
INTRODUCTION
Cancer immunotherapies have revolutionized and improved cancer treatment through boosting the patients’ anti-tumor immune responses. Particularly, the success of immune checkpoint inhibitors has highlighted the importance of an effective anti-cancer immune response in cancer patients. However, the clinical outcome is still very limited and the majority of cancer patients has no benefit by these inhibitors [1, 2]. Therefore, it is important and urgent to clarify mechanisms that cause resistance and to develop a way to further enhance and improve the patients’ immune response [2].
Recently, several studies of cancer immunotherapy revealed that T cells recognizing cancer-specific antigens (neoantigens and shared antigens) may have significant impact on the clinical response. For instance, the higher numbers of somatic mutations may increase a chance to generate highly immunogenic neoantigens that induce/activate T cells, which are thought to drive the clinical effects of immune checkpoint inhibitors [1, 3–5]. In addition, programmed death-ligand 1 (PD-L1) expression on cancer cells was considered to reflect the activity of cancer-antigen specific T cells expressing PD-1 molecules; the interaction of these two molecules is in fact a target of PD-1/PD-L1 blockade [1, 6–8]. Furthermore, the success of adoptive therapy using autologous tumor-infiltrating lymphocytes (TILs) was correlated with the presence of T cells targeting cancer-specific antigens [9].
To enhance a patient’s T cell-mediated anti-tumor immune responses for further improvement of cancer immunotherapy, two approaches are currently investigated. First, peptide vaccines targeting cancer-specific antigens, neoantigens and shared antigens, can activate or induce antigen-specific T cells in cancer patients. As only neoantigens are non-self peptides derived from somatic non-synonymous mutations in cancer cells, they are potentially more immunogenic [10]. Given that the adoptive transfer of patient-derived neoantigen-specific T cells already showed positive clinical results [11, 12], the use of neoantigen vaccines might be an auspicious opportunity to induce or expand mutation-specific T cells in cancer patients [13]. However, recent studies suggest that patient-derived mutation-specific T cells might be functionally impaired because the cells are epigenetically imprinted to revert into an inactive state even after transient activation [14, 15]. This would favor a second approach using the transfer of cancer-specific T cell receptor (TCR) genes into T cells that can be isolated from the patients’ peripheral blood [16]. Preclinical studies have shown that the adoptive transfer of neoantigen-specific TCR-engineered T cells can be effective against large and long-established solid tumors [17, 18]. In order to render this strategy applicable for clinical use, we focused on refining a protocol that allows the effective and prompt identification of cancer neoantigen-specific TCRs using T cells derived from blood of human leucocyte antigen (HLA)-matched healthy donors [19]. Because the reactivation of existing T cells or the priming of neoantigen-specific T cells in a patient might be impaired, we propose this pipeline for the efficient generation of neoantigen-specific T cells and the identification of their TCRs to develop personalized and cancer-specific adoptive immunotherapies using TCR-engineered T cells especially for progressive tumor or for bulky tumor which needs urgent treatments.
RESULTS
A time-efficient protocol to prime and screen neoantitgen-specific T cells from third-party donors
We first established the pipeline for the rapid identification of antigen-specific T cells from healthy donors’ peripheral blood mononuclear cells (PBMCs) to meet the purpose of clinical application using TCR-engineered T cells (Figure 1). After identification of candidate neoantigens by means of whole-exome and RNA sequencing, we prepared matured dendritic cells (DCs) loaded with neoantigen peptides as antigen presenting cells. To induce neoantigen-specific T cells, we initiated priming of autologous CD8+ T cells by incubation with peptide-pulsed DCs (day 1). Antigen-depending T cell stimulation was fostered by supplementing the culture with low amounts of IL-7 and IL-15 three days after the initial priming step (and subsequently every 2 days) [20]. Finally, the time period for inducing antigen-specific T cells was significantly shortened by harvesting the T cells already after the initial priming. Therefore, on day 11, neoantigen-specific CD8+ T cells were stained using peptide-HLA dextramers and we enriched the population by fluorescence activated cell sorting (FACS). Using RNAs from these sorted T cells, we completed TCR sequencing within next 3 days to identify TCR alpha (TCRA) and beta (TCRB) chains. Notably, the screening process to determine TCRA and TCRB genes of potentially neoantigen-specific TCRs was completed only within two weeks after initiating the priming of CD8+ T cells.
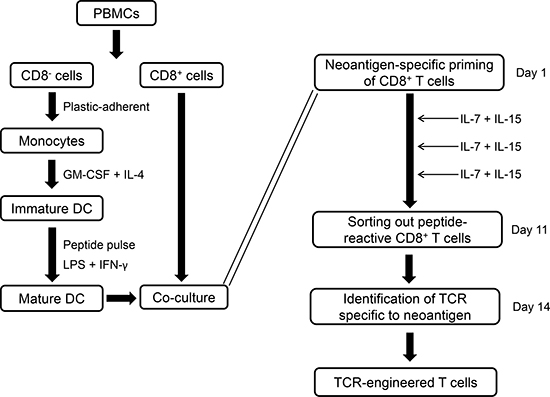
Figure 1: Screening of neoantigens which induce specific cytotoxic T lymphocytes (CTLs). Workflow of rapid screening protocol of neoantigen-reactive CTLs from donors’ blood. PBMCs: peripheral blood mononuclear cells, DCs: dendritic cells.
Neoantigen-specific CD8+ T cells can be generated from HLA-matched third-party donors
To verify that the established protocol is suited to induce neoantigen-specific CD8+ T cells, we performed an analysis using the esophageal cancer cell line TE-8 and primary material obtained from an ovarian cancer patient. In our analyses, we focused either on HLA-A*24:02 (TE-8) or HLA-A*02:01 (ovarian cancer), which are the most common HLA class I types in either the Japanese or the Caucasian population, respectively.
