.
Presurgical anti-estrogen treatment often suppresses cell proliferation in ER+ breast tumors [28]. To confirm the growth-suppressive effects of presurgical letrozole, we measured tumor cell proliferation by Ki67 IHC. Tumor Ki67 scores were not significantly different between baseline biopsies and surgical specimens from patients who did not receive presurgical treatment (Arm A). In contrast, presurgical letrozole significantly decreased Ki67 score in Arm B (Figure 5A and Supplementary Figure 6). Presurgical letrozole also induced a trend towards decreased tumor PR levels (p = 0.06), reflecting reduced ER transcriptional activity, while tumors from untreated patients showed no notable difference between baseline and surgical specimens (Figure 5B and Supplementary Figure 6). Letrozole did not appreciably alter ER expression (Supplementary Figures 6-7).
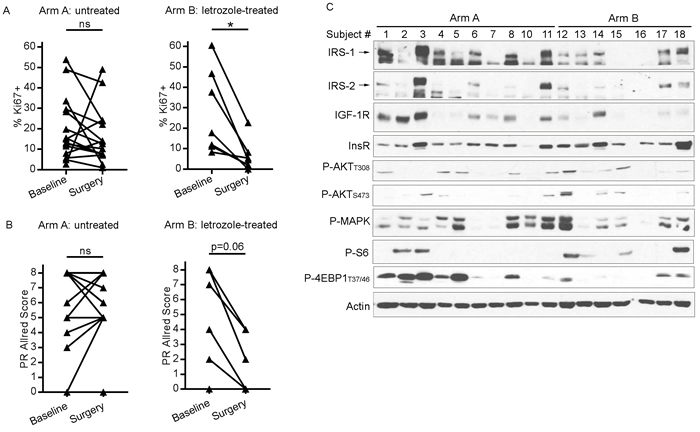
Figure 5: Presurgical estrogen deprivation in patients with ER+ breast cancer decreases tumor cell proliferation and IGF-1R/IRS-1/IRS-2 expression. A/B) Formalin-fixed, paraffin-embedded diagnostic tumor biopsies (baseline) and surgical specimens [post-letrozole (Arm B) or untreated (Arm A)] were analyzed by IHC using antibodies against Ki67 A. or PR B. *p<0.05 by Wilcoxon test. ns- not significant. C. Lysates from frozen surgical tumor specimens were analyzed by immunoblot using the indicated antibodies.
We further evaluated surgical tumor specimens by immunoblot analysis of lysates. Post-letrozole tumors showed lower IGF-1R, IRS-1, and IRS-2 levels than tumors from untreated patients (Figure 5C); similar differences were detected by IGF-1R IHC (Supplementary Figures 6-7). Letrozole-treated tumors also showed decreased phosphorylation of 4EBP1 and MAPK compared to untreated tumors (Figure 5C), suggesting that estrogen deprivation suppresses mTORC1 and MEK activation. These findings from human tumors collectively suggest that 1) ER drives the expression of IGF-1R, IRS-1, and IRS-2, and 2) IGF-1R/InsR kinase activity is required for feedback activation of PI3K/AKT upon inhibition of mTORC1, providing mechanistic insight into the effects of mTORC1 and ER inhibition on signaling in ER+ breast cancer.
Discussion
Herein, we provide evidence from ER+ breast cancer cell lines and human tumors implicating ER in signaling responses to mTORC1 inhibition. mTORC1 inhibitors generally have a cytostatic effect on ER+ breast cancer cells [13, 14], which is thought to be due in part to a loss of negative feedback signaling to activators upstream of PI3K including RTKs and IRS-1 [18-21], driving activation of PI3K/AKT to promote cell survival. We demonstrated that mTORC1 inhibition with RAD001 (everolimus) induces IGF-1R/InsR/IRS-1/IRS-2-dependent activation of PI3K/AKT signaling in ER+ breast cancer cells and human tumors treated ex vivo. RAD001 synergized with the anti-estrogen fulv to inhibit growth in 4/4 ER+ breast cancer cell lines. Inhibition of ER with fulv, RNAi (in vitro), or estrogen deprivation (AI treatment of patients) decreased mTORC1 inhibitor-induced PI3K/AKT activation in endocrine-sensitive cells and tumors. In most cases, anti-estrogen treatments decreased expression of IGF-1R/IRS-1/IRS-2 signaling components, which may be the predominant mechanism by which ER drives such feedback signaling (Figure 6). However, it is also possible that ER-mediated non-genomic signaling, such as via IGF-1R/InsR [22, 29], drives PI3K/AKT activation in response to mTORC1 inhibition.
