
INTRODUCTION
The aryl hydrocarbon receptor interacting protein (AIP) is a highly conserved co-chaperone with a poorly characterized tumor suppressor function [1–3]. Germline mutations in the AIP gene (AIPmuts) resulting in a truncated and/or unstable AIP protein are the most common genetic cause of pituitary adenomas affecting teenagers and young adults, presenting either as familial isolated pituitary adenoma (FIPA) or as simplex cases of acromegaly or gigantism [4–7]. Despite the ubiquitous expression of the AIP protein, no other tumor types have been consistently associated with AIPmuts. This apparent tissue selectivity could perhaps be explained by a tumor suppressor function that is due to pituitary-specific interactions, rather than a tissue-specific expression pattern.
The human AIP protein (UniProt O00170) is a 37 kDa/330 amino acid protein composed of an amino (N)-terminal peptidyl-prolyl cis-trans isomerase (PPIase)-like domain, with no enzymatic activity, and a carboxyl (C)-terminal tetratricopeptide repeat (TPR) domain containing three TPR motifs and a C-terminal alpha 7 helix [8–12]. The best-known function of AIP is to form part, together with the heat-shock protein HSP90 and the co-chaperone translationally-controlled tumor protein (TCTP), of the multiprotein complex that regulates the nuclear translocation of the aryl hydrocarbon receptor (AHR) [8, 13–19]. In addition to HSP90, AIP also serves as a co-chaperone for other molecular chaperones, such as HSPA8 and mitochondrial import receptor subunit TOM20 homolog (TOMM20) [11, 20–22]. It has been proposed that the AIP/HSPA8 complex would bind unfolded mitochondrial pre-proteins; AIP should facilitate the transfer of such pre-proteins to TOMM20, enabling their folding and mitochondrial translocation [20].
Nevertheless, AIP displays a promiscuous repertoire of molecular interactions, including at least two viral proteins, and a variety of human proteins, (Table 1), but none of them has an obvious pituitary-specific function (reviewed in [23]) [24–26]). To elucidate the organ-specific physiological function of AIP in the pituitary gland, and to explore the role of AIP in the pathways that drive pituitary tumorigenesis, we aimed to identify the molecular partners of AIP in the pituitary gland using a proteomic approach.
Table 1: Proven and putative human interacting partners of AIP (human proteins only)*
UniProt entry |
Protein name (gene) |
Experimental method |
References |
|---|---|---|---|
P60709 |
Actin, cytoplasmic 1 (ACTB)¶ |
AC-MS |
[64] |
Q9UL18 |
Argonaute RISC catalytic component 1 (AGO1) |
AC-MS, co-IP |
[95] |
P35869 |
Aryl hydrocarbon receptor (AHR) |
Co-IP |
|
RC |
|||
2H |
|||
P27540 |
Aryl hydrocarbon receptor nuclear translocator (ARNT)† |
Co-IP |
[64] |
RC |
|||
2H |
[9] |
||
O15392 |
Baculoviral IAP repeat-containing protein 5 (BIRC5) |
AC-MS, co-IP |
[21] |
Q9BXL7 |
Caspase recruitment domain-containing protein 11 (CARD11) |
2H, co-IP |
[99] |
Q96HB5 |
Coiled-coil domain containing protein 120 (CCDC120) |
AC-MS |
[100] |
Q16543 |
Hsp90 co-chaperone cell division cycle 37 (CDC37) |
AC-L, AC-MS |
[101] |
P50750 |
Cyclin-dependent kinase 9 (CDK9) |
AC-MS |
|
Q9BXN2 |
C-type lectin domain family 7, member A (CLEC7A) |
2H |
[102] |
P68400 |
Casein kinase 2, alpha 1 polypeptide (CSNK2A1) |
BA |
[103] |
Q9NVR5 |
Protein kintoun (DNAAF2) |
AC-L, AC-MS |
[101] |
P00533 |
Epidermal growth factor receptor (EGFR) |
PCA |
[104] |
P41091 |
Eukaryotic translation initiation factor 2 subunit 3 (EIF2S3) |
Co-F |
[105] |
P03372 |
Oestrogen receptor (ESR1) |
Co-IP |
[106] |
Q9UK99 |
F-box only protein 3 (FBXO3) |
Co-IP, AC-MS |
[7] |
P50395 |
Rab GDP dissociation inhibitor beta (GDIB) |
Co-F |
[107] |
Q14344 |
Guanine nucleotide-binding protein subunit alpha 13 (GNA13) |
RC, 2H |
[108] |
P50148 |
Guanine nucleotide-binding protein G(q) subunit alpha (GNAQ) |
RC |
[108] |
P11142 |
Heat shock cognate 71 kDa protein (HSPA8) |
Co-IP |
[20] |
P07900 |
Heat shock protein 90-alpha (HSP90AA1) |
AC-L, AC-MS |
[101] |
RC |
|||
P08238 |
Heat shock protein 90-beta (HSP90AB1) |
AC-L, AC-MS |
[101] |
Co-IP |
|||
P38646 |
Stress-70 protein, mitochondrial (HSPA9) |
Co-F |
[105] |
Q9Y6K9 |
NF-kappa-B essential modulator (IKBKG) |
2H |
[112] |
Q92985 |
Interferon regulatory factor 7 (IRF7) |
AC-MS, RC |
[113] |
Co-IP |
[114] |
||
P11279 |
Lysosome-associated membrane glycoprotein 1 (LAMP1) |
Co-F |
[105] |
Q9NZR2 |
Low-density lipoprotein receptor-related protein 1B (LRP1B) |
2H, co-IP |
[115] |
Q9NXB0 |
Meckel syndrome type 1 protein (MKS1) |
PL-MS |
[116] |
UniProt entry |
Protein name (gene) |
Experimental method |
References |
Q6IA69 |
Glutamine-dependent NAD(+) synthetase (NADSYN1) |
AC-L, AC-MS |
[101] |
Q86SG6 |
Serine/threonine-protein kinase Nek8 (NEK8) |
PL-MS |
[116] |
Q8WWR8 |
Sialidase-4 (NEU4) |
AC-MS |
[100] |
O75161 |
Nephrocystin-4 (NPHP4) |
PL-MS |
[116] |
P08235 |
Mineralocorticoid receptor (NR3C2) |
Co-IP |
[111] |
O75665 |
Oral-facial-digital syndrome 1 protein (OFD1) |
PL-MS |
[116] |
P27815 |
cAMP-specific 3’,5’-cyclic phosphodiesterase 4A (PDE4A) |
2H |
|
Co-IP |
[117] |
||
O00408 |
cGMP-dependent 3’,5’-cyclic phosphodiesterase 2A (PDE2A) |
2H, co-IP, co-loc |
[118] |
Q15181 |
Inorganic pyrophosphatase (PPA1) |
Co-F |
[119] |
Q9H2U2 |
Inorganic pyrophosphatase 2, mitochondrial (PPA2) |
Co-F |
[119] |
Q07869 |
Peroxisome proliferator-activated receptor alpha (PPARA) |
Co-IP, RC |
[120] |
O75170 |
Serine/threonine-protein phosphatase 6 regulatory subunit 2 (PPP6R2) |
PL-MS |
[121] |
O60809 |
PRAME family member 10 (PRAMEF10) |
AC-MS |
[100] |
Q5VTA0 |
PRAME family member 10 (PRAMEF17) |
AC-MS |
[100] |
P48147 |
Prolyl endopeptidase (PREP) |
Co-F |
[119] |
Q15185 |
Prostaglandin E synthase 3 (PTGES3) |
AC-L, AC-MS |
[101] |
Co-IP |
[111] |
||
P07949 |
Rearranged during transfection tyrosine-kinase receptor (RET) |
Co-IP, PCA |
[122] |
P31948 |
Stress-induced phosphoprotein 1 (STIP1) |
RC |
[52] |
Q9Y2Z0 |
Suppressor of G2 allele of SKP1 homolog (SUGT1) |
AC-L, AC-MS |
[101] |
P13385 |
Teratocarcinoma-derived growth factor 1 (TDGF1) |
AC-MS |
[100] |
Q59H18 |
TNNI3 interacting kinase (TNNI3K) |
2H |
[123] |
Q15388 |
Mitochondrial import receptor subunit TOM20 homolog (TOMM20) |
2H, co-IP |
[20] |
RC |
[11] |
||
P10828 |
Thyroid hormone receptor beta (THRB) |
2H |
[124] |
P0CG48 |
Polyubiquitin C (UBC) |
AC-MS |
|
Q6PHR2 |
Serine/threonine-protein kinase ULK3 (ULK3) |
AC-MS |
[100] |
O94966 |
Ubiquitin specific peptidase 19 (USP19) |
AC-L, AC-MS |
[101] |
P07947 |
Tyrosine-protein kinase Yes (YES1) |
AC-MS |
[100] |
Abbreviations: 2H: two-hybrid assay, AC-L: Affinity capture-luminescence, AC-MS: affinity capture (pull-down)-mass spectrometry, BA: biochemical activity (an interaction is inferred from the biochemical effect of one protein upon another), co-F: co-fractionation, co-IP: co-immunoprecipitation (affinity capture-Western blot), co-loc: co-localization, PCA: protein-fragment complementation assay, PL-MS: proximity label-mass spectrometry, RC: reconstituted complex (an interaction is detected between two proteins in vitro). Detailed explanation of all the experimental methods: http://wiki.thebiogrid.org/doku.php/experimental_systems.
*Includes proteins detected either by direct protein-protein interaction experiments or as part of datasets from high-throughput proteomic studies reported in the literature and/or in the Biological General Repository for Interaction Datasets [26].
¶Contradictive results, a direct interaction between AIP and ACTB was disproven by a different study [65].
†Contradictive results, a direct interaction between AIP and ARNT was disproven by different studies [8, 16, 109].
§These studies did not specify the isoform of HSP90 analyzed or used a peptide that is common to HSP90AA1 and HSP90AB1.
RESULTS
Candidate AIP partners and differential interactions among AIP variants
We synthesized N-terminally glutathione-S-transferase (GST)-tagged AIP proteins, including the human wild-type (WT) protein and the variants p.C238Y, p.K266A, p.A299V, p.R304* and p.R304Q (Table 2), and a GST-only negative control. Using these proteins as baits, pull-down experiments were performed in lysates from rat somatotropinoma-derived cells (GH3). Tandem mass tags were used to label the bound proteins, which were pooled together and analyzed by mass spectrometry (MS). Results were filtered for significance, normalized against the negative control, and compared against the WT experiment. Human homologues of these candidate AIP partners were identified and grouped in signaling pathways.