In TE-8, we identified a total number of 312 non-synonymous mutations (Supplementary Figure 1). Although irrelevant for this exemplary analysis, it should be noted that no germline control was available for this established cell line and we were therefore unable to distinguish somatic mutations from rare germline variations. Subsequently, using a list of these possible mutations, we predicted the binding affinity of peptides including amino acid substitutions to a HLA-A2402 molecule and identified 81 neoantigen epitopes which showed IC50 of 500 nM or lower. We then confirmed the expression of RNA corresponding to 33 neoantigen epitopes with RNA sequencing data (Supplementary Table 1). This number of neoantigen epitopes was further narrowed to those showing an IC50 of 50 nM or lower to select peptides that were used to test induction of neoantigen-specific CD8+ T cells using peripheral blood from healthy donors. After priming of CD8+ T cells, we detected neoantigen-specific CD8+ T cells by staining with HLA-dextramers loaded with a neoantigen peptide derived from the missense substitution L143F in dpy-19 like 4 gene (DPY19L4L143F, LYPEFIASI, Figure 2A). The proportion of CD8+HLA-dextramer+ T cells was 0.62% (4,986 cells). We subsequently performed TCR sequencing and determined the entire sequences of TCRA and TCRB chains with highly variable complementarity determining region 3 (CDR3) sequences in these sorted T cells (Figure 2B).
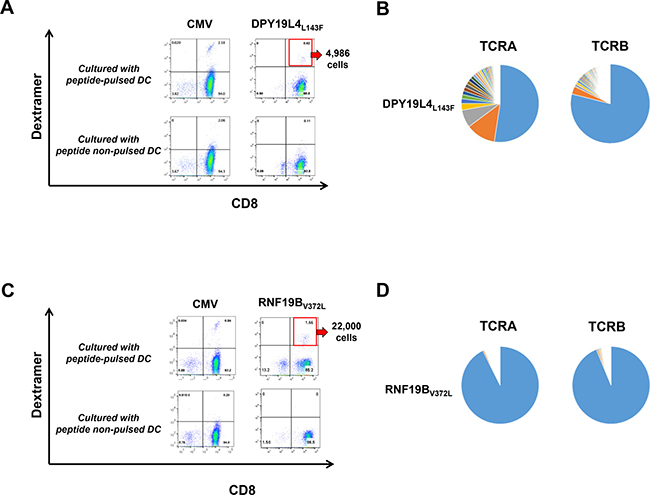
Figure 2: Identification of neoantigen-specific T cells using our rapid screening protocol. (A) Peptide-HLA dextramer staining for CD8+ T cells co-cultured with autologous DCs with or without mutated peptide-pulse of DPY19L4L143F, which was identified in the COSMIC database. The number of sorted CD8+HLA-dextramer+ T cells was 4,986. (B) Frequency distributions of TCRA and TCRB sequences of CD8+ T cells reactive to DPY19L4L143F peptide. After sorting the dextramer-positive cell population, TCR sequencing was performed. Each pie chart represents the frequency of unique TCRA and TCRB CDR3 sequences of sorted CD8+HLA-dextramer+ T cells. (C) Peptide-HLA dextramer staining for CD8+ T cells co-cultured with autologous DCs with or without mutated RNF19BV372L peptide pulse, which was identified in an ovarian cancer patient. The number of sorted CD8+HLA-dextramer+ T cells was 22,000. (D) Frequency distributions of TCRA and TCRB sequences of CD8+ T cells reactive to RNF19BV372L peptide. After sorting the dextramer-positive cell population, TCR sequencing was performed. Each pie chart represents the frequency of unique TCRA and TCRB CDR3 sequences of sorted CD8+HLA-dextramer+ T cells. Antigen peptide of CMV (cytomegalovirus) pp65 for HLA-A*24:02 or HLA-A*02:01 were used as a positive control.
As a second example with higher clinical relevance, we aimed at generating neoantigen-specific CD8+ T cells against a mutation in an ovarian cancer sample. The material was isolated from a 64 year-old patient diagnosed with stage 3C clear cell ovarian carcinoma. We found a total of 80 non-synonymous mutations and calculated the affinity of potential neoantigen epitopes to a HLA-A0201 molecule (Supplementary Tables 2, 3). Among 32 candidate epitope peptides that showed a peptide-HLA affinity of 500 nM or lower and were expressed in tumor cells, we narrowed down the candidate neoepitopes (IC50 of 50 nM or lower) for testing their ability to induce CD8+ T cells isolated from a healthy donor (Supplementary Table 3). We successfully identified CD8+ T cells which were primed against a neoantigen peptide derived from RNF19BV372L (V372L mutation in ring finger protein 19B gene, MLIGIPVYV, Figure 2C). The proportion of CD8+HLA-dextramer+ T cells was 1.66% (22,000 cells). We subsequently analyzed their TCR repertoire (Figure 2D).
Interestingly, in both experiments, even a short expansion time of 11 days was sufficient to stimulate the outgrowth of single T cell clone which TCRA and TCRB sequences dominated the TCR repertoire (Supplementary Table 4). Both TCR pairs were molecularly cloned and used for further analysis.