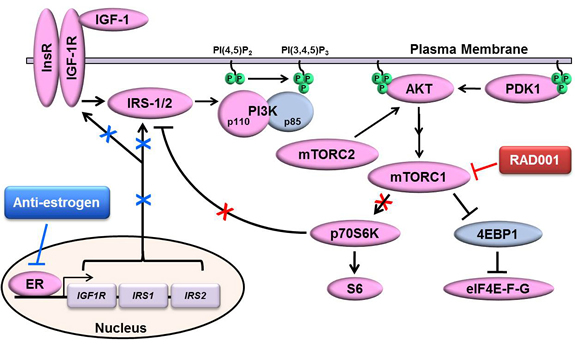
Figure 6: Proposed model of ER-mediated control of mTORC1 inhibitor-induced activation of PI3K/AKT. Depicted is the canonical signaling pathway in which ligand-activated IGF-1R/InsR homo- and hetero-dimers phosphorylate IRS-1/2 at Tyr sites, creating docking sites for p85/PI3K that derepress p110/PI3K. p110/PI3K converts PIP2 to PIP3, and PIP3 enables recruitment of PH domain-containing proteins (e.g., AKT, PDK1) to the cytoplasmic face of the plasma membrane. PDK1 and mTORC2 phosphorylate AKT, and activated AKT in turn signals through the TSC1/2 complex to activate mTORC1. mTORC1 and its phospho-activated substrate p70S6K phosphorylate IRS-1/2 at Ser residues to promote proteasomal degradation of IRS-1/2, creating a negative feedback loop. Inhibition of mTORC1 with RAD001 blocks such negative feedback, leading to increased IRS-1/2/PI3K/AKT activation. ER drives the expression of IGF-1R and IRS-1/2, which are downregulated by anti-estrogen treatment. Thus, anti-estrogens mitigate mTORC1 inhibitor-induced activation of PI3K/AKT in ER+ breast cancer cells.
The extensive crosstalk between the ER and PI3K/AKT/mTORC1 pathways provides rationale to target these pathways in ER+ breast cancer. However, there is a lack of understanding of the reciprocity of this crosstalk. There are numerous examples of PI3K/AKT/mTORC1 signaling regulating ER activity, including AKT-, mTOR-, and p70S6K-mediated phosphorylation and activation of ER, and AKT-mediated phosphorylation of c-Jun, which complexes with ER [30-36]. However, the mechanism(s) by which ER affects PI3K/AKT/mTORC1 signaling remain to be fully elucidated. In a positive feedback model, ER promotes the transcription of genes encoding RTKs, ligands, and signaling adaptors, which are predicted to increase PI3K pathway activation. Anti-estrogen treatment downregulated IGF-1R, IRS-1, and IRS-2 levels in ER+ breast cancer cells and human tumors (Figures 2C, 5C, Supplementary Figure 2A, Supplementary Figure 4, Supplementary Figure 5, Supplementary Figure 7C/7D), although effects varied between biological systems. Accordingly, clinical evidence suggests that ER function sustains PI3K activation. In patients with ER+ breast cancer, 6 months of neoadjuvant treatment with letrozole +/- cyclophosphamide reduces P-AKTS473 and P-mTORS2448 tumor levels, which correlate with improved response and disease outcome [37]. In a second study, 14 d of neoadjuvant letrozole modestly decreased P-S6 levels [38]. Similarly, we showed that presurgical letrozole decreased P-4EBP1 levels (Figure 5C). Hence, anti-estrogens may suppress ER+ breast cancer growth in part by decreasing PI3K/AKT/mTOR signaling. The findings presented herein closely align with a positive feedback model, and mechanistically show how blocking ER can abrogate activation of PI3K/AKT in response to mTORC1 inhibition.