Table 2: AIP variants selected for the study
Variant* |
Minor allele frequency (ExAc)* |
Location in protein |
Clinical data |
Experimental data |
Classification |
|---|---|---|---|---|---|
c.713G>A, p.C238Y (chr11:67257854G>A) |
0.000008 |
TPR2 domain |
Detected in one FIPA family with three cases of acromegaly (1.4% of the AIPmut positive cases in our cohort) [6, 90, 130, 131]. |
Results in an unstable protein, probably due to abnormal packaging of the alpha and beta-helices in the second TPR motif [7, 11], with reduced ability for blocking cell proliferation [90], as well as complete disruption the AIP-PDE4A5 interaction [30, 131]. This variant is unable to rescue the lethality caused by CG1847 knockout in a fruit fly model [132]. |
Pathogenic |
c.796_797delinsGC, p.K266A (chr11:67258267_67258268delinsGC) |
NA |
TPR3 domain |
Not detected in patients. |
Disrupts the interaction of AIP with HSP90. Loss of HSP90 binding significantly reduces the ability of AIP to interact with AHR, but it is unknown if this affects other signaling pathways [55]. |
Experimental |
c.896C>T, p.A299V (chr11:67258367C>T) |
0.000428 |
TPR3 domain |
Clinical evaluation of multiple carriers does not support a pathogenic role for this rare SNP [131]. It was found in trans with a truncating AIP mutation in two subjects without pituitary adenomas. Detected in 0.8% of the individuals in our cohort (one patient and four unaffected members of a single FIPA family) [6]. |
Although in silico analysis predicted a possible disruption of protein folding, this variant results in a stable protein, displaying only slight reduction in PDE4A5 binding [7, 30, 131]. Acting similarly to the wild-type protein, this variant is able to rescue the lethality in a CG1847 knockout fruit fly model [132]. |
Non-pathogenic |
c.910C>T, p.R304* (chr11:67258381C>T) |
0.000017 |
TPR3 domain |
Most common AIP mutation associated with familial and sporadic pituitary adenomas (35.9% of all the AIPmut positive cases in our cohort) [6]. Founder effect in the Northern Irish [133, 134] and Northern Italian [135] populations. |
This nonsense mutation is translated into a truncated, unstable protein, resulting in half the normal total AIP content in cells from heterozygous carriers [7, 136]. Causes complete disruption of the PDE4A5 binding and loss of the ability of the mutant AIP to block cell proliferation [30, 90], and impaired ability to suppress cAMP signaling in response to forskolin [2]. |
Pathogenic |
c.911G>A, p.R304Q (chr11:67258382G>A) |
0.001458** |
TPR3 domain |
Found for the first time in an apparently sporadic case of Cushing’s disease [137], and subsequently in several young-onset and familial cases, this variant is not rare among AIP pituitary adenoma patients (second most common in our cohort, found in 7.9% of the AIPmut cases) [6]. Two subjects in ExAc carry this variant in homozygosis. |
Relatively conservative, changing a longer side chain, positively charged amino acid, to a slightly shorter, uncharged, hydrophilic one at the C-terminal alpha-7 helix [11]. The protein displays normal half-life [7]. Partially disrupts PDE4A5 binding [30]. Retains the ability to rescue the lethality caused by CG1847 knockout in the fruit fly [132]. |
Variant of unknown significance |
NA, not available.
*All the variants are annotated in the GRCh37/hg19 human genome assembly using the reference transcript NM_003977.3.
**Includes two homozygous individuals.
The manually validated qualitative MS results (Supplementary Table 1) accounted for a total of 514 different peptides, matching 154 proteins in addition to AIP itself. After applying selection filters, 30 proteins were identified as candidate partners for WT AIP (Table 3). Pathway analysis identified “Remodeling of the epithelial adherens junction” as the top canonical pathway (Figure 1A), including AIP and 19 of its candidate partners within a single network (Figure 1B). As expected from its previously reported unstable behavior [7], the synthetic p.C238Y AIP protein formed aggregates that precluded its use in our experiments. The repertoire of binding peptides varied among the pull-down experiments for the rest of the AIP variants; quantitative differences in the intensity values of the peptides were interpreted as differential binding of the corresponding proteins for each bait AIP protein (Figure 1C and Supplementary Table 2). Proteins selected for further validation experiments (co-immunoprecipitation [co-IP] and co-localization) included both novel AIP interacting partners and known partners with apparent lost interactions in the pull-down experiments for AIP mutants.
Table 3: Candidate AIP partners and peptides identified by quantitative MS and their human homologues
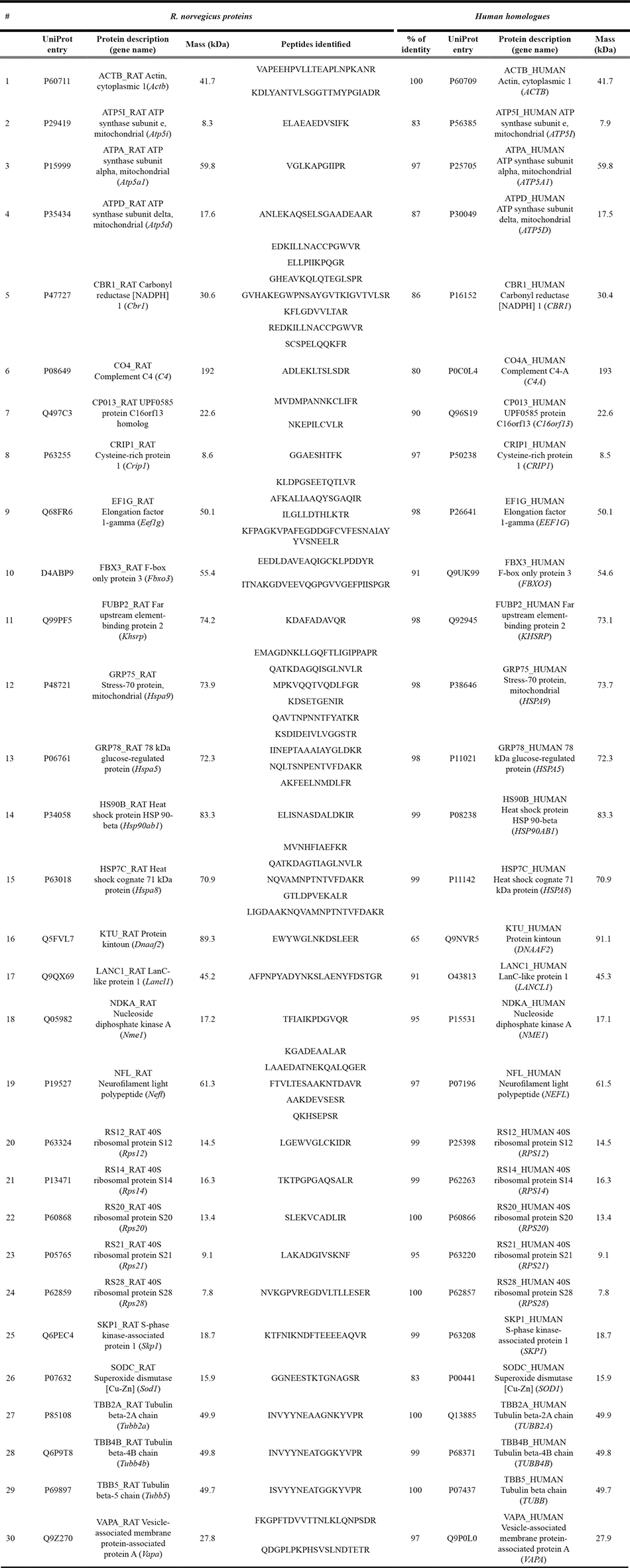
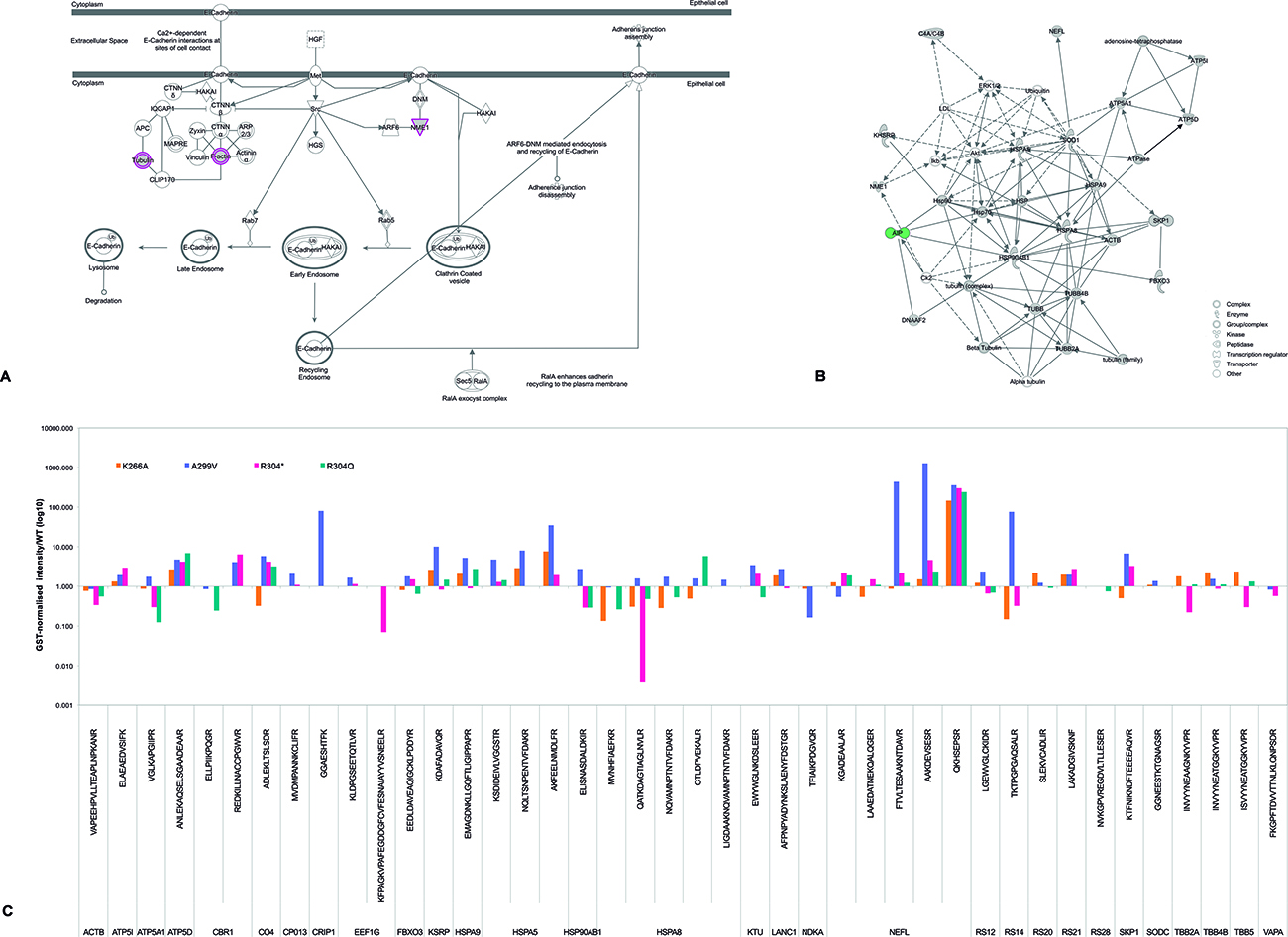
Figure 1: AIP candidate interacting partners: signaling pathways and differential peptide repertoires in pull-down experiments. (A) The top signaling pathway reported by Ingenuity Pathway Analysis when analyzing the 30 candidate interacting partners identified for WT AIP (as listed in Table 3) was “Remodeling of the epithelial adherens junction” (P < 0.0001). This pathway included five candidate partners (ACTB, NME1, TUBB, TUBB2A, and tubulin beta-4B chain [TUBB4B]) out of the total of 68 proteins reported for such pathway by the platform used (overlap: 7.4%). Proteins in this pathway represent multiprotein complexes, with cadherins as central components, mediating cell-cell adhesion and intercellular communication, and regulating cell shape and polarity. Other top canonical pathways reported by this analysis were “Regulation of eIF4 and p70S6K signaling” (5/157 proteins [3.2%], P < 0.0001), “EIF2 signaling” (5/194 [2.6%], P < 0.0001), “mTOR signaling” (5/199 [2.5%], P < 0.0001) and “Protein ubiquitination pathway” (5/255 [2.5%], P < 0.0001). Proteins marked in pink are among the AIP candidate interacting partners identified by our pull-down experiments. F-actin refers to “filamentous actin” (i.e. a polymer of actin molecules, including ACTB among other isoforms) which forms part of the cytoskeleton. (B) Nineteen of the AIP WT candidate partners were grouped together in a single network, either due to functional relationships or direct binding. (C) Schematic representation of the pull-down data presented in Supplementary Table 2 (in a logarithmic scale for easy overview) including only proteins present in the AIP WT experiment. Proteins whose peptides were underrepresented in the AIP variant protein pull-down experiments were interpreted as impaired or lost interactions.
