
INTRODUCTION
Dysregulated cell cycle progression leads to uneven distribution of the genetic information between the two daughter cells, which contributes to genomic instability and ultimately, cancer development. Recent work established that two related, multi-component E3 ubiquitin ligase enzymes, the Anaphase Promoting Complex (APC) and the Skp1-Cullin1-F-box complex (SCF), are the major driving forces governing proper cell cycle progression [1-4]. APC is active from the late G2 phase to mid-G1 phase, and is responsible for degradation of mitotic cyclins, securin and geminin [5, 6]. On the other hand, SCF is thought to be active from the late G1 phase until the G2 phase and mediates the ubiquitination and destruction of G1 cyclins and Cdk inhibitors [1, 7]. SCF consists of the adaptor protein Skp1, the scaffold protein Cul1, the ring-finger protein Rbx1, as well as a variable component that is responsible for substrate recognition known as the F-box protein. The human genome encodes 68 putative F-box proteins, thereby providing sufficient flexibility for substrate specificity [8]. Most of the physiological functions of these putative F-box proteins remain unknown. The well-characterized F-box proteins include Skp2, Cdc4/Fbw7, and β-TRCP1, which targets p27 [9], cyclin E [10], and Cdc25A [11], respectively, for ubiquitination and degradation. In all cases, proper phosphorylation of the substrate is required for its interaction with the F-box proteins.
Fbw7 is a tumor suppressor
Loss of Fbw7 is frequently observed in various types of tumors including breast cancer, colon cancer [12] and T-cell acute lymphoblastic leukemia (T-ALL) [13]. It has been documented that tissue-specific deletion of Fbw7 in mouse T cells results in the development of T-ALL [14-16], suggesting that Fbw7 is a novel tumor suppressor in T-ALL. However, the exact molecular mechanisms by which Fbw7 exerts its anti-tumor activity are still unknown [4]. We previously discovered that Fbw7 regulates the degradation of c-Jun in a GSK3 phosphorylation-dependent manner [17]. Our work assigned a biological significance to the v-Jun S243F point mutation and also underscored the importance of Fbw7 in tumor suppression [17]. In addition to the turnover of cyclin E [10] and c-Jun, Fbw7 is also involved in the degradation of c-Myc [18, 19], and the Notch-1 protein [20] (Figure 1), all of which have been reported to possess oncogenic functions and are frequently found to be overexpressed in various human cancers, including leukemia. Consistent with frequent loss of Fbw7, overexpression of c-Myc, c-Jun and Notch-1 is closely associated with the development of T-ALL. Besides accelerating cell growth [21], overexpression of either c-Jun, c-Myc or Notch-1 results in cell death through upregulation of the pro-apoptotic protein Bim-1 [22]. However, despite the ever-growing list of Fbw7 ubiquitin substrates (Figure 1), it remains unclear how Fbw7-deficient cells evade cell death in the setting of upregulated c-Jun, c-Myc or Notch-1 (Figure 2A).
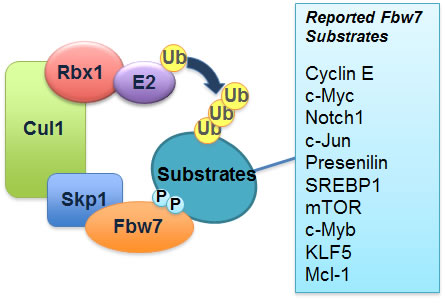
Figure 1: Schematic illustration of the SCFFbw7 E3 ubiquitin ligase complex and a list of its identified downstream ubiquitin substrates.