Clinical and preclinical data also support a negative feedback model of ER-PI3K/AKT/mTORC1 crosstalk in which these pathways antagonize each other. ER and PR levels are inversely correlated with markers of PI3K/AKT/mTOR activation in primary ER+ breast tumors [5, 39]. Adaptation to long-term ER inhibition (with fulv or hormone deprivation) increases PI3K/AKT/mTORC1 activity in ER+ breast cancer cells [5, 40-42]. PI3K inhibitor treatment of patients with ER+ breast cancer for ≥14 d induces upregulation of tumor ER levels and activity [43], which Toska et al. recently demonstrated is mediated by loss of AKT-induced epigenetic silencing of ER activity [44]. Furthermore, we previously showed that adaptation of ER+ breast cancer cells to long-term PI3K inhibition induces upregulation of ER and PR [45]. Thus, substantial data support seemingly conflicting models of positive and negative feedback between the ER and PI3K/AKT/mTORC1 pathways, resolution of which will require further study.
AKT phosphorylation at both T308 and S473 is required for maximal kinase activation [46]. Phosphorylation of T308 is mediated by the PI3K effector PDK1, while S473 phosphorylation is mediated by mTORC2 [47, 48]. The effects of mTORC1 inhibition on AKT phosphorylation varied between cell lines: RAD001 drastically increased both P-AKTT308 and P-AKTS473 in ZR75-1 and MCF-7 cells, but only robustly increased P-AKTS473 in HCC-1428 cells (Figure 1B and Supplementary Figure 2). These observations indicate cell line-specific differences in PI3K-PDK1-mTORC2-AKT programming, and suggest that inhibition of ER and mTORC1 affect different feedback circuits upstream of T308 and/or S473. AKT has 3 human isoforms (AKT1, AKT2, and AKT3) with overlapping and distinct biological functions [46], and similar amino acid motifs surrounding T308 and S473. Since phospho-AKT antibodies do not distinguish between isoforms, it is possible that isoform-specific differences in AKT phosphorylation contributed to the observed differences between cell lines.
In 2012, the U.S. FDA approved everolimus (RAD001) as the first PI3K/AKT/mTOR pathway inhibitor for the treatment of advanced ER+ breast cancer in combination with the steroidal AI exemestane following progression on a non-steroidal AI. The BOLERO-2 study showed that combined exemestane/everolimus therapy increased median PFS from 4.1 to 10.6 months compared to exemestane/placebo [6]. An exploratory subgroup analysis revealed that patients with cancer that had progressed during or within 12 months after the end of (neo)adjuvant AI therapy, rendering exemestane +/- everolimus the first-line therapy for their advanced disease, experienced even greater benefit from the addition of everolimus: exemestane/everolimus increased median PFS to 15.2 months from 4.2 months with exemestane/placebo [49]. Lingering questions remain regarding 1) the optimal time in the disease course to introduce everolimus, 2) the need for continued anti-estrogen therapy for anti-estrogen-resistant disease in the context of mTORC1 inhibition with everolimus, and 3) the clinical significance of mTORC1 inhibitor-induced feedback activation of upstream pathways (i.e., RTKs, PI3K, AKT). Herein, we provide evidence from preclinical models and functional human tumor studies demonstrating a role for ER in RAD001-induced PI3K/AKT activation in endocrine-sensitive ER+ breast cancer. These findings provide rationale for anti-estrogen co-treatment in the setting of endocrine-sensitive ER+ disease being treated with an mTORC1 inhibitor; such drug combinations are currently being tested clinically in the adjuvant setting [12].
Materials and Methods
Cell lines and RNA interference
Parental cell lines were obtained from ATCC and cultured in DMEM (Gibco) with 10% FBS (Hyclone) for <3 months before analysis. Long-term estrogen-deprived (LTED) cells were generated through culture in phenol red-free DMEM with 10% dextran-charcoal-treated FBS (DCC-FBS) for 3 months. Cells resistant to the anti-estrogen fulvestrant (fulv; MCF-7/FR, T47D/FR) were gifts from Matthew Ellis (Washington University), and maintained in DMEM/10% FBS with 1 µM fulv. LTED and FR cells were authenticated by STR genotyping (Univ. of Vermont Advanced Genome Technologies Core). Cells were transfected with siRNA targeting IGF-1R (#GS3480), InsR (#GS3643), IRS-1 (#GS3667), IRS-2 (#GS8660), or non-silencing control (#1027310) from Qiagen using Lipofectamine RNAiMAX per manufacturer’s instructions (Life Technologies). Cells were treated with phenol red-free DMEM containing 10% dextran-charcoal-treated FBS (DCC-FBS; Hyclone), RAD001, OSI-906 (Selleck Chemicals), fulv (Tocris Bioscience), IGF-1 (R&D Systems), or 17β-estradiol (E2; Sigma) as indicated.