AIP interacts with multiple molecular chaperones of the HSP70 and HSP90 families
Two of the best-known molecular partners of AIP, the heat-shock proteins HSP90 and HSPA8 (HSC70), were detected in the pull-down experiments. Co-IP experiments for AIP and both the inducible (HSPA90AA1 [HSP90-alpha]) and the constitutive (HSP90AB1 [HSP90-beta]) isoforms of HSP90 [27] and with HSPA8 were carried out and confirmed (Figure 2A–2C). Loss of HSPA8 binding to the AIP mutant p.R304* (Figure 2D) was also confirmed, supporting the reliability of our approach with pull-down experiments and comparative analysis for assuming loss of protein-protein interactions. Novel interactions of AIP with two other members of the HSP70 family, HSPA5 (also known as 78 kDa glucose-regulated protein [GRP-78]) and HSPA9 (mitochondrial HSP70, GRP-75, mortalin), were also detected by pull-down and confirmed by co-IP (Figure 2E and 2F). Co-localization of AIP and HSPA9 in the mitochondrial network was verified by immunocytofluorescence (Figure 2G).
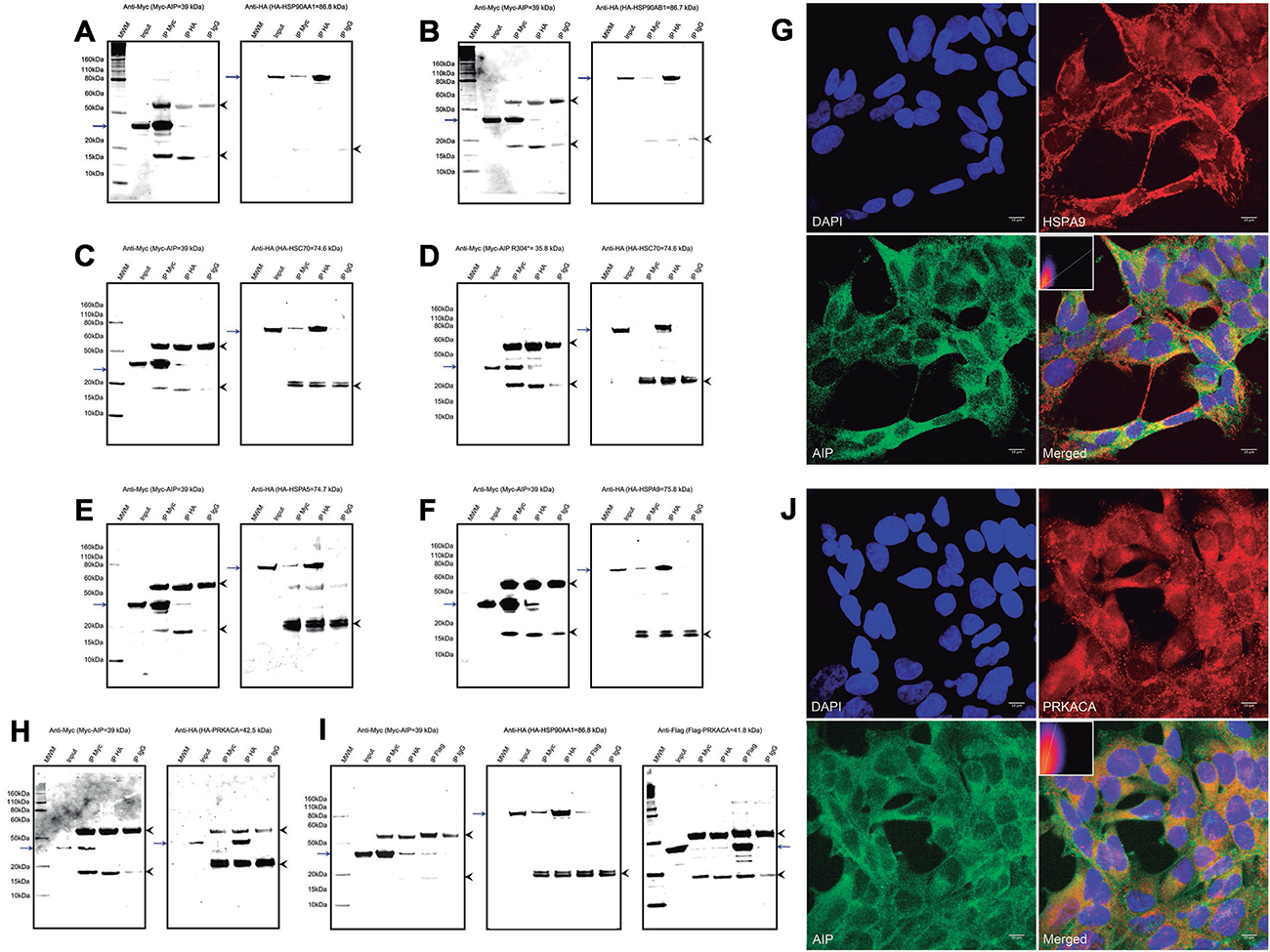
Figure 2: AIP interacts with multiple proteins from the HSP90 and HSP70 families of molecular chaperones. WT AIP interactions with HSP90AA1 (A) and HSP90AB1 (B) were confirmed by co-IP, validating the experimental procedures. Also by co-IP, HSPA8 interacted with WT AIP (C), but not with the p.R304* mutant (D), as predicted by the pull-down experiment results. Interactions with two novel molecular chaperones were validated by co-IP: HSPA5 (E) and HSPA9 (F). Co-localization of AIP and HSPA9 (G) was confirmed in the mitochondrial network, with a Pearson’s R-value of 0.72. (H) AIP does not co-immunoprecipitate with PRKACA, ruling out direct interaction of these two proteins. (I) However, in the presence of HSP90AB1, the three proteins co-immunoprecipitate with the anti-Myc, anti-HA and anti-Flag antibodies. (J) Areas of co-localization for AIP and PRKACA were detected in the cytoplasm, although with a weaker correlation compared with HSPA9, for a Pearson’s R-value of 0.39. For the co-IP experiments a-f and h, the left panels represent anti-Myc western blot (WB) membranes and the right panels, anti-HA WB membranes. For the co-IP experiment presented in i the additional panel at the extreme right presents an anti-Flag WB membrane. The blue arrows in all the panels represent the protein of interest in each WB membrane (Myc-AIP on the left panels, and HA-tagged proteins on the right panels, plus Flag-PRKACA in experiment i). The arrowheads in all the panels point out the heavy (top) and light (bottom) chains of mouse immunoglobulins. IP: immunoprecipitation, MWM: molecular weight marker. The immunocytofluorescence images (G and J) are reconstructions of representative images of the z-stacks obtained, at a 63× magnification. Top left: nuclei (DAPI), top right: HSPA9 (G) or PRKACA (J), bottom left: AIP and bottom right: merged image. Inserts in the merged images present 2-D intensity histograms corresponding to the co-localization calculations.
The pituitary-specific function of AIP could occur via chaperone client proteins
Some pituitary-specific effects of AIP could be exerted via indirect interactions with client proteins of molecular chaperones. Therefore, a possible indirect interaction of AIP with the cyclic adenosine monophosphate (cAMP)-dependent protein kinase catalytic subunit alpha (PRKACA), an HSP90 client protein with an important role in the somatotroph cell function, was explored. AIP and PRKACA did not interact directly (Figure 2H), but when a triple co-IP experiment with AIP, PRKACA and HSP90AA1 was performed, the three proteins were successfully co-immunoprecipitated (Figure 2H–2I), and cytoplasmic co-localization of AIP and PRKACA was observed (Figure 2J).
AIP is involved in cytoskeletal organization
Multiple cytoskeletal proteins were identified in the pull-down experiments. Although these proteins are abundantly expressed in every cell type and could therefore represent experimental artifacts, some of them displayed differential binding between WT and mutant AIP proteins, suggesting the relevance of these potential interactions. Beta-actin (ACTB) was underrepresented in the pull-down experiments for the AIP variants p.R304Q and p.R304* (normalized intensity values of 0.3 and 0.6, compared with the WT protein). Validation of this interaction was attempted but, although the co-IP reactions were positive in both directions, bands for the co-immunoprecipitated proteins were also observed in the mouse IgG negative control, indicating a non-specific binding of ACTB to this immunoglobulin (Figure 3A). The co-IP experiment between cytoskeletal protein neurofilament light polypeptide (NEFL) and AIP failed to prove the interaction (Figure 3B). In the HEK293 cells, ACTB and AIP displayed different distribution patterns, although some small areas of perinuclear co-localization were observed (Figure 3C), while areas of co-localization for AIP and NEFL were observed in only a few cells (Figure 3D). Interactions of AIP with ACTB and NEFL cannot be discarded, but the experimental artifacts and/or the lack of consistency between co-IP and co-localization complicate data interpretation.
Figure 3: Interactions of AIP with cytoskeletal proteins, the cytoskeletal organizer and tumor suppressor NME1, and the enzyme SOD1. (A) A co-IP experiment for AIP and ACTB showed inconclusive results, as the protein was precipitated by the IgG negative control. (B) Co-IP for AIP and NEFL rendered negative results. (C) ACTB and AIP displayed different distribution patterns in the cell and co-localized only in small perinuclear areas, for a Pearson’s R-value of 0.17. (D) Likewise, only small areas of co-localization in a few cells were identified for AIP and NEFL, for a Pearson’s R-value of 0.38. Positive co-IP experiments were obtained for TUBB (E) and TUBB2A (F), although the band representing HA-TUBB when immunoprecipitating with an anti-Myc antibody was weak. (G) AIP and TUBB2A clearly co-localize in the cytoplasm, particularly in the perinuclear area (Pearson’s R-value of 0.54). (H) Positive co-IP of AIP and NME1 was observed, although only when the proteins were detected using the anti-Myc antibody. (I) Likewise, co-IP of AIP and SOD1 was observed only when detection was performed using the anti-Myc antibody, but not when using the anti-HA antibody. For the co-IP experiments a, b, e, f and h, the left panels represent anti-Myc WB membranes and the right panels, anti-HA WB membranes. The blue arrows in all the panels represent the protein of interest in each WB membrane (Myc-AIP on the left panels, and HA-tagged proteins on the right panels). The arrowheads in all the panels point out the heavy (top) and light (bottom) chains of mouse immunoglobulins. IP: immunoprecipitation, MWM: molecular weight marker. The immunocytofluorescence images (c, d and g) are reconstructions of representative images of the z-stacks obtained, at a 63× magnification. Top left: nuclei (DAPI), top right: ACTB (c), NEFL (d), or TUBB2A (g); bottom left: AIP and bottom right: merged image. Inserts in the merged images present 2-D intensity histograms corresponding to the co-localization calculations.