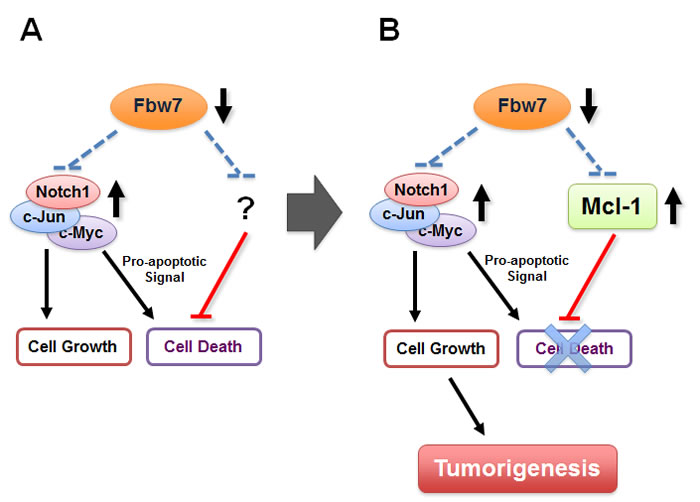
Figure 2: Fbw7 participates in the regulation of cellular apoptosis by targeting the pro-survival factor Mcl-1 for ubiquitination and destruction.
A. Loss of Fbw7 leads to the elevated expression of c-Myc, c-Jun, and the Notch-1 protein, all of which possess oncogenic functions and are frequently found to be overexpressed in various human cancers, including leukemia. Besides promoting cell growth, it has been shown that overexpression of either c-Jun, c-Myc or Notch-1 provokes cellular apoptosis. However, it remains unclear how Fbw7-deficient cells evade programmed cell death in the setting of upregulated c-Jun, c-Myc or Notch-1.
B. We recently reported that loss of Fbw7 also leads to a significant elevation in Mcl-1 expression, which suppresses the induction of apoptosis by inactivating the pro-apoptotic function of many BH3 only proteins including Bim, Bax and Bak
The Mcl-1 oncoprotein is found to be frequently overexpressed in leukemia
Expression of the anti-apoptotic protein Mcl-1 is frequently elevated in various human tumors including leukemia, but the underlying mechanisms causing its elevation are not fully understood [23, 24]. Mcl-1 is a pro-survival member of the Bcl-2 family of proteins, which can suppress apoptosis by interacting with and suppressing the activities of pro-apoptotic proteins including Bim, Bax and Bak [25]. However, unlike other Bcl-2 family members, the Mcl-1 protein is extremely unstable, having a very short half-life [26]. The rapid induction and destruction of Mcl-1 has been proposed as a molecular mechanism for cells to switch into either the survival or apoptotic pathways in response to various stresses [27, 28]. Although GSK3 phosphorylation is reported to regulate Mcl-1 stability directly [26], little is known about the upstream E3 ubiquitin ligase that targets phosphorylated Mcl-1 for destruction. As illustrated in Figure 2B, we and others recently reported that Fbw7 targets Mcl-1 for ubiquitination and destruction in a GSK3-dependent manner [29, 30]. Therefore, our studies suggest that the simultaneous elevation of the pro-survival factor Mcl-1 provides a protection mechanism allowing Fbw7-deficient cells to evade apoptosis, thus providing a novel molecular mechanism for the tumor suppression function of Fbw7 (Figure 2B). Moreover, Mcl-1 has been demonstrated to play a key role in regulating the cellular apoptosis of T cells, but not other tissue types such as liver cells [27, 31]. Therefore, our studies also provide the possible mechanism for why loss of Fbw7 is very frequently observed in T-ALL patients.