Sulforhodamine B (SRB) assay
Cells were seeded in triplicate at 3,000-5,000/well in 96-well plates. The following day, cells were treated as indicated for 5 d. Relative numbers of adherent cells were determined by SRB staining [50]. IC50 values were determined using Calcusyn software (Biosoft), where the mean Abs490 value of triplicate wells was used. For drug synergy studies, cells were treated with fulv and RAD001 at fixed ratios of IC50 values. The combination indices (CI) for the drug combination at the IC25, IC50, and IC75 were determined by the Median Effect method [51] using Calcusyn software. CI<1 is indicative of synergy.
Patients and ex vivo tumor culture
Seventeen patients with stage I-III invasive ER+/HER2- breast cancer were recruited to clinical study NCT02010021. The clinical study protocol was approved by the Dartmouth College Institutional Review Board. All patients provided signed informed consent. This study was conducted in accordance with good clinical practice and all applicable regulatory requirements, including the 1996 version of the Declaration of Helsinki. Tumors were required to have ≥50% ER+ cancer cells, and be HER2- [immunohistochemistry (IHC) 0-1+, or with a FISH ratio of <1.8 if IHC is 2+ or if IHC had not been done]. The first 10 patients received no presurgical anti-cancer therapy (Arm A), and an interim analysis was performed. The next 7 patients were presurgically treated with the AI letrozole for 10-21 d (Arm B). Primary tumors were surgically resected as standard of care, and tumor fragments for research use were then dissected from the surgical specimen within 1.5 h of removal from the patient. Tumor fragments were transported in serum-free DMEM on ice. Using a 1-mm biopsy punch device (Miltex), ~40 viable tumor cores were dissected from each tumor specimen within 30 min. Tumor cores (~10 per treatment group) were cultured ex vivo in serum-free DMEM for 5 h, then treated +/- 20 nM RAD001, 4 µM OSI-906, or the combination for 1 h. Tumor cores were then snap-frozen, lysed, and analyzed by immunoblot.
Immunoprecipitation and immunoblotting
Immunoblotting of lysates from cells and frozen tumor fragments was performed as described [52]. Immunoprecipitation of p85 from cell lysates was performed using Dynal protein-G beads (Invitrogen) and p85 antibody (Abcam) as described [53]. Following SDS-PAGE, proteins were transferred to nitrocellulose membranes, and even protein loading was visually confirmed by Ponceau S staining. Blots were probed with antibodies against P-AKTS473, P-AKTT308, AKT, P-p70S6KT389, P-S6S240/244, P-IGF-1RβY1135/6/P-InsRβY1150/1, IGF-1Rβ, InsRβ, IRS-1, IRS-2, actin, vinculin (Cell Signaling), p85 (Abcam), or ER (Santa Cruz). HRP-labeled secondary antibodies (GE Healthcare) and ECL substrate (Pierce) were used for signal detection. Densitometry analysis of immunoblot film was performed using ImageJ software, and relative signal values were analyzed by ANOVA followed by Bonferroni multiple comparison-adjusted post-hoc test between groups.
Immunohistochemistry (IHC)
Five-micron sections of formalin-fixed, paraffin-embedded (FFPE) tumor tissues acquired from patients at baseline (diagnostic biopsy) and at the time of surgery (untreated or after presurgical letrozole) were used for IHC staining with antibodies against Ki67 (Biocare Medical), ER (Cell Marque, SP1), PR (Leica Biosystems, # NCL-l-PGR-312), or IGF-1R (Cell Signaling, #3027). IHC evaluation was performed by a breast pathologist blinded to treatment arm; details are provided in Supplementary Information. In Arm A, two patients had bilateral tumors, and two patients had two ipsilateral tumors; all were analyzed by IHC.