Two isotypes of beta tubulins were under-represented in the pull-down experiment for the mutant p.R304*, compared with WT AIP: tubulin beta chain (TUBB) and tubulin beta-2A chain (TUBB2A, normalized intensity values of 0.2 and 0.3, respectively). Positive co-IP experiments were obtained for TUBB and TUBB2A proteins with AIP, although the band corresponding to TUBB in the anti-Myc immunoprecipitation was weak (Figure 3E–3F). In the confocal immunocytofluorescence images, TUBB2A appeared distributed in cytoplasmic filaments, while AIP had predominantly cytoplasmic diffuse distribution, with clear overlap of both proteins in the cytoplasmic microtubule network (Figure 3G). In addition, a novel interaction between AIP and the cytoskeletal organizer/tumor suppressor NME/NM23 nucleoside diphosphate kinase 1 (NME1) was identified by the pull-down experiment, and binding was reduced for AIP p.A299V (normalized intensity value: 0.2) and lost for the variants p.R304* and p.R304Q. This interaction was validated by co-IP, although only when the anti-Myc antibody was used for protein detection (Figure 3H).
Interaction of AIP with SOD1
A novel interaction between AIP and the mitochondrial protein superoxide dismutase [Cu-Zn] (SOD1), absent for the variants p.R304* and p.R340Q, was confirmed by co-IP, although a band for SOD1 was not observed in the anti-Myc IP when attempting detection with an anti-HA antibody (Figure 3H), suggesting a weak interaction.
Negative validation experiments
Negative co-IP experiments were obtained for the candidate AIP partners vesicle-associated membrane protein-associated protein A (VAPA) and carbonyl reductase [NADPH] 1 (CBR1), while inconclusive results (due to immunoprecipitation of the protein of interest with the negative control IgG) were observed for elongation factor 1-gamma (EEF1G). CBR1 binds glutathione and EEF1G contains a GST domain [28]. Therefore, it is likely that these two proteins were pulled down due to the use of a GST/glutathione method and not due to specific AIP binding.
Differential expression of AIP interacting partners in AIPmut positive somatotropinomas
We performed microarray gene expression analysis on samples from AIPmut positive and negative human pituitary adenomas and normal pituitaries. We aimed to determine whether the identified AIP interacting partners played a role in pituitary tumorigenesis, independently of AIPmuts, as well as to determine how the expression profile could be affected by AIP deficiency. Comparison of the gene expression profiles of the AIP candidate partners identified in the pull-down experiments revealed that members of the beta tubulin family were differentially expressed. In particular, TUBB2B was found significantly downregulated in AIPmut positive (-8.3-fold change, P = 0.0043) and AIPmut negative (-5.2-fold change, P = 0.0464) somatotropinomas, compared with the normal pituitaries. Likewise, TUBB6B (tubulin beta 6 class V) was significantly downregulated in AIPmut negative non-functioning pituitary adenomas (NFPAs, -8.4-fold change, P = 0.0057 vs. normal pituitary). By contrast, TUBB1 (tubulin beta 1 class VI) was upregulated in AIPmut positive somatotropinomas (5.16-fold change, P = 0.0078 vs. normal pituitary). In addition, upregulation of NME1 (2.16-fold change, P = 0.03) and HSPA9 (2.37-fold change, P = 0.05) was observed in AIPmut positive somatotropinomas, compared with normal pituitary. There were no significant differences in these results in AIPmut positive compared with AIPmut negative pituitary adenomas. Expression of the rest of the AIP partners identified by pull-down was not statistically significantly differentially expressed in AIPmut positive, AIPmut negative or sporadic somatotropinomas compared with the normal pituitaries. Our full microarray datasets have been deposited at the National Center for Biotechnology Information’s Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo, accession number GSE63357).
AIP overexpression does not affect AIP interacting partners at the transcriptional level
In contrast to the findings of the microarray analysis, overexpression of WT AIP in HEK293 and GH3 cells (Supplementary Figure 1A and 1B, respectively) displayed no effect on the expression of the interacting partners HSPA5, HSPA8, HSPA9, HSP90AA1, HSPA90AB1, NME1, SOD1, TUBB and TUBB2A at the RNA level at 24 hours post-transfection.
Overexpressed AIP interacting partners have no effect on GH3 cell proliferation
Individual overexpression of the AIP interacting partners HSPA5, HSPA8, HSPA9, HSP90AA1, HSPA90AB1, NME1, SOD1, TUBB and TUBB2A had no effect on the proliferation of GH3 cells at 24 and 48 h compared with the empty vector control (Supplementary Figure 1C). We also compared the results at the two time points within each experimental condition, finding that overexpression of TUBB2A resulted in reduced cell count at 48 h; this effect was not observed for the rest of the genes studied.
DISCUSSION
We have explored the interactions of AIP with other proteins in pituitary cells, and identified six novel molecular partners for this co-chaperone: HSPA5, HSPA9, TUBB, TUBB2A, NME1 and SOD1. Thus, our results expand the list of possible signaling pathways implicated in the pituitary-specific tumor suppressior function of AIP. Deregulation of the cyclic 3′-5′-adenosine monophosphate/protein kinase A pathway (cAMP pathway) is a well-known initiator of somatotroph cell tumorigenesis [29]. It has recently been suggested that the anti-tumorigenic function of AIP occurs through a regulatory effect on such pathway, mediated by G-inhibitory proteins (Giα2/Giα3), very likely coupled to somatostatin receptors (SSTRs) [2, 3], and probably also via phosphodiesterase function regulation [30]. However, while pituitary adenomas/hyperplasia caused by genetic alterations in components of the cAMP pathway (such as GNAS in the McCune-Albright syndrome and PRKAR1A in Carney complex) share similar molecular, histopathological and clinical characteristics, AIPmut-related pituitary adenomas display distinctive features that point to the involvement of additional independent signaling pathways [1, 6, 31].
We have demonstrated interactions between AIP and four different heat shock proteins: two previously known, HSP90 and HSPA8, and two novel molecular partners, HSPA5 and HSPA9. The latter ones have specific subcellular distributions, in sharp contrast with the wide cytoplasmic and nuclear distribution of HSPA8 and HSP90. HSPA5 is a molecular chaperone resident in the endoplasmic reticulum involved in protein quality control and in the unfolded protein response [32]. This cellular response triggers the degradation of unfolded proteins and eliminates cells subjected to prolonged stress but, if sustained, it may elicit the selective proliferation of transformed cells with anti-apoptotic and angiogenic abilities [32–36]. HSPA5 overexpression in cancer is associated with poor prognosis and drug resistance, and represents a novel therapeutic target [37–39]. No previous data exist on the role of HSPA5 on pituitary function, but in a rat gliosarcoma cell line HSPA5 expression is induced by cAMP-responsive protein kinases [40]. If such kinases stimulate HSPA5 expression also in the pituitary gland, this chaperone could amplify the pro-tumorigenic effect of the pathway and the interaction with AIP could regulate its subcellular localization.
Originally identified as a mitochondrial chaperone, HSPA9 is also present in other subcellular localizations, particularly in neoplastic tissues [41–43]. This heat-shock protein is involved in the translocation of proteins from the cytosol into the mitochondrial matrix, in the folding of such imported proteins and in the response to oxidative stress [44, 45]. Deletion of the HSPA9 locus (5q31.1) is a frequent finding in myeloid malignancies [46]. In other settings, HSPA9 exhibits oncogenic properties, making it a potential therapeutic target for cancer. For instance, overexpressed HSPA9 can sequester TP53 in the cytoplasm [43, 47], inhibit CDKN1A [48, 49], and activate hTERT [50]. Besides the multiple studies addressing the HSPA9 functions in normal and neoplastic cells, little is known about the co-chaperones that regulate its subcellular localization and function, and its expression pattern in the pituitary gland has not been assessed. AIP is a direct partner for TOMM20, located on the mitochondrial surface, and thus the novel interaction with HSPA9 expands the regulatory role of AIP in two different steps of the mitochondrial protein-import machinery [51].
The highly conserved and abundant chaperone HSP90 regulates a wide variety of cellular pathways via interactions with multiple client proteins [52, 53]. When overexpressed in ACTH-secreting pituitary adenomas, HSP90 impairs the sensitivity of the glucocorticoid receptor and therefore adrenal-pituitary negative feedback [54]. However, the role of HSP90 in AIPmut-associated pituitary adenomas has not been explored. It has previously been shown that the highly conserved AIP residues G272 and K266 are required for the two proteins to interact [55, 56], but under our experimental conditions, AIP p.K266A did not impair the binding, while mutations affecting the C-terminal alpha-helix of the protein (p.R304* and p.R304Q) did so. Although these variants do not affect the HSP90 binding site directly, an abnormal C-terminal alpha-helix could possibly alter the folding of the third TPR motif, affecting the three-dimensional structure of the TPR-binding site and thus the interaction with HSP90. Previously reported co-IP experiments [57], and now our data, indicate that, despite the fact that the PPIase domain of AIP can also bind HSP90 [58], the integrity of not only the TPR motifs but also of the C-terminal alpha-helix is crucial for this interaction to occur.
Via interactions with multiple co-chaperones, HSPA8 coordinates the dynamics of clathrin-coated vesicles, influences the cell cycle through cyclin D1 and is implicated in protein translocation and quality control [59, 60]. In the absence of the aryl hydrocarbon receptor (AHR), AIP preferentially binds HSPA8 instead of HSP90 and contributes to the prevention of the aggregation of cytosolic proteins, such as mitochondrial pre-proteins synthesized in the cytoplasm, which are then transferred to TOMM20, enabling their folding and mitochondrial translocation [20–22]. HSPA8 was the most consistently represented AIP partner among pull-down experiments, and it was strikingly affected by AIP p.R304*. The physiological importance of this direct interaction between HSPA8 and AIP in the pituitary gland remains undetermined.
The TPR domain of AIP displays binding sites for the MEEVD conserved sequence of HSP90, for the C-terminal motif IEEVD of HSPA8, and EDDVE of TOMM20 [11]. Interestingly, neither HSPA5, nor HSPA9 contain such conserved motifs, and none of the AIP variants studied disturbed these interactions. Given the prominent role of these chaperones in protein folding, a role for AIP as a mediator of the protein quality control system in the pituitary gland could be expected. Both HSP90 and HSPA8 are ubiquitously expressed, but their interacting partners include proteins with tissue-specific functions and/or expression patterns. The interaction between HSP90 and PRKACA was reported previously as part of a high throughput proteomic study, but it was not further functionally or structurally characterized [61]. Our data here support this interaction. The HSP90/AIP complex could feasibly regulate PKA localization, and it could possibly also interact with the regulatory subunits of PKA, as it has recently been proposed [62].