The Fbw7-deficient T-ALL cells are “addicted” to high expression levels of Mcl-1
Our studies for the first time provide firm experimental evidence for a potential role for the Fbw7 tumor suppressor in the modulation of the apoptotic pathway by governing Mcl-1 ubiquitination and destruction. Therefore, loss of Fbw7 not only provides a growth advantage by upregulating the c-Jun and c-Myc oncoproteins, but also leads to elevated Mcl-1 expression as a protection mechanism allowing Fbw7-deficient cells to evade possible apoptosis induced by high c-Jun and c-Myc expression (Figure 2B). However, this intricate balance to evade apoptosis seems to depend on high levels of the Mcl-1 oncoprotein. As a result, compared to WT-Fbw7 cells, Fbw7-deficient T-ALL cells are much more sensitive to the Mcl-1 inhibitor, sorafenib (Figure 3). Sorafenib is a multi-kinase inhibitor reported to suppress B-Raf, PDGF receptor and VEGF receptor kinase activities. Although its ability to repress Mcl-1 has been attributed to inactivating MAPK kinase and/or activating GSK-3 [32], the exact mechanism remains unclear. Nevertheless, this data suggests that Fbw7-deficient T-ALL cell lines might require high levels of the Mcl-1 oncoprotein to evade apoptosis, a phenotype that has been described previously as “oncogene addiction” [33]. Our studies thus provide a basis for personalized medicine for T-ALL patients as well as the rationale for developing specific Mcl-1 antagonists, or agents that significantly reduce Mcl-1 expression, to treat T-ALL patients.
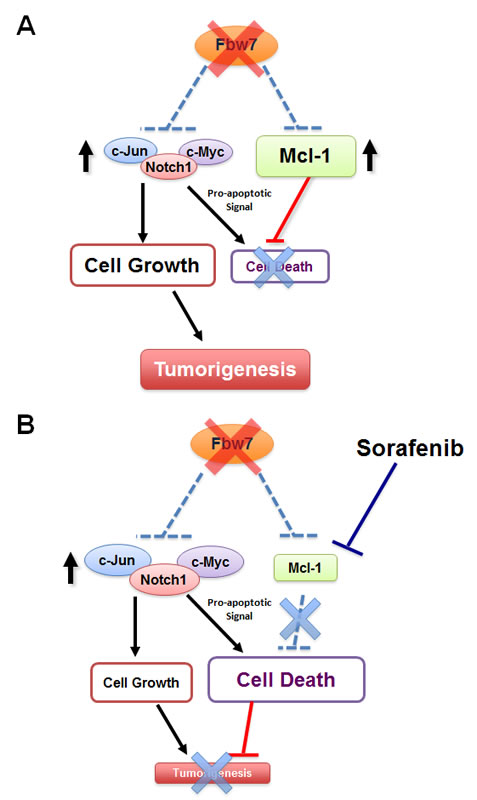
Figure 3: Fbw7-deficient T-ALL cells are “addicted” to high levels of Mcl-1 expression and are particularly sensitive to the Mcl-1 antagonist sorafenib.
A. In unstressed Fbw7-deficient T-ALL cells, induced expression of the pro-survival factor Mcl-1 balances the pro-apoptotic effects of elevated c-Jun, c-Myc and Notch-1 oncoproteins. However, the cells become “addicted” to high expression levels of the Mcl-1 oncoprotein.
B. When Fbw7-deficient cells are treated with the Mcl-1 antagonist, sorafenib, the pro-survival function of Mcl-1 is inhibited. On the other hand, loss of Fbw7 leads to elevated expression of c-Jun, c-Myc and Notch-1, which provokes cellular apoptosis. When the anti-apoptotic effect of Mcl-1 is inhibited, cells undergo programmed cell death. Therefore, compared to the WT-Fbw7 T-ALL cells, Fbw7-deficient T-ALL cells are very sensitive to Mcl-1 antagonists.
The Fbw7-deficient T-ALL cells are resistant to ABT-737 treatment
The BH3 mimetic ABT-737 is a specific pan-inhibitor of the Bcl-2 family of anti-apoptotic proteins, which is reported to effectively kill leukemia cells, presumably by disrupting the Bcl2/Bax complex and inducing the Bak-dependent apoptotic pathway [34]. However, leukemia cells with elevated Mcl-1 expression are found to be refractory to ABT-737 treatment [35, 36] primarily because ABT-737 fails to inactivate Mcl-1 due to a low binding affinity [34]. Consistent with this, we found that Fbw7-deficient T-ALL cells, which displayed a significant increase in Mcl-1 expression, are much more resistant than Fbw7-WT T-ALL cells in response to ABT-737 (Figure 4). We further showed that depletion of Mcl-1, or reintroduction of Fbw7 into Fbw7-deficient T-ALL cells, restored ABT-737 sensitivity. This suggests that increased Mcl-1 expression is the determining factor that confers Fbw7-deficient cells resistance to ABT-737. Although it warrants further investigation, this work indicates that Fbw7-deficient T-ALL patients may not respond well to ABT-737 treatment.