Abbreviations
ER- estrogen receptor alpha; HER3- human epidermal growth factor receptor 3; IGF-1R- insulin-like growth factor-1 receptor; IHC- immunohistochemistry; InsR- insulin receptor; IRS-1- insulin receptor substrate-1; IRS-1- insulin receptor substrate-2; MAPK- mitogen-activated protein kinase (also called ERK1/2); MEK- mitogen-activated protein kinase kinase; mTOR- mechanistic target of rapamycin; mTORC1- mTOR complex I; p70S6K- ribosomal protein S6 kinase beta-1; PI3K- phosphatidylinositol 3-kinase; PFS- progression-free survival; PR- progesterone receptor; Raptor- regulatory-associated protein of mTOR; RTK- receptor tyrosine kinase; SRB- Sulforhodamine B
Authors' Contributions
WY and TWM designed the preclinical studies. WY, VC, NAT and TWM conducted preclinical experiments, and collected, analyzed, and interpreted data. GNS, TWM, and JG designed the clinical study. GNS directed and recruited patients to the clinical study. JDM procured clinical tissue specimens, and scored IHC specimens. WY performed ex vivo analyses of fresh clinical specimens. WY and TWM wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgments
We thank the following Norris Cotton Cancer Center Shared Resources for assistance: Pathology, Biostatistics, and the Office of Clinical Research. We thank patients and family members involved in the NCT02010021 clinical study.
Funding
Financial support was provided by the American Cancer Society (RSG-13-292-01-TBE to TWM) and NIH (Dartmouth College Norris Cotton Cancer Center Support Grant P30CA023108).
Conflicts of interest
The authors declare that they have no competing interests.
References
1. Goss PE, Ingle JN, Pritchard KI, Robert NJ, Muss H, Gralow J, Gelmon K, Whelan T, Strasser-Weippl K, Rubin S, Sturtz K, Wolff AC, Winer E, et al. Extending Aromatase-Inhibitor Adjuvant Therapy to 10 Years. The New England journal of medicine. 2016; 375: 209-19.
2. Ferlay JS, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Internat J Cancer. 2010: 2893-917.
3. Davies C, Godwin J, Gray R, Clarke M, Cutter D, Darby S, McGale P, Pan HC, Taylor C, Wang YC, Dowsett M, Ingle J, Peto R, et al, and Early Breast Cancer Trialists’ Collaborative Group. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet. 2011; 378: 771-84.
4. Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Molecular cell. 2010; 40: 310-22.
5. Miller TW, Hennessy BT, Gonzalez-Angulo AM, Fox EM, Mills GB, Chen H, Higham C, Garcia-Echeverria C, Shyr Y, Arteaga CL. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor-positive human breast cancer. The Journal of clinical investigation. 2010; 120: 2406-13.
6. Baselga J, Campone M, Piccart M, Burris HA 3rd, Rugo HS, Sahmoud T, Noguchi S, Gnant M, Pritchard KI, Lebrun F, Beck JT, Ito Y, Yardley D, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012; 366: 520-9.
7. Yardley DA, Noguchi S, Pritchard KI, Burris HA 3rd, Baselga J, Gnant M, Hortobagyi GN, Campone M, Pistilli B, Piccart M, Melichar B, Petrakova K, Arena FP, et al. Everolimus plus exemestane in postmenopausal patients with HR(+) breast cancer: BOLERO-2 final progression-free survival analysis. Advances in therapy. 2013; 30: 870-84.
8. Piccart M, Hortobagyi GN, Campone M, Pritchard KI, Lebrun F, Ito Y, Noguchi S, Perez A, Rugo HS, Deleu I, Burris HA, Provencher L, Neven P, et al. Everolimus plus exemestane for hormone-receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: overall survival results from BOLERO-2. Ann Oncol. 2014; 25: 2357-62.
9. Bachelot T, Bourgier C, Cropet C, Ray-Coquard I, Ferrero JM, Freyer G, Abadie-Lacourtoisie S, Eymard JC, Debled M, Spaeth D, Legouffe E, Allouache D, El Kouri C, et al. Randomized Phase II Trial of Everolimus in Combination With Tamoxifen in Patients With Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Metastatic Breast Cancer With Prior Exposure to Aromatase Inhibitors: A GINECO Study. J Clin Oncol. 2012.
10. Ejlertsen B, Jerusalem GH, Hurvitz SA, De Boer RH, Taran T, Sahmoud T, Burris HA 3rd. BOLERO-6: Phase II study of everolimus plus exemestane versus everolimus or capecitabine monotherapy in HR+, HER2- advanced breast cancer. J Clin Oncol. 2013; 31: abstr TPS660.