The second group of AIP partners identified is composed of cytoskeletal proteins. While very weak or negative binding of the PPIase-like domain of AIP to dynein was found previously [63], a different study demonstrated that the AIP-mediated cytoplasmic localization of the AHR requires the anchoring of the complex to actin filaments [64]. The latter study showed direct binding of AIP to ACTB when AHR is inactive, but a different group reported conflicting results regarding such an interaction [65]. Unfortunately, we cannot shed light on this issue, due to non-specific binding of ACTB to IgG, although perinuclear co-localization was seen on immunostaining. Likewise, the interaction of AIP with NEFL could not be clearly validated or ruled out (negative co-IP, despite positive co-localization in some cells).
Composed of heterodimers of various alpha and beta-tubulins, microtubules are structures involved in cell movement, intracellular transport (including vesicle trafficking) and cell division and are targets of anti-mitotic drugs [66–69]. In the pituitary gland, the cytoskeletal network plays a crucial role in regulating the function of signal transduction. It has been suggested that low levels of filamin A, a cytoskeletal-associated protein, could represent a post-receptor mechanism of pharmacological resistance in pituitary adenomas, as this scaffolding protein stabilizes the SSTR2 and dopamine 2 receptor, linking them to their intracellular effectors [69, 70]. We validated direct interactions of AIP with two isotypes of tubulins: TUBB, widely expressed, and TUBB2A, the major isotype in neural tissue [67]. Interestingly, two isotypes of beta tubulin were also deregulated (TUBB1 with five-fold upregulation and TUBB2B with eight-fold downregulation) in AIP-mutated somatotroph tumors, compared with normal pituitaries. These results point towards a role for AIP as a regulator of the microtubule network, with possible implications for post-receptor signal transduction and hormone secretion.
NME1 (granzyme A-activated DNase, metastasis inhibition factor nm23), the first metastasis suppressor gene identified [71–73], is a tumor suppressor that negatively regulates cell migration/motility and inhibits the cell cycle through downregulation of cyclin B, an effect inhibited by cAMP [74]. This protein phosphorylates kinase suppressor of Ras 1 (KSR1), thereby inactivating the RAS/RAF/MEK/ERK signaling pathway [75]. NME1 mRNA levels are high in cell lines with low metastatic potential [71], while LOH involving the NME1 locus (17q21.3) has the opposite effect [76]. Low NME1 expression correlates with metastasis and poor clinical prognosis in different human epithelial cancer types [77]. Moreover, NME1 knockdown in various human cancer cell lines disrupts E-cadherin-mediated cell adhesion, leading to nuclear translocation of beta-catenin [78], while its overexpression inhibits the metastatic potential of TP53-deficient cells [73]. NME1 co-localizes with E-cadherin in epithelial cancer cell lines, suggesting a possible role on the stabilization of the adherens junctions [78]. In pituitary adenomas, NME1 expression inversely correlates with tumor extension into the cavernous sinus [79], but there are no data available on its expression pattern in different pituitary adenoma subtypes. Since AIPmut positive pituitary adenomas are frequently invasive, the interaction with AIP should have a regulatory effect on this kinase [80].
Interestingly, “Remodeling of the epithelial adherens junction” was identified as the top molecular pathway encompassing our candidate AIP interacting partners. Loss of the integrity of epithelial adherens junctions results in epithelial-mesenchymal transition (EMT), a process by which polarized epithelial cells develop increased migratory capacity, invasiveness, resistance to apoptosis and increased production of extracellular matrix, ultimately acquiring a mesenchymal cell phenotype [81]. Loss of expression of E-cadherin, the main constituent of the adherens junctions, is a hallmark of EMT. In somatotropinomas, E-cadherin expression correlates positively with GH and IGF-1 secretion and and with somatostatin analogue treatment, and negatively with tumor size and invasiveness [82]. Interestingly, the characteristics of somatotropinomas with low E-cadherin expression recapitulate the phenotype described in the AIPmut positive setting (unpublished data from our group). A function of AIP as a regulator of the adherens junctions in the somatotroph cells, suggested by our results, would explain why somatotroph cells acquire an EMT phenotype in the setting of AIP deficiency.
Finally, we found that AIP is able to bind the enzyme SOD1, required for the conversion of superoxide to hydrogen peroxide, a mechanism that counteracts oxidative stress [83]. Rat somatotropinoma-derived GH3 cells secrete SOD1, which, via activation of a muscarinic M1 receptor, reduces the activity of the MAPK1 signaling pathway, by inhibiting MAPK3 phosphorylation, reducing cell proliferation [84, 85]. On the other hand, accumulation of free radicals as a result of defective enzyme function can lead to neurotoxicity, as it happens in SOD1 mutation associated familial amyotrophic lateral sclerosis, or to malignant transformation in different tissues [83]. Molecular chaperones such as HSP70 and HSPA8 regulate SOD1 function and prevent its aggregation [86]. We hypothesize that AIP could form part of this complex, therefore regulating the activity of SOD1.
It was not completely unexpected to find that AIP overexpression has no effect on the expression of its interacting partners at the transcriptional level. Besides the fact that AIP does not have a known direct transcriptional effect, this co-chaperone is highly expressed in the somatotroph cells under basal conditions. Therefore, it is unlikely that AIP overexpression should have any acute transcriptional effects, even if indirect. However, the role of AIP overexpression under certain stimuli, for instance, in somatotropinomas treated with somatostatin analogues, might have a role on the posttranscriptional regulation of other proteins. Likewise, overexpression of multiple AIP interacting partners had no effect on the proliferation of GH3 cells. Overexpression of TUBB2A had a small, but significant effect on reducing cell proliferation at 48h compared with the same condition at 24 h. HSPA5, HSPA8, HSPA9, HSP90AA1, HSPA90AB1, NME1, SOD1, TUBB and TUBB2A are highly expressed in the cells and have no known direct oncogenic effects.
We acknowledge the shortcomings of our study. Ideally, the pull-down experiments should had been done in lysates from human pituitary cells, but the lack of commercially available human pituitary cell lines and the difficulty to obtain fresh normal pituitary tissue from autopsy specimens in a quantity enough as to optimize and perform the experiments precluded the use of such material. Although a rat pituitary cell line was used for the pull-down experiments, we overcame this disadvantage by validating our results using human proteins. Finally, concerning the validation experiments, it would be desirable, but extremely impractical, to validate all the protein interactions identified. Interactions of AIP with novel partners have been fully validated, but it is possible that some of the other proteins identified (and not validated) could represent true interactions, providing opportunity for further experiments. While the finding of new chaperones interacting with AIP has shed light on the repertoire of interacting partners of AIP, it has also increased the number of chaperone-client proteins that could be regulated by the co-chaperone activity of AIP. Although mapping all these possible indirect interactions would be complicated, future studies could concentrate on determining which interactions of these chaperones in the somatotroph cells are mediated by AIP.
CONCLUSIONS
Several novel protein partners were identified and validated for AIP in somatotroph cells. Interactions with HSP70 family members HSPA5 and HSPA9 expand the repertoire of heat-shock proteins that could be modulated by AIP, opening a new window for possible anti-tumorigenic functions of AIP as a regulator of stress-induced heat-shock protein functions. AIP also binds SOD1, an anti-oxidative protein with anti-proliferative potential. In addition, novel molecular interactions with the cytoskeletal proteins TUBB and TUBB2A and the cytoskeletal regulator NME1 indicate a possible role for AIP as a regulator of cytoskeletal organization and on the integrity of the adherens junction, which might be a novel mechanism for the complex tumor suppressor function of AIP in the pituitary gland. The study of this mechanism could be the focus of future studies.
METHODS
Protein synthesis and pull-down assays
The human AIP variants listed in Table 1 were obtained by site-directed mutagenesis (QuikChange II XL kit, Agilent Technologies, Santa Clara, CA, USA, 200521) from the p-THREE-E-AIP_WT plasmid [7]. Under IPTG induction, WT and mutant GST-AIP proteins were produced in BL21-PLyss E. coli and subjected to affinity (Glutathione Sepharose 4 Fast Flow, GE Healthcare, Little Chalfont, UK, 17-5132-02) and gel filtration chromatography. GST was obtained likewise and used as a negative control. Ten micrograms of each synthetic protein were used as baits for individual GST pull-down experiments against 2 mg of total protein from GH3 cells (ECACC, Porton Down, UK, 87012603), and eluates from four independent experiments for each bait protein were pooled together for MS analysis, as reported before [7].
Quantitative MS and identification of candidate AIP partners
Tandem mass tagging (TMTsixplex Label Reagent Set, Thermo Fisher Scientific, Waltham, MA, USA, 90061) [87], and ion trap tandem MS (Orbitrap Velos Pro, Thermo Fisher Scientific) of the pull-down eluates were carried out at the King’s College London Denmark Hill Proteomics Facility, according to their standard procedures. Data were processed using the Proteome Discoverer version 1.3.0.339 (Thermo Fisher Scientific) software, and the sequences identified were searched in the UniProt database [12] via the Mascot v.2.2 platform [88]. Results were qualitatively analyzed and the MS fragmentation spectra were manually validated using the Scaffold 3.6 (Proteome Software) software. Only peptides with Mascot score ≥20 (to filter out non-significant results) and valid MS spectrum were considered for further analysis. The intensity value of each peptide in the GST experiment was subtracted from the value obtained for the same peptide in the rest of the experiments, and the normalized value for each of the mutants was divided by the normalized value for the WT experiment. Differential binding results are presented as fold-change, considering the results for the WT experiment in each case as 1. Candidate partners were selected based on the Mascot score, the number of unique peptides identified per protein (≥3 peptides was interpreted as highly probable candidates) and the differential intensity values among experiments. The human homologues of the candidate peptides selected were identified using the Blast tool in the UniProt database; the best match was selected in each case. The Ingenuity Pathways Analysis platform [89] was used for grouping the identified proteins in signaling pathways. Results for proteins involved in the ubiquitin-proteasome pathway have been reported elsewhere [7].
Validation of protein-protein interactions
Plasmids containing the coding sequence for HSC70, HSP90AA1 and HSP90AB1 were a kind gift from Prof. Paul Chapple (Barts and The London School of Medicine). The coding sequence for SOD1 was cloned from human cDNA from HEK293 (human embryo kidney, ECACC 85120602) cells. For the rest of the candidate partners, plasmids were obtained from a repository (PlasmID DNA Resource Core, Harvard Medical School). All the sequences were sub-cloned into the pSF-CMV-NH2-HA-EKT-NcoI plasmid (Oxford Genetics, Oxford, UK, OG93) to express N-terminally HA-tagged proteins. The plasmids pcDNA3.0-Myc-WT_AIP and pcDNA3.0-Myc-AIP-R304* were used to express N-terminally Myc-tagged WT and p.R304* AIP, respectively [90], and the plasmid pSF-CMV-NH2-HA-EKT-Nco1-PRKACA was subjected to site-directed mutagenesis to obtain an N-terminal Flag tag. The final constructs are detailed in Supplementary Table 3.