TCRs isolated from DPY19L4L143F-reactive T cells of third party donors are mutation-specific
Next, we generated a retroviral vector encoding for the DPY19L4L143F-TCR genes and transduced T cells derived from peripheral blood of a healthy donor (DPY19L4L143F TCR-engineered T cells). DPY19L4L143F TCR-engineered T cells showed binding of specific dextramers loaded with the mutant but not with the wild-type DPY19L4 peptide (Figure 3A). Furthermore, we confirmed antigen-specific function of DPY19L4L143F TCR-engineered T cells by co-culture with C1R lymphoblastoid cells stably expressing either HLA-A*24:02 or HLA-A*02:01 and loaded with titrated amounts of either mutant or wild-type DPY19L4 peptide. Activation of TCR-engineered T cells was determined by measuring IFN-γ secretion using ELISPOT (Figure 3B) or ELISA (Supplementary Figure 2A) and by analyzing CD137 upregulation by FACS (Supplementary Figure 2B). The functional characterization substantiated the exquisite specificity of the isolated DPY19L4L143F-TCR because C1R cells were only recognized when (i) expressing HLA-A*24:02 and (ii) loaded with the mutant DPY19L4 peptide. The wild-type DPY19L4 peptide and HLA-A*02:01 were not sufficient to stimulate function of TCR-engineered T cells.
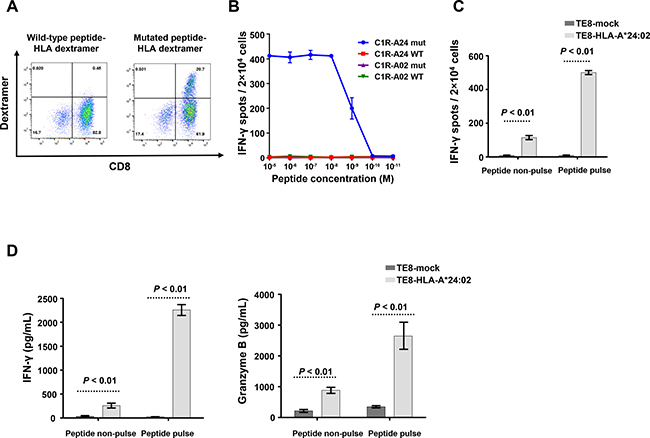
Figure 3: In vitro analysis of DPY19L4L143F TCR-engineered T cells. (A) Flow cytometric analysis of HLA-A*24:02 dextramer with wild-type or mutated peptide on DPY19L4L143F TCR-engineered T cells. (B) IFN-γ ELISPOT assay on DPY19L4L143F TCR-engineered T cells co-cultured with C1R-A24/A02 cells loaded with graded amounts of peptide. (C) IFN-γ ELISPOT assay on DPY19L4L143F TCR-engineered T cells co-cultured with HLA-A*24:02- or mock-transfected TE-8 cells. (D) ELISA assays for IFN-γ, and granzyme B on DPY19L4L143F TCR-engineered T cells co-cultured with HLA-A*24:02- or mock-transfected TE-8 cells.
To test whether endogenously processed antigen can be recognized, we incubated DPY19L4L143F TCR-engineered T cells together with TE-8 cancer cells that were reported to express the HLA-A*24:02 allele [21]. However, HLA expression could not be verified by FACS and surface presentation of endogenously processed DPY19L4L143F antigen had to be restored by transfection of TE-8 cancer cells with an HLA-A*24:02 vector (Supplementary Figure 3). Hence, DPY19L4L143F TCR-engineered T cells secreted IFN-γ only when incubated with HLA-A*24:02-transfected TE-8 cells, whereas mock-transfected TE-8 cells could not trigger T cell activation (Figure 3C, 3D). The TCR-engineered T cells also secreted the cytolytic molecule granzyme B (Figure 3D). In addition, when we pulsed HLA-A*24:02-transfected TE-8 cells with the mutant peptide, IFN-γ and granzyme B secretion was further enhanced (Figure 3C, 3D). These results indicate that DPY19L4L143F TCR-engineered T cells recognized the endogenously-expressed mutated peptide in the HLA-A2402-restricted manner and showed cytotoxic activity.
To further explore the cytotoxic activity of T cells engineered with the DPY19L4L143F-TCR, we made use of HLA-A*24:02-positive TE-11 esophageal cancer cells since we could not establish TE-8 cells that stably express HLA-A*24:02 (Supplementary Figure 3). Direct killing of TE-11 cancer cells was only observed after loading with DPY19L4L143F peptide (cell viability was reduced to 27.5%, Supplementary Movie 1). The cell viability of TE-11 cancer cells that were not loaded with peptide was only marginally impaired (reduced to 73.1%, Supplementary Movie 2).
TCRs isolated from RNF19BV372L-reactive T cells recognizes the neoantigen peptide and its wild-type analog
To analyze the TCR chains that were identified after priming of T cells against the RNF19BV372L mutation, we constructed a retroviral vector encoding the RNF19BV372L-TCR genes and generated TCR-engineered T cells (RNF19BV372L TCR-engineered T cells). In contrast to the analysis of the DPY19L4L143F-TCR, RNF19BV372L TCR-engineered T cells bound dextramers irrespective of whether the HLAs were loaded with mutant or wild-type RNF19BV372L peptide (Figure 4A). IFN-γ ELISPOT assay also revealed that RNF19BV372L TCR-engineered T cells secreted IFN-γ at the similar levels when the antigen-presentation cells were pulsed with the wild-type and mutated peptides although the recognition of these peptides by RNF19BV372L TCR-engineered T cells were confirmed to occur on an HLA-A0201-restricted manner (Figure 4B and Supplementary Figure 4). These results substantiate the potential risk that neoantigen-specific TCR-engineered T cells may be cross-reactive to the wild-type analog of neoantigen peptides and calls for judicious selection of neoantigen for T cell priming.