11. Ellard SL, Clemons M, Gelmon KA, Norris B, Kennecke H, Chia S, Pritchard K, Eisen A, Vandenberg T, Taylor M, Sauerbrei E, Mishaeli M, Huntsman D, et al. Randomized phase II study comparing two schedules of everolimus in patients with recurrent/metastatic breast cancer: NCIC Clinical Trials Group IND.163. J Clin Oncol. 2009; 27: 4536-41.
12. Chavez-Mac Gregor M, Barlow WE, Pusztai L, Goetz MP, Rastogi P, Ganz PA, Mamounas EP, Paik S, Bandos S, Gralow J, Lew DL, Hortobagyi GN. Phase III randomized, placebo-controlled clinical trial evaluating the use of adjuvant endocrine therapy +/- one year of everolimus in patients with high-risk, hormone receptor (HR) positive and HER2-negative breast cancer (BC): SWOG/NRG/Alliance S1207 (NCT01674140). J Clin Oncol. 2015; 33: abstr TPS637.
13. Boulay A, Rudloff J, Ye J, Zumstein-Mecker S, O’Reilly T, Evans DB, Chen S, Lane HA. Dual inhibition of mTOR and estrogen receptor signaling in vitro induces cell death in models of breast cancer. Clin Cancer Res. 2005; 11: 5319-28.
14. Martin LA, Pancholi S, Farmer I, Guest S, Ribas R, Weigel MT, Thornhill AM, Ghazoui Z, A’Hern R, Evans DB, Lane HA, Johnston SR, Dowsett M. Effectiveness and molecular interactions of the clinically active mTORC1 inhibitor everolimus in combination with tamoxifen or letrozole in vitro and in vivo. Breast Cancer Res. 2012; 14: R132.
15. Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nature reviews Genetics. 2006; 7: 606-19.
16. Miller TW, Balko JM, Arteaga CL. Phosphatidylinositol 3-kinase and antiestrogen resistance in breast cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011; 29: 4452-61.
17. Alayev A, Salamon RS, Berger SM, Schwartz NS, Cuesta R, Snyder RB, Holz MK. mTORC1 directly phosphorylates and activates ERalpha upon estrogen stimulation. Oncogene. 2016; 35: 3535-43.
18. Miller TW, Forbes JT, Shah C, Wyatt SK, Manning HC, Olivares MG, Sanchez V, Dugger TC, de Matos Granja N, Narasanna A, Cook RS, Kennedy JP, Lindsley CW, et al. Inhibition of mammalian target of rapamycin is required for optimal antitumor effect of HER2 inhibitors against HER2-overexpressing cancer cells. Clin Cancer Res. 2009; 15: 7266-76.
19. O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, Baselga J, Rosen N. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006; 66: 1500-8.
20. Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, Egia A, Sasaki AT, Thomas G, Kozma SC, Papa A, Nardella C, Cantley LC, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. Journal of Clinical Investigation. 2008; 118: 3065-74.
21. Wan X, Harkavy B, Shen N, Grohar P, Helman LJ. Rapamycin induces feedback activation of Akt signaling through an IGF-1R-dependent mechanism. Oncogene. 2007; 26: 1932-40.
22. Miller TW, Perez-Torres M, Narasanna A, Guix M, Stal O, Perez-Tenorio G, Gonzalez-Angulo AM, Hennessy BT, Mills GB, Kennedy JP, Lindsley CW, Arteaga CL. Loss of Phosphatase and Tensin homologue deleted on chromosome 10 engages ErbB3 and insulin-like growth factor-I receptor signaling to promote antiestrogen resistance in breast cancer. Cancer Res. 2009; 69: 4192-201.
23. Fox EM, Miller TW, Balko JM, Kuba MG, Sanchez V, Smith RA, Liu S, Gonzalez-Angulo AM, Mills GB, Ye F, Shyr Y, Manning HC, Buck E, et al. A kinome-wide screen identifies the insulin/IGF-I receptor pathway as a mechanism of escape from hormone dependence in breast cancer. Cancer Res. 2011; 71: 6773-84.
24. Chandarlapaty S, Sawai A, Scaltriti M, Rodrik-Outmezguine V, Grbovic-Huezo O, Serra V, Majumder PK, Baselga J, Rosen N. AKT inhibition relieves feedback suppression of receptor tyrosine kinase expression and activity. Cancer cell. 2011; 19: 58-71.