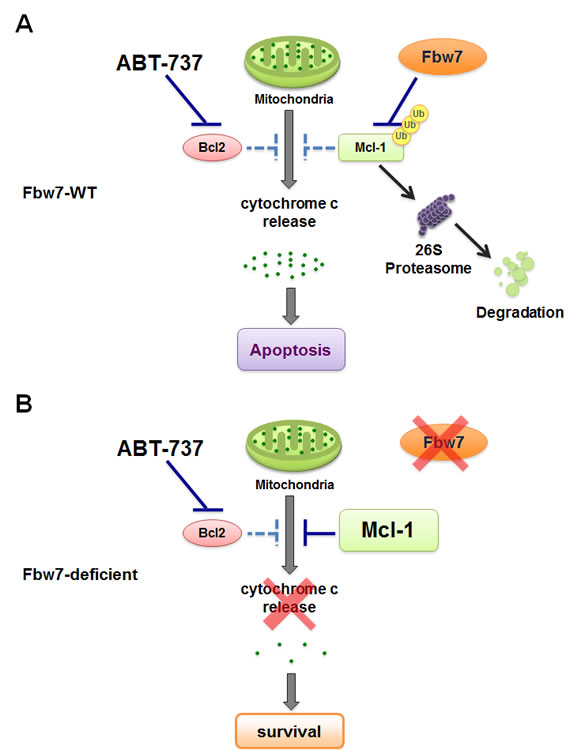
Figure 4: Loss of Fbw7 leads to elevated Mcl-1 expression, which confers increased resistance to the pan-Bcl-2 inhibitor ABT-737.
A. In T-ALL cells with WT-Fbw7 genetic background, Mcl-1 stability is governed by Fbw7 in a GSK3-dependent manner. Inhibition of the Bcl-2 family of proteins with ABT-737 efficiently triggers apoptosis by inducing cytochrome c release.
B. Due to structural differences, the pan-Bcl-2 inhibitor ABT-737 cannot efficiently inactivate Mcl-1 as it does to the rest of the Bcl-2 family of proteins. Therefore, loss of Fbw7 leads to elevated Mcl-1 expression, subsequently resulting in increased resistance to ABT-737.
Fbw7 is the physiological E3 ligase that targets Mcl-1 for ubiquitination in T-ALL
Besides Fbw7, other E3 ubiquitin ligases including c-Mule [37] and β-TRCP [38] have also been implicated in Mcl-1 stability control. However, we found that although depletion of c-Mule leads to Mcl-1 upregulation in T-ALL, this regulation is not contingent upon GSK3-dependent phosphorylation of Mcl-1 [37, 38]. Most importantly, unlike the frequent loss of Fbw7 found in T-ALL, no correlation was found between the expression of c-Mule and Mcl-1 in various T-ALL cell lines. These results exclude a physiological role for c-Mule in regulating Mcl-1 in T-ALL cells. Additionally, although ectopic expression of β-TRCP promotes Mcl-1 destruction, no β-TRCP-dependent induction of Mcl-1 ubiquitination was observed. Furthermore, depletion of endogenous Fbw7, but not endogenous β-TRCP, leads to a significant induction of Mcl-1, rejecting the notion that β-TRCP physiologically control Mcl-1 abundance in T-ALL cell lines. Consistent with this finding, array CGH analysis demonstrated a high frequency of Fbw7 loss [13], but not simultaneous loss of β-TRCP1 and β-TRCP2 in the T-ALL cell lines. These data together support the hypothesis that the Fbw7 tumor suppressor, which is frequently lost in T-ALL, is the physiological E3 ubiquitin ligase for Mcl-1. However, it remains unclear whether in other tissue types or cellular context, other than Fbw7, c-Mule or β-TRCP will be the major force that governs Mcl-1 stability.