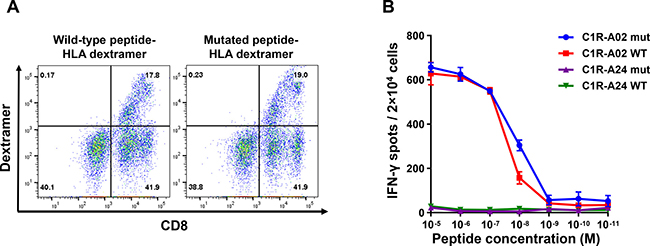
Figure 4: RNF19BV372L TCR-engineered T cells cross-react towards the wild-type peptide. (A) Flow cytometric analysis of HLA-A*02:01 dextramer with wild-type or mutated peptide on RNF19BV372L TCR-engineered T cells. (B) IFN-γ ELISPOT assay on RNF19BV372L TCR-engineered T cells co-cultured with C1R-A24/A02 cells loaded with graded amounts of peptide.
DISCUSSION
Identification of human tumor antigens and immune checkpoint molecules significantly contributed to the better understanding of tumor immunology [22–24]. These findings were translated into the applied medicine, led to the development of effective immune checkpoint inhibitors, cancer peptide vaccine and adoptive cell transfer therapy (e.g. TIL infusion therapy) that have revolutionized cancer treatment [25–28]. In particular, several types of immune checkpoint inhibitor emerged as a novel cancer treatment after the first approval of a fully humanized antibody against cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) for treatment of advanced melanoma and showed significant survival benefit in various types of cancer [2, 29]. However, recent meta-analysis of clinical data made it clear that only a subset of patients responded to immune checkpoint inhibitors, and the majority of patients had no benefit and some of them suffered from severe immune-related adverse reactions. Therefore, it is crucial to develop a new strategy to enhance the host anti-tumor immune response for further improvement of clinical outcomes in cancer immunotherapies.
In this study, we developed a time-efficient approach to identify neoantigen-specific TCRs that can be applied to neoantigen-specific TCR-engineered T cell therapy. Our approach has several major advantages. First, our protocol requires only two weeks in the process from the beginning of T cells priming with possible immunogenic neoantigen peptides to identification of neoantigen-specific TCRs (Figure 1). We previously established the protocol for in vitro induction of T cells specific to shared antigens (oncoantigens) by three stimulations of peptide-pulsed DCs [30]. We reduced the number of stimulations with peptide-pulsed DCs to one and confirmed that the single stimulation with peptide-pulsed DCs seemed to be sufficient to identify neoantigen-specific TCRs in combination with our TCR sequencing method. As a result, we could significantly shorten the duration to identify neoantigen-reactive TCRs. We demonstrated that TCR sequencing of directly-sorted multimer-positive T cells was able to rapidly identify TCRA and TCRB pairs of dominant clones, using our TCR sequencing method previously developed in various disease conditions including several types of cancer [31–39].
Secondly, our approach use T cells isolated from healthy donors to induce neoantntigen-specific T cells. T cells obtained from healthy donors may have broader T cell repertoire and have a higher chance to induce neoantigen-specific T cells since cancer patients who are heavily treated with anti-cancer drugs, particularly cytotoxic agents, are likely to have severe damage in their immune system and have smaller diversity of T cells [19]. Recently, Lu et al. developed a unique approach on TILs co-cultured with tandem minigene-transfected antigen presenting cells to isolate neoantigen-specific TCRs. They performed single-cell RNA sequencing analysis on T cells to identify a pair of TCRA and TCRB sequences of neoantigen-specific T cells [40]. However, this approach requires 3–6 weeks of TIL culture and a large number of TILs need to be examined by single-cell analysis. In our protocol, although peripheral blood T cells of healthy donors might yield the lower proportion of neoantigen-specific T cells compared with TILs, we could identify a dominant pair of TCRA and TCRB of FACS-sorted dextramer-positive T cells. Hence, we believe that our protocol is time-efficient and cost-efficient for establishing neoantigen-specific TCR-engineered T cells. Because a time is very limited for patients with progressive or advanced tumors, how to identify neoantigen-specific TCR sequences is critically important. Since adoptive therapy using expanded TILs, some of which were shown to react on neoantigens, revealed encouraging clinical outcome for melanoma and other types of solid tumor [11, 12], TCR-engineered T cells targeting neoantigens are also considered to be a promising therapy for advanced cancer.
Our data also indicate that the current protocol would benefit from further improvements. In order to avoid autoimmune side effects when using neoantigen-specific TCRs, the TCRs should not recognize the wild-type analogs of mutated antigens as shown in RNF19BV372L found in the ovarian cancer sample. TCRs distinguish neoantigens from its corresponding wild-type peptides if the mutation change the recognition site for the TCR, where T cells can directly interact [41]. The neoantigens with amino acid changes in the HLA anchor position, usually position 2 and 9 for HLA-A*02:01, relies on significant differences in binding affinity between wild-type and mutant peptides [42]. However, both wild-type and mutant of the RNF19B peptides (V372L in position 2 of the epitope) are predicted to have similarly high HLA-binding affinity, and each TCR that is raised against the mutant peptide will probably be reactive against its wild-type counterpart. Our results indicate that a TCR recognizing neoantigen is not always mutated-peptide specific, and we should apply additional selection criterion to demarcate false-positive neoantigens such as RNF19BV372L. This would help to clearly earmark the most promising neoantigen epitopes for in vitro induction of specific T cells.
In conclusion, the developed protocol facilitates the generation of neoantigen-specific TCRs in a time frame that will allow a patient to benefit from subsequent immunotherapy using TCR-engineered T cells. When considering a small precursor frequency of neoantigen-specific T cells in the blood of third party donors and a judicious selection of neoantigen peptides for stimulation, the protocol will have clear advantages over other strategies that rely on reactivation or induction of T cells in a tolerant repertoire.