25. Miller TW, Perez-Torres M, Narasanna A, Guix M, Stal O, Perez-Tenorio G, Gonzalez-Angulo AM, Hennessy BT, Mills GB, Kennedy JP, Lindsley CW, Arteaga CL. Loss of Phosphatase and Tensin homologue deleted on chromosome 10 engages ErbB3 and insulin-like growth factor-I receptor signaling to promote antiestrogen resistance in breast cancer. Cancer research. 2009; 69: 4192-201.
26. Haruta T, Uno T, Kawahara J, Takano A, Egawa K, Sharma PM, Olefsky JM, Kobayashi M. A rapamycin-sensitive pathway down-regulates insulin signaling via phosphorylation and proteasomal degradation of insulin receptor substrate-1. Mol Endocrinol. 2000; 14: 783-94.
27. Miller WR, Larionov A, Anderson TJ, Evans DB, Dixon JM. Changes in breast cancer transcriptional profiles after treatment with the aromatase inhibitor, letrozole. Pharmacogenet Genomics. 2007; 17: 813-26.
28. Geisler J, Detre S, Berntsen H, Ottestad L, Lindtjorn B, Dowsett M, Einstein Lonning P. Influence of neoadjuvant anastrozole (Arimidex) on intratumoral estrogen levels and proliferation markers in patients with locally advanced breast cancer. Clin Cancer Res. 2001; 7: 1230-6.
29. Song RX, Fan P, Yue W, Chen Y, Santen RJ. Role of receptor complexes in the extranuclear actions of estrogen receptor alpha in breast cancer. Endocrine-related cancer. 2006; 13 Suppl 1: S3-13.
30. Miller TW, Balko JM, Arteaga CL. Phosphatidylinositol 3-Kinase and Antiestrogen Resistance in Breast Cancer. J Clin Oncol. 2011; 29: 4452-61.
31. Logan SK, Falasca M, Hu P, Schlessinger J. Phosphatidylinositol 3-kinase mediates epidermal growth factor-induced activation of the c-Jun N-terminal kinase signaling pathway. Molecular and cellular biology. 1997; 17: 5784-90.
32. DeNardo DG, Cuba VL, Kim H, Wu K, Lee AV, Brown PH. Estrogen receptor DNA binding is not required for estrogen-induced breast cell growth. Mol Cell Endocrinol. 2007; 277: 13-25.
33. Petz LN, Ziegler YS, Loven MA, Nardulli AM. Estrogen receptor alpha and activating protein-1 mediate estrogen responsiveness of the progesterone receptor gene in MCF-7 breast cancer cells. Endocrinology. 2002; 143: 4583-91.
34. Alayev A, Salamon RS, Berger SM, Schwartz NS, Cuesta R, Snyder RB, Holz MK. mTORC1 directly phosphorylates and activates ER alpha upon estrogen stimulation. Oncogene. 2016; 35: 3535-43.
35. Yamnik RL, Digilova A, Davis DC, Brodt ZN, Murphy CJ, Holz MK. S6 Kinase 1 Regulates Estrogen Receptor alpha in Control of Breast Cancer Cell Proliferation. Journal of Biological Chemistry. 2009; 284: 6361-9.
36. Becker MA, Ibrahim YH, Cui X, Lee AV, Yee D. The IGF Pathway Regulates ER alpha through a S6K1-Dependent Mechanism in Breast Cancer Cells. Mol Endocrinol. 2011; 25: 516-28.
37. Generali D, Fox SB, Brizzi MP, Allevi G, Bonardi S, Aguggini S, Milani M, Bersiga A, Campo L, Dionisio R, Vergoni F, Giardini R, Dogliotti L, et al. Down-regulation of phosphatidylinositol 3’-kinase/AKT/molecular target of rapamycin metabolic pathway by primary letrozole-based therapy in human breast cancer. Clin Cancer Res. 2008; 14: 2673-80.
38. Baselga J, Semiglazov V, van Dam P, Manikhas A, Bellet M, Mayordomo J, Campone M, Kubista E, Greil R, Bianchi G, Steinseifer J, Molloy B, Tokaji E, et al. Phase II randomized study of neoadjuvant everolimus plus letrozole compared with placebo plus letrozole in patients with estrogen receptor-positive breast cancer. J Clin Oncol. 2009; 27: 2630-7.