Whether the PTEN/PI3K/Akt axis is the upstream signaling pathway that governs Fbw7-mediated ubiquitination of Mcl-1
Our work indicates that loss of Fbw7 contributes to Mcl-1 overexpression in T-ALL. However, only 20-30% of T-ALL cases are estimated to possess inactive Fbw7 [13], thus it is critical to understand how Fbw7-mediated Mcl-1 ubiquitination is physiologically regulated in vivo, and how Mcl-1 expression is aberrantly elevated in WT-Fbw7 genetic background, which accounts for 70-80% of T-ALL cases. To this end, we found that in WT-Fbw7 genetic backgrounds, loss of the PTEN tumor suppressor, which leads to inactivation of GSK3 kinase, also results in elevated Mcl-1 expression in a similar fashion to Fbw7-deficiency (data not shown). The PTEN phosphatase is a negative regulator of the PI3K/Akt signaling pathway and loss of PTEN is frequently observed in many types of tumors including T-ALL [13, 39-41]. Since our recent studies clearly demonstrated that GSK3 plays a critical role for Fbw7-mediated Mcl-1 ubiquitination, we hypothesize that any aberrant inactivation of GSK3, including loss of PTEN activity [42], might phenocopy Fbw7 deficiency, resulting in elevated Mcl-1 expression (Figure 5). Looking forward, it is thus critical to determine, especially in the setting of T-ALL, the molecular mechanisms by which the PTEN/PI3K/Akt/GSK3 axis participates in Fbw7-mediated destruction of Mcl-1, and to evaluate whether Akt or mTOR inhibitors could be a novel anti-leukemia therapeutic option to trigger apoptosis by decreasing Mcl-1 protein abundance, especially for those who are PTEN-deficient.
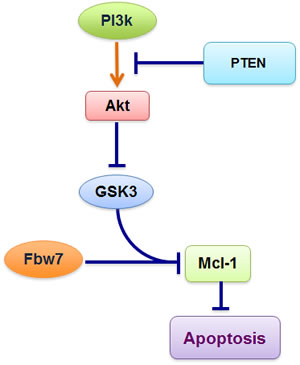
Figure 5: Schematic illustration of the potential role for the PTEN/PI3K/Akt signaling axis in governing Fbw7-mediated Mcl-1 ubiquitination and destruction. Loss of PTEN, which leads to aberrant activation of the PI3K/Akt signaling cascade, results in inactivation of GSK3 and a subsequent increase in Mcl-1 expression.
CONFLICT OF INTEREST
The authors have no conflict of interests to declare.
ACKNOWLEDGEMENTS
We thank Lixin Wan and Daming Gao for critical reading of the manuscript, and the other members of the Wei laboratory for useful discussions. W.W. is a DOD Prostate Research Program New Investigator. Our studies of Mcl-1 destruction were supported in part by the Massachusetts Life Science Center New Investigator award (W.W.), and by NIH grant GM089763 to W.W.
REFERENCES
1. Nakayama KI, Hatakeyama S, Nakayama K. Regulation of the cell cycle at the G1-S transition by proteolysis of cyclin E and p27Kip1. Biochem Biophys Res Commun 2001; 282:853-860.
2. Ang XL, Harper JW. Interwoven ubiquitination oscillators and control of cell cycle transitions. Sci STKE 2004; 2004:pe31.
3. Wei W, Ayad NG, Wan Y, Zhang GJ, Kirschner MW, Kaelin WG, Jr. Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature 2004; 428:194-198.
4. Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer 2008; 8:83-93.