Each of the plasmids was co-transfected with pcDNA3.0-Myc-WT_AIP plasmid in 10 × 106 HEK293 cells for a total of 20 μg of plasmid DNA, with 1 μl of Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA, 11668027) per μg of DNA, according to the manufacturer’s instructions. Cells were harvested one day later by trypsinization and resuspended in 1.5 ml of lysis buffer, composed of 150 mM NaCl, 10 mM Tris-Cl pH 7.5, 10% v/v glycerol, 1% v/v IGEPAL CA-630 (Sigma-Aldrich, St. Louis, MO, USA, I8896) and 1 tablet per 50 ml Complete Protease Inhibitor Cocktail (Roche, Basel, Switzerland, 11836145001). After cleared by centrifugation, lysates were cleaned up by incubation with 50 μl of Protein G Sepharose 4 Fast Flow (GE Healthcare 17-0618-01), and divided in thirds for incubation with 5 μg of anti-Myc (Sigma-Aldrich M4439), or anti-HA mouse (Sigma-Aldrich H3663) monoclonal antibodies or mouse anti-IgG (Sigma-Aldrich I5381), as appropriate. Co-IP was performed as previously described [7], and the eluates were resolved by denaturing polyacrylamide gel electrophoresis and mouse anti-Myc and rabbit anti-HA (Sigma-Aldrich H6908) Western blot. A mouse monoclonal anti-ACTB antibody (Sigma-Aldrich A1978) was used for ACTB immunoprecipitation. Detection was performed in an Odyssey Infrared Imaging System (LI-COR, Lincoln, NE, USA) after incubation with secondary infrared fluorescent antibodies (LI-COR 926-68180 and 926-32211). All the co-IP experiments were performed at least twice for confirmation.
Co-localization
For co-localization experiments, 5 × 104 HEK293 cells per well were plated on an 8-well chamber slide (Thermo Fisher Scientific 154534) and grown for 48 h. Cells were pre-fixed for 2 min with 4% formaldehyde in PBS added to the medium, fixed for 10 more minutes after medium removal, washed thrice with PBS, permeabilized for 20 min at room temperature (0.1% triton X-100 in PBS), blocked for 1 h (10% normal goat serum [VECTOR Laboratories, Burlingame, CA, USA, S-1000] in permeabilization buffer) and incubated overnight with primary antibodies against AIP (mouse monoclonal 1:500 v/v [Novus, Littleton CO, USA, NB100-127], or rabbit polyclonal 1:100 v/v [NBP1-31347], as appropriate) and one of the following: rabbit polyclonal anti-PRKACA (Proteintech, Rosemont, IL, USA, 5388-1-AP, 1:50 v/v), rabbit monoclonal anti-GRP75 (Cell Signalling Technology, Danvers, MA, USA, 3593, 1:50 v/v), rabbit monoclonal anti-NEFL (Cell Signalling Technology 2837, 1:100 v/v) or mouse monoclonal anti-TUBB2A (Abnova, Taipei, Taiwan, H00007280-M03, 1:100 v/v). For actin staining, ActinGreen488 anti-ACTB probe (Invitrogen R37110) was used, following the manufacturer’s protocol, after incubation with the primary anti-AIP antibody. The cells were washed, incubated with 1:500 v/v fluorescent secondary antibodies (Alexa Fluor 488 Goat Anti-Mouse IgG [H+L] green and Alexa Fluor 568 Goat Anti-Rabbit IgG [H+L] orange, Invitrogen A-11029 and A-11036, respectively), washed again, and mounted with 4’,6-diamidino-2-phenylindole-containing mounting medium (UltraCruz Hard-set Mounting Medium, Santa Cruz Biotechnology, Dallas, TX, USA, sc-359850) for confocal microscopy analysis in an LSM 510 (Mark 4) Laser Scanning Confocal Microscope (Zeiss, Oberkochen, Germany). Z-stack images were obtained and visualized using the ImageJ version 2.0.0-rc-54/1.51g software [91]. Co-localization was analyzed on representative images with the Coloc 2 plugin, using the Costes threshold regression method and using the Pearson’s correlation coefficient to quantify co-localization [92–94].
Microarray expression analysis
Total RNA was extracted from five normal pituitaries (obtained from autopsies) as well as from twenty freshly frozen pituitary adenoma samples, including six somatotropinomas from patients carrying germline AIP variants (c.910C>T, p.R304* in three cases, and c.911G>A, p.R340Q, c.100–1025_279_357del, p.A34_K93del and c.100–18C>T in one patient each), seven AIPmut negative somatotropinomas and seven clinically NFPAs, using the RNeasy plus mini kit (Qiagen, Hilden, Germany 74134). Patients were recruited as part of our cohort of pituitary adenoma patients and provided signed informed consent [6]. Global gene expression analysis was performed using Human Gene Chip HG-U133 Plus 2.0 arrays (Affymetrix, Santa Clara, CA, USA). A double cut-off of false discovery rate <0.05 and fold change of ≥2 was used to identify differentially expressed genes. The Ingenuity Pathway Analysis platform was used to analyze differentially expressed genes, and results were validated by RT-qPCR. Results were analyzed separately for each subgroup of samples and gene expression profiles were compared to seek differences between AIPmut positive and negative somatotropinomas, between each tumor type and the normal pituitary or between functioning tumors (somatotropinomas) and NFPAs. Data are presented only for genes with significant differential expression corresponding to protein families that were present also in the pull-down assays.
Expression of AIP interacting partners under AIP overexpression
For AIP overexpression experiments, 2.5 × 105 HEK 293 or GH3 cells per well were plated in 12-well plates and transfected 24h later with 1μg of either pcDNA3.1(-) (empty vector), pcDNA3.0-Myc-WT_AIP, or an equivalent volume of water, and 2 μl of TurboFect (Thermo Fisher Scientific R0531) per well, according to the manufacturer’s instructions, in triplicate; each experiment was repeated at least twice for confirmation. RNA was extracted 24 hours later, as described before, and 500 ng of total RNA were reverse-transcribed using the SuperScriptIII First-Strand Synthesis Super Mix (Thermo Fisher Scientific 11752250), following the manufacturer’s protocol. Quantitative PCR was performed in a final volume of 10 μl, with 5 ng of cDNA per reaction, in triplicate, using the TaqMan Fast Advanced Master Mix (Thermo Fisher Scientific 4444557). The FAM-MGB labelled TaqMan expression assays Rn00565250_m1 (Hspa5), Rn00821191_g1 (Hspa8), Rn01402372_g1 (Hspa9), Rn00822023_g1 (Hsp90aa1), Rn01511686_g1 (Hsp90ab1), Rn00821755_g1 (Nme1), Rn00566938_m1 (Sod1), Rn00597407_m1 (Tubb5), and Rn01774446_m1 (Tubb2a), were used for experiments in GH cells, and Hs99999174_m1 (HSPA5), Hs03044880_gH (HSPA8), Hs00945584_m1 (HSPA9), Hs00743767_sH HSP90AA1), Hs00607336_gH (HSP90AB1), Hs00533490_m1 (SOD1), Hs02621161_s1 (NME1), Hs00742828_s1 (TUBB), Hs00742533_s1 (TUBB2A), were used for HEK293 cells; Hs066610222_m1 (AIP) was used for both sets of experiments. The VIC-TAMRA labelled endogenous controls Rn00667869_m1 (Actb) and 4325788 (ACTB) were used for GH3 a and HEK293 cells, respectively. Reactions were carried out using the ViiA 7 Real-Time PCR System (Thermo Fisher Scientific) and results were analyzed via the 2-DDCT method, normalized against empty vector control, and compared among experimental conditions.
Cell proliferation assays under overexpression of AIP interacting partners
For cell proliferation experiments, 2.5 × 104 GH3 cells per well were plated in 96-well plates and transfected 24 h later with 200 ng of HA-tagged plasmids to overexpress AIP interacting partners (Supplementary Table 3), empty vector or water, and 4 μl of Turbo Fect per well, in triplicate. Cell proliferation assays were performed at 24 and 48 h after transfection, using the CyQUANT Direct Cell Proliferation Assay (Thermo Fisher Scientific C35011), according to the manufacturer’s instructions. Fluorescence was detected using a FLUOstar Omega plate reader (BMG Labtech, Offenburg, Germany), normalized against blanks (cell culture medium only) and then against empty vector control, and compared among experimental conditions. Experiments were performed at least twice, for confirmation.
Abbreviations
AIP: aryl hydrocarbon receptor interacting protein; AIP mutation: AIPmut; C: carboxyl; cAMP: cyclic 3′-5′-adenosine monophosphate; co-IP: co-immunoprecipitation; FIPA: familial isolated pituitary adenoma; GST: glutathione-S-transferase; IP: immunoprecipitation; MS: mass spectrometry; MWM: molecular weight marker; N: amino; NFPAs: non-functioning pituitary adenomas; PPIase: peptidyl-prolyl cis-trans isomerase; SSTRs: somatostatin receptors; TPR: tetratricopeptide; WB: western blot; WT: wild-type.
Author contributions
Laura C. Hernández-Ramírez: synthesized and purified proteins, performed pull-down, mass spectrometry, co-immunoprecipitation and co-localization experiments, wrote the manuscript.
Rhodri M. L. Morgan: generated and produced expression plasmids, synthesized and purified proteins, wrote the manuscript.
Sayka Barry: performed global expression experiments and data analysis, wrote the manuscript.
Fulvio D’Acquisto: designed and supervised pull-down experiments and filtering of candidate AIP partners, wrote the manuscript.
Chrisostomos Prodromou: designed and supervised protein synthesis and chromatography, wrote the manuscript.
Márta Korbonits: designed and supervised the project, collected pituitary adenoma samples, wrote the manuscript.
CONFLICTS OF INTEREST
The authors have nothing to disclose.
FUNDING
L.C.H.R. was supported by grants from the National Council of Science and Technology and the Secretariat of Public Education from the Mexican Government. C.P. is funded by the Wellcome Trust grant 095605/Z11/Z. M.K. received grants from the Medical Research Council of the United Kingdom, Wellcome Trust, National Institute of Health Research, Barts and The London Charity, Royal Society, and Pfizer.
REFERENCES
1. Chahal HS, Trivellin G, Leontiou CA, Alband N, Fowkes RC, Tahir A, Igreja SC, Chapple JP, Jordan S, Lupp A, Schulz S, Ansorge O, Karavitaki N, et al. Somatostatin analogs modulate AIP in somatotroph adenomas: the role of the ZAC1 pathway. J Clin Endocrinol Metab. 2012; 97:E1411–20. https://doi.org/10.1210/jc.2012-1111.
2. Formosa R, Xuereb-Anastasi A, Vassallo J. Aip regulates cAMP signalling and GH secretion in GH3 cells. Endocr Relat Cancer. 2013; 20:495–505. https://doi.org/10.1530/ERC-13-0043.
3. Tuominen I, Heliovaara E, Raitila A, Rautiainen MR, Mehine M, Katainen R, Donner I, Aittomaki V, Lehtonen HJ, Ahlsten M, Kivipelto L, Schalin-Jantti C, Arola J, et al. AIP inactivation leads to pituitary tumorigenesis through defective Galpha-cAMP signaling. Oncogene. 2015; 34:1174–84. https://doi.org/10.1038/onc.2014.50.