39. Creighton CJ, Fu X, Hennessy BT, Casa AJ, Zhang Y, Gonzalez-Angulo AM, Lluch A, Gray JW, Brown PH, Hilsenbeck SG, Osborne CK, Mills GB, Lee AV, et al. Proteomic and transcriptomic profiling reveals a link between the PI3K pathway and lower estrogen-receptor (ER) levels and activity in ER+ breast cancer. Breast Cancer Res. 2010; 12: R40.
40. Fan M, Yan PS, Hartman-Frey C, Chen L, Paik H, Oyer SL, Salisbury JD, Cheng AS, Li L, Abbosh PH, Huang TH, Nephew KP. Diverse gene expression and DNA methylation profiles correlate with differential adaptation of breast cancer cells to the antiestrogens tamoxifen and fulvestrant. Cancer Res. 2006; 66: 11954-66.
41. Frogne T, Benjaminsen RV, Sonne-Hansen K, Sorensen BS, Nexo E, Laenkholm AV, Rasmussen LM, Riese DJ 2nd, de Cremoux P, Stenvang J, Lykkesfeldt AE. Activation of ErbB3, EGFR and Erk is essential for growth of human breast cancer cell lines with acquired resistance to fulvestrant. Breast Cancer Res Treat. 2009; 114: 263-75.
42. Massarweh S, Osborne CK, Jiang S, Wakeling AE, Rimawi M, Mohsin SK, Hilsenbeck S, Schiff R. Mechanisms of tumor regression and resistance to estrogen deprivation and fulvestrant in a model of estrogen receptor-positive, HER-2/neu-positive breast cancer. Cancer Res. 2006; 66: 8266-73.
43. Bosch A, Li Z, Bergamaschi A, Ellis H, Toska E, Prat A, Tao JJ, Spratt DE, Viola-Villegas NT, Castel P, Minuesa G, Morse N, Rodon J, et al. PI3K inhibition results in enhanced estrogen receptor function and dependence in hormone receptor-positive breast cancer. Sci Transl Med. 2015; 7: 283ra51.
44. Toska E, Osmanbeyoglu HU, Castel P, Chan C, Hendrickson RC, Elkabets M, Dickler MN, Scaltriti M, Leslie CS, Armstrong SA, Baselga J. PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D. Science. 2017; 355: 1324-9.
45. Fox EM, Kuba MG, Miller TW, Davies BR, Arteaga CL. Autocrine IGF-I/insulin receptor axis compensates for inhibition of AKT in ER-positive breast cancer cells with resistance to estrogen deprivation. Breast Cancer Res. 2013; 15: R55.
46. Manning BD, Toker A. AKT/PKB Signaling: Navigating the Network. Cell. 2017; 169: 381-405.
47. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005; 307: 1098-101.
48. Alessi DR, Deak M, Casamayor A, Caudwell FB, Morrice N, Norman DG, Gaffney P, Reese CB, MacDougall CN, Harbison D, Ashworth A, Bownes M. 3-phosphoinositide-dependent protein kinase-1 (PDK1): structural and functional homology with the Drosophila DSTPK61 kinase. Current Biology. 1997; 7: 776-89.
49. Beck JT, Hortobagyi GN, Campone M, Lebrun F, Deleu I, Rugo HS, Pistilli B, Masuda N, Hart L, Melichar B, Dakhil S, Geberth M, Nunzi M, et al. Everolimus plus exemestane as first-line therapy in HR+, HER2(-) advanced breast cancer in BOLERO-2. Breast Cancer Res Tr. 2014; 143: 459-67.
50. Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nature protocols. 2006; 1: 1112-6.
51. Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984; 22: 27-55.
52. Dillon LM, Bean JR, Yang W, Shee K, Symonds LK, Balko JM, McDonald WH, Liu S, Gonzalez-Angulo AM, Mills GB, Arteaga CL, Miller TW. P-REX1 creates a positive feedback loop to activate growth factor receptor, PI3K/AKT and MEK/ERK signaling in breast cancer. Oncogene. 2015; 34: 3968-76.
53. Engelman JA, Janne PA, Mermel C, Pearlberg J, Mukohara T, Fleet C, Cichowski K, Johnson BE, Cantley LC. ErbB-3 mediates phosphoinositide 3-kinase activity in gefitinib-sensitive non-small cell lung cancer cell lines. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102: 3788-93.