5. Peters JM. The anaphase-promoting complex: proteolysis in mitosis and beyond. Mol Cell 2002; 9:931-943.
6. Harper JW, Burton JL, Solomon MJ. The anaphase-promoting complex: it’s not just for mitosis any more. Genes Dev 2002; 16:2179-2206.
7. Nakayama KI, Nakayama K. Regulation of the cell cycle by SCF-type ubiquitin ligases. Semin Cell Dev Biol 2005; 16:323-333.
8. Jin J, Cardozo T, Lovering RC, Elledge SJ, Pagano M, Harper JW. Systematic analysis and nomenclature of mammalian F-box proteins. Genes Dev 2004; 18:2573-2580.
9. Carrano AC, Eytan E, Hershko A, Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol 1999; 1:193-199.
10. Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW, Elledge SJ. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science 2001; 294:173-177.
11. Busino L, Donzelli M, Chiesa M, Guardavaccaro D, Ganoth D, Dorrello NV, Hershko A, Pagano M, Draetta GF. Degradation of Cdc25A by beta-TrCP during S phase and in response to DNA damage. Nature 2003; 426:87-91.
12. Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, Silliman N, Szabo S, Dezso Z, Ustyanksky V, Nikolskaya T, Nikolsky Y, Karchin R, Wilson PA, Kaminker JS, Zhang Z, Croshaw R, Willis J, Dawson D, Shipitsin M, Willson JK, Sukumar S, Polyak K, Park BH, Pethiyagoda CL, Pant PV, Ballinger DG, Sparks AB, Hartigan J, Smith DR, Suh E, Papadopoulos N, Buckhaults P, Markowitz SD, Parmigiani G, Kinzler KW, Velculescu VE, Vogelstein B. The genomic landscapes of human breast and colorectal cancers. Science 2007; 318:1108-1113.
13. Maser RS, Choudhury B, Campbell PJ, Feng B, Wong KK, Protopopov A, O’Neil J, Gutierrez A, Ivanova E, Perna I, Lin E, Mani V, Jiang S, McNamara K, Zaghlul S, Edkins S, Stevens C, Brennan C, Martin ES, Wiedemeyer R, Kabbarah O, Nogueira C, Histen G, Aster J, Mansour M, Duke V, Foroni L, Fielding AK, Goldstone AH, Rowe JM, Wang YA, Look AT, Stratton MR, Chin L, Futreal PA, DePinho RA. Chromosomally unstable mouse tumours have genomic alterations similar to diverse human cancers. Nature 2007; 447:966-971.
14. Thompson BJ, Buonamici S, Sulis ML, Palomero T, Vilimas T, Basso G, Ferrando A, Aifantis I. The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J Exp Med 2007; 204:1825-1835.
15. Onoyama I, Tsunematsu R, Matsumoto A, Kimura T, de Alboran IM, Nakayama K, Nakayama KI. Conditional inactivation of Fbxw7 impairs cell-cycle exit during T cell differentiation and results in lymphomatogenesis. J Exp Med 2007; 204:2875-2888.
16. Mao JH, Perez-Losada J, Wu D, Delrosario R, Tsunematsu R, Nakayama KI, Brown K, Bryson S, Balmain A. Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature 2004; 432:775-779.
17. Wei W, Jin J, Schlisio S, Harper JW, Kaelin WG, Jr. The v-Jun point mutation allows c-Jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer Cell 2005; 8:25-33.
18. Welcker M, Orian A, Jin J, Grim JA, Harper JW, Eisenman RN, Clurman BE. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci U S A 2004; 101:9085-9090.
19. Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, Ishida N, Okumura F, Nakayama K, Nakayama KI. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. Embo J 2004; 23:2116-2125.
20. Gupta-Rossi N, Le Bail O, Gonen H, Brou C, Logeat F, Six E, Ciechanover A, Israel A. Functional interaction between SEL-10, an F-box protein, and the nuclear form of activated Notch1 receptor. J Biol Chem 2001; 276:34371-34378.
21. Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol 2002; 4:E131-136.
22. Sanchez I, Yuan J. A convoluted way to die. Neuron 2001; 29:563-566.
23. Cory S, Huang DC, Adams JM. The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene 2003; 22:8590-8607.
24. Akgul C. Mcl-1 is a potential therapeutic target in multiple types of cancer. Cell Mol Life Sci 2009; 66:1326-1336.
25. Willis SN, Adams JM. Life in the balance: how BH3-only proteins induce apoptosis. Curr Opin Cell Biol 2005; 17:617-625.
26. Maurer U, Charvet C, Wagman AS, Dejardin E, Green DR. Glycogen synthase kinase-3 regulates mitochondrial outer membrane permeabilization and apoptosis by destabilization of MCL-1. Mol Cell 2006; 21:749-760.
27. Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature 2003; 426:671-676.
28. Nijhawan D, Fang M, Traer E, Zhong Q, Gao W, Du F, Wang X. Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev 2003; 17:1475-1486.
29. Inuzuka H, Shaik S, Onoyama I, Gao D, Tseng A, Maser RS, Zhai B, Wan L, Gutierrez A, Lau AW, Xiao Y, Christie AL, Aster J, Settleman J, Gygi SP, Kung AL, Look T, Nakayama KI, DePinho RA, Wei W. SCFFBW7 regulates cellular apoptosis by targeting MCL1 for ubiquitylation and destruction. Nature 2011; 471:104-109.
30. Wertz IE, Kusam S, Lam C, Okamoto T, Sandoval W, Anderson DJ, Helgason E, Ernst JA, Eby M, Liu J, Belmont LD, Kaminker JS, O’Rourke KM, Pujara K, Kohli PB, Johnson AR, Chiu ML, Lill JR, Jackson PK, Fairbrother WJ, Seshagiri S, Ludlam MJ, Leong KG, Dueber EC, Maecker H, Huang DC, Dixit VM. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 2011; 471:110-114.
31. Opferman JT, Iwasaki H, Ong CC, Suh H, Mizuno S, Akashi K, Korsmeyer SJ. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science 2005; 307:1101-1104.
32. Panka DJ, Cho DC, Atkins MB, Mier JW. GSK-3beta inhibition enhances sorafenib-induced apoptosis in melanoma cell lines. J Biol Chem 2008; 283:726-732.
33. Sharma SV, Settleman J. Oncogene addiction: setting the stage for molecularly targeted cancer therapy. Genes Dev 2007; 21:3214-3231.
34. Cragg MS, Harris C, Strasser A, Scott CL. Unleashing the power of inhibitors of oncogenic kinases through BH3 mimetics. Nat Rev Cancer 2009; 9:321-326.
35. Konopleva M, Watt J, Contractor R, Tsao T, Harris D, Estrov Z, Bornmann W, Kantarjian H, Viallet J, Samudio I, Andreeff M. Mechanisms of antileukemic activity of the novel Bcl-2 homology domain-3 mimetic GX15-070 (obatoclax). Cancer Res 2008; 68:3413-3420.
36. van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, Willis SN, Scott CL, Day CL, Cory S, Adams JM, Roberts AW, Huang DC. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 2006; 10:389-399.
37. Zhong Q, Gao W, Du F, Wang X. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 2005; 121:1085-1095.
38. Ding Q, He X, Hsu JM, Xia W, Chen CT, Li LY, Lee DF, Liu JC, Zhong Q, Wang X, Hung MC. Degradation of Mcl-1 by beta-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol Cell Biol 2007; 27:4006-4017.
39. Mao JH, Kim IJ, Wu D, Climent J, Kang HC, DelRosario R, Balmain A. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science 2008; 321:1499-1502.
40. Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell 2008; 133:403-414.
41. Carracedo A, Pandolfi PP. The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene 2008; 27:5527-5541.
42. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell 2007; 129:1261-1274.