MATERIALS AND METHODS
Cell lines
C1R (lacking HLA-A and HLA-B, B lymphoblast) were purchased from American Type Culture Collection (Rockville, MD). TE-8 (HLA-A*24:02, esophageal cancer) and TE-11 (HLA-A*24:02, esophageal cancer) cells were provided by the Cell Resource Center for Biomedical Research Institute of Development, Aging and Cancer at Tohoku University. C1R cells stably expressing HLA-A*24:02 (C1R-A24) or HLA-A*02:01 (C1R-A02) were prepared by the transfection of the vectors encoding HLA-A*24:02 or HLA-A*02:01 gene. All cells were cultured under the recommendations of their respective depositors.
Screening of potential neoantigens
To screen potential neoantigens in TE-8 cancer cell lines having HLA-A*24:02, we used somatic mutation information from the COSMIC database [43]. Among the somatic mutations, we selected non-synonymous mutations, which the expression of mutant RNA were detected at least 1× read using RNA-sequencing bam data obtained from CCLE database, for the further analysis of neoantigen prediction. HLA genotype information was obtained from previous reports [44].
From an ovarian cancer sample having HLA-A*02:01, genomic DNAs and total RNAs were extracted using AllPrep DNA/RNA mini kit (Qiagen, Valencia, CA). As germline control DNAs, genomic DNAs were extracted from PBMCs. Whole-exome libraries were prepared from 1,000 ng of genomic DNAs using SureSelectXT Human All Exon V5 kit (Agilent Technologies, Santa Clara, CA) and the prepared whole-exome libraries were sequenced by 100-bp paired-end reads on HiSeq2500 Sequencer (Illumina, San Diego, CA). Sequence alignment and mutation calling were performed using our in-house pipelines described previously [45]. Briefly, the sequence reads were mapped to the human reference genome GRCh37/hg19 using Burrows-Wheeler Aligner (BWA) (v0.7.10). Possible PCR duplicated reads were removed using Picard tool (http://broadinstitute.github.io/picard/), and reads with a mapping quality of <30 and with mismatches of more than 5% of sequence reads were also excluded. Finally, somatic variants (single nucleotide variations (SNVs) and indels) were called with the following parameters, (i) base quality of ≥15, (ii) sequence depth of ≥ 10, (iii) variant depth of ≥4, (iv) variant frequency in tumor of ≥10%, (v) variant frequency in normal of <2%, and (vi) Fisher p value of <0.05. HLA class I genotypes of the tumor were determined by OptiType algorithm using whole-exome sequence data of normal DNAs [45]. Potential neoantigens were predicted for each non-synonymous mutation by testing all 8- to 11-mer peptides harboring the amino acid substitution using NetMHCv3.4 software as described previously [35]. We selected candidates whose predicted binding affinity to HLA-A*24:02 or HLA-A*02:01 was < 500 nM.
The study protocol for this process was approved by the Institutional Review Board of the University of Chicago (approval number 13–0797 and 13–0526) and Saitama Medical University International Medical Center (approval number 15–225). All patients provided written informed consents.
Induction of neoantigen-specific cytotoxic T lymphocytes (CTLs)
The research protocol for collecting and using PBMCs from healthy donors was approved by the Institutional Review Board of the University of Chicago (approval number 13–0797 and 15–1738). To induce neoantigen-specific CTLs, we first collected PBMCs from healthy donors by means of Vacutainer CPT Cell Preparation Tube (BD Biosciences, San Jose, CA). Neontigen-derived peptides (more than 95% purity) were purchased from Innopep (San Diego, CA). CD8+ T cells were separated using Dynabeads CD8 Positive Isolation Kit (Thermo Fisher Scientific, Carlsbad, CA). Monocyte-derived immature DCs were enriched using plastic adherence method, and were cultured in CellGro DC (Cellgenix, Freiburg, Germany) containing 1% Human AB serum (ABS), 1,000 U/mL GM-CSF (R&D Systems, Minneapolis, MN) and 500 U/mL IL-4 (R&D Systems) for 72 h in Primaria 6-well plate (Corning, Inc., Corning, NY) [30]. Then, 10 ng/mL LPS (Sigma-Aldrich, ST. Louis, MO) and 100 U/mL IFN-γ (PeproTech, Rocky Hill, NJ) were added in the culture to induce the maturation of DCs. DCs were pulsed with 20 μg/mL of the respective synthesized peptides for 16 h at 37° C, and treated with 30 μg/mL of mitomycin C (MMC, Sigma-Aldrich) at 37° C for 30 min. Following washing out the residual peptides and MMC, DCs were cultured with autologous CD8+ T cells in 0.5 mL of CellGro DC/5% ABS supplemented with 30 ng/mL IL-21 (R&D Systems) on 48-well plate (each well contained 1.0 × 105 peptide-pulsed DCs, 5 × 105 CD8+ T cells) on day 1 [46, 47]. Three days later (on days 4), 5 ng/mL IL-7 (R&D Systems) and 5 ng/mL IL-15 (Novoprotein, Summit, NJ) were added in the culture media. On days 6, the cultures were transferred to 12-well plate with CellGro DC/5% ABS containing 5 ng/mL IL-7 and 5 ng/mL IL-15 [48]. On days 8, cultures were supplemented with CellGro DC/5% ABS containing 10 ng/mL IL-7 and 10 ng/mL IL-15. On days 11, neoantigen-specific T cells were assessed using specific dextramer (Immudex, Copenhagen, Denmark) by flow cytometry analysis.