4. Vierimaa O, Georgitsi M, Lehtonen R, Vahteristo P, Kokko A, Raitila A, Tuppurainen K, Ebeling TM, Salmela PI, Paschke R, Gundogdu S, De Menis E, Makinen MJ, et al. Pituitary adenoma predisposition caused by germline mutations in the AIP gene. Science. 2006; 312:1228–30. https://doi.org/10.1126/science.1126100.
5. Daly AF, Tichomirowa MA, Petrossians P, Heliovaara E, Jaffrain-Rea ML, Barlier A, Naves LA, Ebeling T, Karhu A, Raappana A, Cazabat L, De Menis E, Montanana CF, et al. Clinical characteristics and therapeutic responses in patients with germ-line AIP mutations and pituitary adenomas: an international collaborative study. J Clin Endocrinol Metab. 2010; 95:E373–83. https://doi.org/10.1210/jc.2009-2556.
6. Hernández-Ramírez LC, Gabrovska P, Dénes J, Stals K, Trivellin G, Tilley D, Ferraù F, Evanson J, Ellard S, Grossman AB, Roncaroli F, Gadelha MR, Korbonits M, et al, and International FIPA Consortium. Landscape of familial isolated and young-onset pituitary adenomas: prospective diagnosis in AIP mutation carriers. J Clin Endocrinol Metab. 2015; 100:E1242–54. https://doi.org/10.1210/jc.2015-1869.
7. Hernández-Ramírez LC, Martucci F, Morgan RM, Trivellin G, Tilley D, Ramos-Guajardo N, Iacovazzo D, D’Acquisto F, Prodromou C, Korbonits M. Rapid proteasomal degradation of mutant proteins is the primary mechanism leading to tumorigenesis in patients with missense AIP mutations. J Clin Endocrinol Metab. 2016; 101:3144–54. https://doi.org/10.1210/jc.2016-1307.
8. Ma Q, Whitlock JP Jr. A novel cytoplasmic protein that interacts with the Ah receptor, contains tetratricopeptide repeat motifs, and augments the transcriptional response to 2,3,7,8-tetrachlorodibenzo-p-dioxin. J Biol Chem. 1997; 272:8878–84. https://doi.org/10.1074/jbc.272.14.8878.
9. Carver LA, LaPres JJ, Jain S, Dunham EE, Bradfield CA. Characterization of the Ah receptor-associated protein, ARA9. J Biol Chem. 1998; 273:33580–7. https://doi.org/10.1074/jbc.273.50.33580.
10. Linnert M, Haupt K, Lin YJ, Kissing S, Paschke AK, Fischer G, Weiwad M, Lucke C. NMR assignments of the FKBP-type PPIase domain of the human aryl-hydrocarbon receptor-interacting protein (AIP). Biomol NMR Assign. 2012; 6:209–12. https://doi.org/10.1007/s12104-012-9359-0.
11. Morgan RM, Hernández-Ramírez LC, Trivellin G, Zhou L, Roe SM, Korbonits M, Prodromou C. Structure of the TPR domain of AIP: Lack of client protein interaction with the C-terminal alpha-7 helix of the TPR domain of AIP is sufficient for pituitary adenoma predisposition. PLoS One. 2012; 7:e53339. https://doi.org/10.1371/journal.pone.0053339.
12. The Uniprot Consortium. UniProt: a hub for protein information (http://www.uniprot.org/). Nucleic Acids Res. 2015; 43:D204–D12. https://doi.org/10.1093/nar/gku989.
13. Perdew GH. Association of the Ah receptor with the 90-kDa heat shock protein. J Biol Chem. 1988; 263:13802–5.
14. Chen HS, Perdew GH. Subunit composition of the heteromeric cytosolic aryl hydrocarbon receptor complex. J Biol Chem. 1994; 269:27554–8.
15. Nair SC, Toran EJ, Rimerman RA, Hjermstad S, Smithgall TE, Smith DF. A pathway of multi-chaperone interactions common to diverse regulatory proteins: estrogen receptor, Fes tyrosine kinase, heat shock transcription factor Hsf1, and the aryl hydrocarbon receptor. Cell Stress Chaperones. 1996; 1:237–50.
16. Carver LA, Bradfield CA. Ligand-dependent interaction of the aryl hydrocarbon receptor with a novel immunophilin homolog in vivo. J Biol Chem. 1997; 272:11452–6. https://doi.org/https://doi.org/10.1074/jbc.272.17.11452.
17. Meyer BK, Pray-Grant MG, Vanden Heuvel JP, Perdew GH. Hepatitis B virus X-associated protein 2 is a subunit of the unliganded aryl hydrocarbon receptor core complex and exhibits transcriptional enhancer activity. Mol Cell Biol. 1998; 18:978–88.
18. Kazlauskas A, Sundstrom S, Poellinger L, Pongratz I. The hsp90 chaperone complex regulates intracellular localization of the dioxin receptor. Mol Cell Biol. 2001; 21:2594–607. https://doi.org/10.1128/MCB.21.7.2594-2607.2001.
19. Ramadoss P, Petrulis JR, Hollingshead BD, Kusnadi A, Perdew GH. Divergent roles of hepatitis B virus X-associated protein 2 (XAP2) in human versus mouse Ah receptor complexes. Biochemistry. 2004; 43:700–9. https://doi.org/10.1021/bi035827v.
20. Yano M, Terada K, Mori M. AIP is a mitochondrial import mediator that binds to both import receptor Tom20 and preproteins. J Cell Biol. 2003; 163:45–56. https://doi.org/10.1083/jcb.200305051.
21. Kang BH, Altieri DC. Regulation of survivin stability by the aryl hydrocarbon receptor-interacting protein. J Biol Chem. 2006; 281:24721–7. https://doi.org/10.1074/jbc.M603175200.
22. Kang BH, Xia F, Pop R, Dohi T, Socolovsky M, Altieri DC. Developmental control of apoptosis by the immunophilin aryl hydrocarbon receptor-interacting protein (AIP) involves mitochondrial import of the survivin protein. J Biol Chem. 2011; 286:16758–67. https://doi.org/10.1074/jbc.M110.210120.
23. Trivellin G, Korbonits M. AIP and its interacting partners. J Endocrinol. 2011; 210:137–55. https://doi.org/10.1530/JOE-11-0054.
24. Kuzhandaivelu N, Cong YS, Inouye C, Yang WM, Seto E. XAP2, a novel hepatitis B virus X-associated protein that inhibits X transactivation. Nucleic Acids Res. 1996; 24:4741–50. https://doi.org/10.1093/nar/24.23.4741.
25. Kashuba E, Kashuba V, Pokrovskaja K, Klein G, Szekely L. Epstein-Barr virus encoded nuclear protein EBNA-3 binds XAP-2, a protein associated with Hepatitis B virus X antigen. Oncogene. 2000; 19:1801–6. https://doi.org/10.1038/sj.onc.1203501.
26. Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, Tyers M. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 2006; 34:D535–D9. https://doi.org/34/suppl_1/D535.
27. Sreedhar AS, Kalmar E, Csermely P, Shen YF. Hsp90 isoforms: functions, expression and clinical importance. FEBS Lett. 2004; 562:11–5. https://doi.org/10.1016/S0014-5793(04)00229-7.
28. Koonin EV, Mushegian AR, Tatusov RL, Altschul SF, Bryant SH, Bork P, Valencia A. Eukaryotic translation elongation factor 1 gamma contains a glutathione transferase domain--study of a diverse, ancient protein superfamily using motif search and structural modeling. Protein Sci. 1994; 3:2045–54. https://doi.org/10.1002/pro.5560031117.
29. Peverelli E, Mantovani G, Lania AG, Spada A. cAMP in the pituitary: an old messenger for multiple signals. J Mol Endocrinol. 2013; 52:R67–77. https://doi.org/10.1530/JME-13-0172.
30. Bolger GB, Bizzi MF, Pinheiro SV, Trivellin G, Smoot L, Accavitti MA, Korbonits M, Ribeiro-Oliveira A Jr. cAMP-specific PDE4 phosphodiesterases and AIP in the pathogenesis of pituitary tumors. Endocr Relat Cancer. 2016; 23:419–31. https://doi.org/10.1530/ERC-15-0205.
31. Jaffrain-Rea ML, Rotondi S, Turchi A, Occhi G, Barlier A, Peverelli E, Rostomyan L, Defilles C, Angelini M, Oliva MA, Ceccato F, Maiorani O, Daly AF, et al. Somatostatin analogues increase AIP expression in somatotropinomas, irrespective of Gsp mutations. Endocr Relat Cancer. 2013; 20:753–66. https://doi.org/10.1530/ERC-12-0322.
32. Ma Y, Hendershot LM. The role of the unfolded protein response in tumour development: friend or foe? Nat Rev Cancer. 2004; 4:966–77. https://doi.org/10.1038/nrc1505.
33. Laitusis AL, Brostrom MA, Brostrom CO. The dynamic role of GRP78/BiP in the coordination of mRNA translation with protein processing. J Biol Chem. 1999; 274:486–93. https://doi.org/10.1074/jbc.274.1.486.
34. Misra UK, Gonzalez-Gronow M, Gawdi G, Pizzo SV. The role of MTJ-1 in cell surface translocation of GRP78, a receptor for alpha 2-macroglobulin-dependent signaling. J Immunol. 2005; 174:2092–7. https://doi.org/10.4049/jimmunol.174.4.2092.
35. Fu Y, Lee AS. Glucose regulated proteins in cancer progression, drug resistance and immunotherapy. Cancer Biol Ther. 2006; 5:741–4. https://doi.org/10.4161/cbt.5.7.2970.
36. Misra UK, Deedwania R, Pizzo SV. Activation and cross-talk between Akt, NF-kappaB, and unfolded protein response signaling in 1-LN prostate cancer cells consequent to ligation of cell surface-associated GRP78. J Biol Chem. 2006; 281:13694–707. https://doi.org/10.1074/jbc.M511694200.
37. Uckun FM, Qazi S, Ozer Z, Garner AL, Pitt J, Ma H, Janda KD. Inducing apoptosis in chemotherapy-resistant B-lineage acute lymphoblastic leukaemia cells by targeting HSPA5, a master regulator of the anti-apoptotic unfolded protein response signalling network. Br J Haematol. 2011; 153:741–52. https://doi.org/10.1111/j.1365-2141.2011.08671.x.
38. Chang YW, Chen HA, Tseng CF, Hong CC, Ma JT, Hung MC, Wu CH, Huang MT, Su JL. De-acetylation and degradation of HSPA5 is critical for E1A metastasis suppression in breast cancer cells. Oncotarget. 2014; 5:10558–70. https://doi.org/10.18632/oncotarget.2510.
39. Booth L, Roberts JL, Tavallai M, Nourbakhsh A, Chuckalovcak J, Carter J, Poklepovic A, Dent P. OSU-03012 and Viagra treatment inhibits the activity of multiple chaperone proteins and disrupts the blood-brain barrier: Implications for anti-cancer therapies. J Cell Physiol. 2015; 230:1982–98. https://doi.org/10.1002/jcp.24977.
40. Chen KD, Chen LY, Huang HL, Lieu CH, Chang YN, Chang MD, Lai YK. Involvement of p38 mitogen-activated protein kinase signaling pathway in the rapid induction of the 78-kDa glucose-regulated protein in 9L rat brain tumor cells. J Biol Chem. 1998; 273:749–55. https://doi.org/10.1074/jbc.273.2.749.