Flow cytometry and antibodies
Peptide-HLA dextramers labeled with allophycocyanin (APC) were purchased from Immudex. To assess the positivity of neoantigen-specific T cells, the cells were incubated with dextamers for 10 min at room temperature and then treated with fluorescein isothiocyanate (FITC)-conjugated anti-human CD8 antibody (clone HIT8a, BD Biosciences) at 4° C for 20 min. To examine the expression of CD137, TCR-engineered T cells were incubated with anti-human CD137 antibody (clone 4B4-1, Miltenyi Biotec, Bergisch Gladbach, Germany). To confirm the expression of HLA-A24, TE-8 and TE-11 cells were stained with anti-human HLA-A24 antibody (clone 17A10, MBL, Woburn, MA). Fluorescence was quantified by flow cytometry (FACS LSRII; Becton Dickinson, San Jose, CA). Data analysis was performed using Flow Jo software (Treestar, Ashland, OR).
TCR sequencing
We determined TCR sequences using the method we previously developed [35, 49]. In brief, we extracted total RNAs from FACS-sorted dextramer-positive T cells. cDNAs with common 5′-RACE adapter were synthesized using SMART library construction kit (Clontech, Mountain View, CA). The fusion PCR was performed to amplify TCRA and TCRB cDNAs using a forward primer corresponding to the SMART adapter sequence and reverse primers corresponding to the constant region of each of TCRA and TCRB. After adding the Illumina index sequences with barcode using the Nextera Index kit (Illumina, San Diego, CA), the prepared libraries were sequenced by 300-bp paired-end reads on the MiSeq (Illumina). Obtained sequence reads were analyzed using Tcrip software [49].
TCR-engineered T cells
Both TCRA and TCRB sequences were codon-optimized and cloned into a retroviral vector, pMP71-PRE as described previously [50]. pMP71-PRE lacks its own replicating ability. To improve TCR surface expression, we employed constant regions of mouse TCR [51]. Transient retroviral supernatants were generated and PBMCs from donors were transduced as described previously [52].
To examine the cytotoxic activity by DPY19L4L143F TCR-engineered T cells, we enriched TCR-engineered T cells using the staining with APC-conjugated anti-mouse TCR beta monoclonal antibody (H57-597, eBioscience, San Diego, CA) at 1:2000 (0.1 μg/mL) followed by the incubation with anti-APC microbeads (Miltenyi Biotec) according to the manufacturer’s instructions.
ELISPOT and ELISA assays
ELISPOT assay was performed using Human IFN-γ ELISpotPRO kit (MABTECH, Cincinnati, OH) according to the manufacturer’s instruction. Briefly, antigen presenting cells were pulsed with respective peptides at 37° C for 20 h, and the residual peptides that did not bind to cells were washed out to prepare peptide-pulsed cells as the stimulator cells. T cells were pre-treated with IL-2 (35 U/mL) for 16 h and then co-incubated with peptide-pulsed stimulator cells (2 × 104 cells/well) at 37° C for 20 h in 96-well plate. The supernatant was transferred into a new 96-well plate for ELISA assay. Spots were captured and analyzed by an automated ELISPOT reader, ImmunoSPOT S4 (Cellular Technology Ltd, Shaker Heights, OH) and the ImmunoSpot Professional Software package, Version 5.1 (Cellular Technology Ltd). To measure the secreted cytokine levels in the supernatant, we used OptEIA Human IFN-γ ELISA set (BD Biosciences), Human Granzyme B ELISA development kit (MABTECH) and read absorbance in a microplate reader at 450/570 nm.
Evaluation of cytotoxic activities of CTLs against cancer cells by time-lapse recording
TCR-engineered T cells were pre-treated with IL-2 (100 U/mL) for 16 h. Target cells were pre-treated with IFN-γ (100 U/mL) for 48 h before experiments. The target cells were incubated with 1 μg/mL of Calcein AM (Dojindo, Kumamoto, Japan) for 30 min. After 3-time wash by PBS, 2 × 104 of target cells were mixed with 4 × 105 TCR-engineered T cells into Lab-Tek Chamber Slide Cover Glass Slide Sterile 16 Well (Thermo Fisher Scientific). We used automated imaging system with high-resolution (EVOS FL Auto 2 Imaging System, Thermo Fisher Scientific).
Statistical analysis
The student’s t test was performed for comparison of the number of IFN-γ positive spots, the concentration of IFN-γ and granzyme B between HLA-A*24:02- or mock-transfected TE-8 cells when co-cultured with DPY19L4L143F TCR-engineered T cells. Statistical analyses were done using GraphPad Prism version 6.0 (GraphPad software, La Jolla, CA). P value of <0.05 was considered to be statistically significant.
Abbreviations
CDR3: complementarity determining region 3; HLA: human leucocyte antigen; PBMCs: peripheral blood mononuclear cells; TCR: T cell receptor; TCRA: T cell receptor alpha; TCRB: T cell receptor beta; TILs: tumor-infiltrating lymphocytes.
Author contributions
Y.N. planned and supervised the entire project; K.K., J.P. provided the study design and the working hypothesis and completed the article; T.K., T.M., Y.I. designed the study, conducted experiments, performed data analysis, and drafted the article; M.L., generated genetically engineered T cells, provided scientific advice and revised the article; T.H., S.Y., M.H., M.Z. conducted experiments and performed data analysis; S.S., K.H. provided clinical samples, provided scientific advice and completed the article.
ACKNOWLEDGMENTS
We would like to thank Drs. Rui Yamaguchi, Seiya Imoto and Satoru Miyano at the University of Tokyo for developing the algorithm of TCR repertoire analysis and helpful supports in data management. The super-computing resource (http://sc.hgc.jp/shirokane.html) was provided by Human Genome Center, the Institute of Medical Science, the University of Tokyo. We also thank Kimberley Borutta for excellent technical support.
CONFLICTS OF INTEREST
Y.N. is a stockholder and an adviser of OncoTherapy Science Inc. J.P. is a scientific adviser of OncoTherapy Science Inc. S.Y., T.H. and M.H. are employees of OncoTherapy Science Inc.
FUNDING
This work was supported partly by a research grant from OncoTherapy Science, Inc.
REFERENCES
1. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, Leming PD, Spigel DR, Antonia SJ, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012; 366:2443–54.
2. Postow MA, Callahan MK, Wolchok JD. Immune Checkpoint Blockade in Cancer Therapy. J Clin Oncol. 2015; 33:1974–82.
3. Yarchoan M, Johnson BA 3rd, Lutz ER, Laheru DA, Jaffee EM. Targeting neoantigens to augment antitumour immunity. Nat Rev Cancer. 2017; 17:209–22.
4. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015; 348:124–8.
5. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015; 160:48–61.
6. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, West AN, Carmona M, Kivork C, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014; 515:568–71.
7. Schalper KA, Velcheti V, Carvajal D, Wimberly H, Brown J, Pusztai L, Rimm DL. In situ tumor PD-L1 mRNA expression is associated with increased TILs and better outcome in breast carcinomas. Clin Cancer Res. 2014; 20:2773–82.
8. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012; 12:252–64.
9. Stevanovic S, Pasetto A, Helman SR, Gartner JJ, Prickett TD, Howie B, Robins HS, Robbins PF, Klebanoff CA, Rosenberg SA, Hinrichs CS. Landscape of immunogenic tumor antigens in successful immunotherapy of virally induced epithelial cancer. Science. 2017; 356:200–5.
10. Blankenstein T, Leisegang M, Uckert W, Schreiber H. Targeting cancer-specific mutations by T cell receptor gene therapy. Curr Opin Immunol. 2015; 33:112–9.
11. Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS, Parkhurst MR, Yang JC, Rosenberg SA. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 2014; 344:641–5.
12. Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, Pasetto A, Zheng Z, Ray S, Groh EM, Kriley IR, Rosenberg SA. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med. 2016; 375:2255–62.
13. Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, Chen C, Olive O, Carter TA, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017; 547:217–21.
14. Schietinger A, Philip M, Krisnawan VE, Chiu EY, Delrow JJ, Basom RS, Lauer P, Brockstedt DG, Knoblaugh SE, Hammerling GJ, Schell TD, Garbi N, Greenberg PD. Tumor-Specific T Cell Dysfunction Is a Dynamic Antigen-Driven Differentiation Program Initiated Early during Tumorigenesis. Immunity. 2016; 45:389–401.
15. Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, Scott AC, Viale A, Lauer P, Merghoub T, Hellmann MD, Wolchok JD, Leslie CS, et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature. 2017; 545:452–6.
16. Schumacher TN. T-cell-receptor gene therapy. Nat Rev Immunol. 2002; 2:512–9.
17. Leisegang M, Engels B, Schreiber K, Yew PY, Kiyotani K, Idel C, Arina A, Duraiswamy J, Weichselbaum RR, Uckert W, Nakamura Y, Schreiber H. Eradication of Large Solid Tumors by Gene Therapy with a T-Cell Receptor Targeting a Single Cancer-Specific Point Mutation. Clin Cancer Res. 2016; 22:2734–43.
18. Leisegang M, Kammertoens T, Uckert W, Blankenstein T. Targeting human melanoma neoantigens by T cell receptor gene therapy. J Clin Invest. 2016; 126:854–8.
19. Stronen E, Toebes M, Kelderman S, van Buuren MM, Yang W, van Rooij N, Donia M, Boschen ML, Lund-Johansen F, Olweus J, Schumacher TN. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science. 2016; 352:1337–41.
20. Geginat J, Sallusto F, Lanzavecchia A. Cytokine-driven proliferation and differentiation of human naive, central memory, and effector memory CD4(+) T cells. J Exp Med. 2001; 194:1711–9.
21. Kikuchi M, Nakao M, Inoue Y, Matsunaga K, Shichijo S, Yamana H, Itoh K. Identification of a SART-1-derived peptide capable of inducing HLA-A24-restricted and tumor-specific cytotoxic T lymphocytes. Int J Cancer. 1999; 81:459–66.
22. van der Bruggen P, Traversari C, Chomez P, Lurquin C, De Plaen E, Van den Eynde B, Knuth A, Boon T. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science. 1991; 254:1643–7.
23. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992; 11:3887–95.
24. Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995; 182:459–65.
25. Guo C, Manjili MH, Subjeck JR, Sarkar D, Fisher PB, Wang XY. Therapeutic cancer vaccines: past, present, and future. Adv Cancer Res. 2013; 119:421–75.
26. Hinrichs CS, Rosenberg SA. Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunol Rev. 2014; 257:56–71.
27. Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry R, Restifo NP, Hubicki AM, Robinson MR, Raffeld M, Duray P, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002; 298:850–4.
28. Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR, Morton KE, Laurencot CM, Steinberg SM, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011; 17:4550–7.
29. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010; 363:711–23.
30. Yoshimura S, Tsunoda T, Osawa R, Harada M, Watanabe T, Hikichi T, Katsuda M, Miyazawa M, Tani M, Iwahashi M, Takeda K, Katagiri T, Nakamura Y, et al. Identification of an HLA-A2-restricted epitope peptide derived from hypoxia-inducible protein 2 (HIG2). PLoS One. 2014; 9:e85267.
31. Liu X, Venkataraman G, Lin J, Kiyotani K, Smith S, Montoya M, Nakamura Y, Kline J. Highly clonal regulatory T-cell population in follicular lymphoma - inverse correlation with the diversity of CD8+ T cells. Oncoimmunology. 2015; 4:e1002728.
32. Jang M, Yew PY, Hasegawa K, Ikeda Y, Fujiwara K, Fleming GF, Nakamura Y, Park JH. Characterization of T cell repertoire of blood, tumor, and ascites in ovarian cancer patients using next generation sequencing. Oncoimmunology. 2015; 4: e1030561.
33. Park JH, Jang M, Tarhan YE, Katagiri T, Sasa M, Miyoshi Y, Kalari KR, Suman VJ, Weinshilboum R, Wang L, Boughey JC, Goetz MP, Nakamura Y. Clonal expansion of antitumor T cells in breast cancer correlates with response to neoadjuvant chemotherapy. Int J Oncol. 2016; 49:471–8.
34. Tamura K, Hazama S, Yamaguchi R, Imoto S, Takenouchi H, Inoue Y, Kanekiyo S, Shindo Y, Miyano S, Nakamura Y, Kiyotani K. Characterization of the T cell repertoire by deep T cell receptor sequencing in tissues and blood from patients with advanced colorectal cancer. Oncol Lett. 2016; 11:3643–9.
35. Choudhury NJ, Kiyotani K, Yap KL, Campanile A, Antic T, Yew PY, Steinberg G, Park JH, Nakamura Y, O’Donnell PH. Low T-cell Receptor Diversity, High Somatic Mutation Burden, and High Neoantigen Load as Predictors of Clinical Outcome in Muscle-invasive Bladder Cancer. Eur Urol Focus. 2016; 2:445–52.
36. Inoue H, Park JH, Kiyotani K, Zewde M, Miyashita A, Jinnin M, Kiniwa Y, Okuyama R, Tanaka R, Fujisawa Y, Kato H, Morita A, Asai J, et al. Intratumoral expression levels of PD-L1, GZMA, and HLA-A along with oligoclonal T cell expansion associate with response to nivolumab in metastatic melanoma. Oncoimmunology. 2016; 5: e1204507.
37. Kato T, Iwasaki T, Uemura M, Nagahara A, Higashihara H, Osuga K, Ikeda Y, Kiyotani K, Park JH, Nonomura N, Nakamura Y. Characterization of the cryoablation-induced immune response in kidney cancer patients. Oncoimmunology. 2017; 6: e1326441.
38. Yew PY, Alachkar H, Yamaguchi R, Kiyotani K, Fang H, Yap KL, Liu HT, Wickrema A, Artz A, van Besien K, Imoto S, Miyano S, Bishop MR, et al. Quantitative characterization of T-cell repertoire in allogeneic hematopoietic stem cell transplant recipients. Bone Marrow Transplant. 2015; 50:1227–34.
39. Alachkar H, Mutonga M, Kato T, Kalluri S, Kakuta Y, Uemura M, Imamura R, Nonomura N, Vujjini V, Alasfar S, Rabb H, Nakamura Y, Alachkar N. Quantitative characterization of T-cell repertoire and biomarkers in kidney transplant rejection. BMC Nephrol. 2016; 17:181.
40. Lu YC, Zheng Z, Robbins PF, Tran E, Prickett TD, Gartner JJ, Li YF, Ray S, Franco Z, Bliskovsky V, Fitzgerald PC, Rosenberg SA. An Efficient Single-Cell RNA-Seq Approach to Identify Neoantigen-Specific T Cell Receptors. Mol Ther. 2017 Oct 28. [Epub ahead of print].
41. Fritsch EF, Rajasagi M, Ott PA, Brusic V, Hacohen N, Wu CJ. HLA-binding properties of tumor neoepitopes in humans. Cancer Immunol Res. 2014; 2:522–9.
42. Parker KC, Bednarek MA, Coligan JE. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J Immunol. 1994; 152:163–75.
43. Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, Kok CY, Jia M, De T, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015; 43:D805–11.
44. Boegel S, Lower M, Bukur T, Sahin U, Castle JC. A catalog of HLA type, HLA expression, and neo-epitope candidates in human cancer cell lines. Oncoimmunology. 2014; 3:e954893.
45. Kiyotani K, Park JH, Inoue H, Husain A, Olugbile S, Zewde M, Nakamura Y, Vigneswaran WT. Integrated analysis of somatic mutations and immune microenvironment in malignant pleural mesothelioma. Oncoimmunology. 2017; 6:e1278330.
46. Li Y, Bleakley M, Yee C. IL-21 influences the frequency, phenotype, and affinity of the antigen-specific CD8 T cell response. J Immunol. 2005; 175:2261–9.
47. Alves NL, Arosa FA, van Lier RA. IL-21 sustains CD28 expression on IL-15-activated human naive CD8+ T cells. J Immunol. 2005; 175:755–62.
48. Wolfl M, Greenberg PD. Antigen-specific activation and cytokine-facilitated expansion of naive, human CD8+ T cells. Nat Protoc. 2014; 9:950–66.
49. Fang H, Yamaguchi R, Liu X, Daigo Y, Yew PY, Tanikawa C, Matsuda K, Imoto S, Miyano S, Nakamura Y. Quantitative T cell repertoire analysis by deep cDNA sequencing of T cell receptor alpha and beta chains using next-generation sequencing (NGS). Oncoimmunology. 2014; 3: e968467.
50. Leisegang M, Turqueti-Neves A, Engels B, Blankenstein T, Schendel DJ, Uckert W, Noessner E. T-cell receptor gene-modified T cells with shared renal cell carcinoma specificity for adoptive T-cell therapy. Clin Cancer Res. 2010; 16:2333–43.
51. Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, Morgan RA. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res. 2006; 66:8878–86.
52. Leisegang M, Engels B, Meyerhuber P, Kieback E, Sommermeyer D, Xue SA, Reuss S, Stauss H, Uckert W. Enhanced functionality of T cell receptor-redirected T cells is defined by the transgene cassette. J Mol Med (Berl). 2008; 86:573–83.