41. Wadhwa R, Yaguchi T, Hasan MK, Taira K, Kaul SC. Mortalin-MPD (mevalonate pyrophosphate decarboxylase) interactions and their role in control of cellular proliferation. Biochem Biophys Res Commun. 2003; 302:735–42. https://doi.org/10.1016/S0006-291X(03)00226-2.
42. Wadhwa R, Takano S, Kaur K, Deocaris CC, Pereira-Smith OM, Reddel RR, Kaul SC. Upregulation of mortalin/mthsp70/Grp75 contributes to human carcinogenesis. Int J Cancer. 2006; 118:2973–80. https://doi.org/10.1002/ijc.21773.
43. Lu WJ, Lee NP, Kaul SC, Lan F, Poon RT, Wadhwa R, Luk JM. Mortalin-p53 interaction in cancer cells is stress dependent and constitutes a selective target for cancer therapy. Cell Death Differ. 2011; 18:1046–56. https://doi.org/10.1038/cdd.2010.177.
44. Kang PJ, Ostermann J, Shilling J, Neupert W, Craig EA, Pfanner N. Requirement for hsp70 in the mitochondrial matrix for translocation and folding of precursor proteins. Nature. 1990; 348:137–43. https://doi.org/10.1038/348137a0.
45. Wadhwa R, Ryu J, Ahn HM, Saxena N, Chaudhary A, Yun CO, Kaul SC. Functional significance of point mutations in stress chaperone mortalin and their relevance to Parkinson disease. J Biol Chem. 2015; 290:8447–56. https://doi.org/10.1074/jbc.M114.627463.
46. Krysiak K, Tibbitts JF, Shao J, Liu T, Ndonwi M, Walter MJ. Reduced levels of Hspa9 attenuate Stat5 activation in mouse B cells. Exp Hematol. 2015; 43:319–30. https://doi.org/10.1016/j.exphem.2014.12.005.
47. Ma Z, Izumi H, Kanai M, Kabuyama Y, Ahn NG, Fukasawa K. Mortalin controls centrosome duplication via modulating centrosomal localization of p53. Oncogene. 2006; 25:5377–90. https://doi.org/10.1038/sj.onc.1209543.
48. Wu PK, Hong SK, Veeranki S, Karkhanis M, Starenki D, Plaza JA, Park JI. A mortalin/HSPA9-mediated switch in tumor-suppressive signaling of Raf/MEK/extracellular signal-regulated kinase. Mol Cell Biol. 2013; 33:4051–67. https://doi.org/10.1128/MCB.00021-13.
49. Starenki D, Hong SK, Lloyd RV, Park JI. Mortalin (GRP75/HSPA9) upregulation promotes survival and proliferation of medullary thyroid carcinoma cells. Oncogene. 2015; 34:4624–34. https://doi.org/10.1038/onc.2014.392.
50. Ryu J, Kaul Z, Yoon AR, Liu Y, Yaguchi T, Na Y, Ahn HM, Gao R, Choi IK, Yun CO, Kaul SC, Wadhwa R. Identification and functional characterization of nuclear mortalin in human carcinogenesis. J Biol Chem. 2014; 289:24832–44. https://doi.org/10.1074/jbc.M114.565929.
51. Baker MJ, Frazier AE, Gulbis JM, Ryan MT. Mitochondrial protein-import machinery: correlating structure with function. Trends Cell Biol. 2007; 17:456–64. https://doi.org/10.1016/j.tcb.2007.07.010.
52. Li J, Richter K, Buchner J. Mixed Hsp90-cochaperone complexes are important for the progression of the reaction cycle. Nat Struct Mol Biol. 2011; 18:61–6. https://doi.org/10.1038/nsmb.1965.
53. Guy NC, García YA, Sivils JC, Galigniana MD, Cox MB. Functions of the Hsp90-binding FKBP immunophilins. Subcell Biochem. 2015; 78:35–68. https://doi.org/10.1007/978-3-319-11731-7_2.
54. Riebold M, Kozany C, Freiburger L, Sattler M, Buchfelder M, Hausch F, Stalla GK, Paez-Pereda M. A C-terminal HSP90 inhibitor restores glucocorticoid sensitivity and relieves a mouse allograft model of Cushing disease. Nat Med. 2015; 21:276–80. https://doi.org/10.1038/nm.3776.
55. Bell DR, Poland A. Binding of aryl hydrocarbon receptor (AhR) to AhR-interacting protein. The role of hsp90. J Biol Chem. 2000; 275:36407–14. https://doi.org/10.1074/jbc.M004236200.
56. Meyer BK, Petrulis JR, Perdew GH. Aryl hydrocarbon (Ah) receptor levels are selectively modulated by hsp90-associated immunophilin homolog XAP2. Cell Stress Chaperones. 2000; 5:243–54. https://doi.org/10.1379/1466-1268(2000)005<0243:AHARLA>2.0.CO;2.
57. Kazlauskas A, Poellinger L, Pongratz I. Two distinct regions of the immunophilin-like protein XAP2 regulate dioxin receptor function and interaction with hsp90. J Biol Chem. 2002; 277:11795–801. https://doi.org/10.1074/jbc.M200053200.
58. Linnert M, Lin YJ, Manns A, Haupt K, Paschke AK, Fischer G, Weiwad M, Lucke C. The FKBP-type domain of the human aryl hydrocarbon receptor-interacting protein reveals an unusual Hsp90 interaction. Biochemistry. 2013; 52:2097–107. https://doi.org/10.1021/bi301649m.
59. Liu T, Daniels CK, Cao S. Comprehensive review on the HSC70 functions, interactions with related molecules and involvement in clinical diseases and therapeutic potential. Pharmacol Ther. 2012; 136:354–74. https://doi.org/10.1016/j.pharmthera.2012.08.014.
60. Truman AW, Kristjansdottir K, Wolfgeher D, Hasin N, Polier S, Zhang H, Perrett S, Prodromou C, Jones GW, Kron SJ. CDK-dependent Hsp70 Phosphorylation controls G1 cyclin abundance and cell-cycle progression. Cell. 2012; 151:1308–18. https://doi.org/10.1016/j.cell.2012.10.051.
61. Varjosalo M, Sacco R, Stukalov A, van Drogen A, Planyavsky M, Hauri S, Aebersold R, Bennett KL, Colinge J, Gstaiger M, Superti-Furga G. Interlaboratory reproducibility of large-scale human protein-complex analysis by standardized AP-MS. Nat Methods. 2013; 10:307–14. https://doi.org/10.1038/nmeth.2400.
62. Schernthaner-Reiter MH, Trivellin G, Hernández-Ramírez LC, Korbonits M, Stratakis C. Interaction between AIP and the cAMP-dependent protein kinase (PKA) pathway in pituitary tumor formation. Endocr Rev. 2015; 36:SAT-427.
63. Galigniana MD, Harrell JM, Murphy PJ, Chinkers M, Radanyi C, Renoir JM, Zhang M, Pratt WB. Binding of hsp90-associated immunophilins to cytoplasmic dynein: direct binding and in vivo evidence that the peptidylprolyl isomerase domain is a dynein interaction domain. Biochemistry. 2002; 41:13602–10. https://doi.org/10.1021/bi020399z.
64. Berg P, Pongratz I. Two parallel pathways mediate cytoplasmic localization of the dioxin (aryl hydrocarbon) receptor. J Biol Chem. 2002; 277:32310–9. https://doi.org/10.1074/jbc.M203351200.
65. Petrulis JR, Kusnadi A, Ramadoss P, Hollingshead B, Perdew GH. The hsp90 Co-chaperone XAP2 alters importin beta recognition of the bipartite nuclear localization signal of the Ah receptor and represses transcriptional activity. J Biol Chem. 2003; 278:2677–85. https://doi.org/10.1074/jbc.M209331200.
66. Akhmanova A, Steinmetz MO. Tracking the ends: a dynamic protein network controls the fate of microtubule tips. Nat Rev Mol Cell Biol. 2008; 9:309–22. https://doi.org/10.1038/nrm2369.
67. Kavallaris M. Microtubules and resistance to tubulin-binding agents. Nat Rev Cancer. 2010; 10:194–204. https://doi.org/10.1038/nrc2803.
68. Garnham CP, Roll-Mecak A. The chemical complexity of cellular microtubules: tubulin post-translational modification enzymes and their roles in tuning microtubule functions. Cytoskeleton (Hoboken). 2012; 69:442–63. https://doi.org/10.1002/cm.21027.
69. Peverelli E, Treppiedi D, Giardino E, Vitali E, Lania AG, Mantovani G. Dopamine and somatostatin analogues resistance of pituitary tumors: Focus on cytoskeleton involvement. Front Endocrinol (Lausanne). 2015; 6:187. https://doi.org/10.3389/fendo.2015.00187.
70. Brunet-de Carvalho N, Picart R, Van de Moortele S, Tougard C, Tixier-Vidal A. Laminin induces formation of neurite-like processes and potentiates prolactin secretion by GH3 rat pituitary cells. Differentiation. 1989; 40:106–18. https://doi.org/10.1111/j.1432-0436.1989.tb00820.x.
71. Steeg PS, Bevilacqua G, Kopper L, Thorgeirsson UP, Talmadge JE, Liotta LA, Sobel ME. Evidence for a novel gene associated with low tumor metastatic potential. J Natl Cancer Inst. 1988; 80:200–4. https://doi.org/10.1093/jnci/80.3.200.
72. Takacs-Vellai K. The metastasis suppressor Nm23 as a modulator of Ras/ERK signaling. J Mol Signal. 2014; 9:4. https://doi.org/10.1186/1750-2187-9-4.
73. Tong Y, Yung LY, Wong YH. Metastasis suppressors Nm23H1 and Nm23H2 differentially regulate neoplastic transformation and tumorigenesis. Cancer Lett. 2015; 361:207–17. https://doi.org/10.1016/j.canlet.2015.02.050.
74. Suhardja A, Kovacs K, Rutka J. Genetic basis of pituitary adenoma invasiveness: a review. J Neurooncol. 2001; 52:195–204. https://doi.org/10.1023/A:1010655419332.
75. Tso PH, Wang Y, Yung LY, Tong Y, Lee MM, Wong YH. RGS19 inhibits Ras signaling through Nm23H1/2-mediated phosphorylation of the kinase suppressor of Ras. Cell Signal. 2013; 25:1064–74. https://doi.org/10.1016/j.cellsig.2013.02.010.
76. Berney CR, Fisher RJ, Yang J, Russell PJ, Crowe PJ. Genomic alterations (LOH, MI) on chromosome 17q21-23 and prognosis of sporadic colorectal cancer. Int J Cancer. 2000; 89:1–7.
77. Boissan M, Lacombe ML. Learning about the functions of NME/NM23: lessons from knockout mice to silencing strategies. Naunyn Schmiedebergs Arch Pharmacol. 2011; 384:421–31. https://doi.org/10.1007/s00210-011-0649-3.
78. Boissan M, De Wever O, Lizarraga F, Wendum D, Poincloux R, Chignard N, Desbois-Mouthon C, Dufour S, Nawrocki-Raby B, Birembaut P, Bracke M, Chavrier P, Gespach C, Lacombe ML. Implication of metastasis suppressor NM23-H1 in maintaining adherens junctions and limiting the invasive potential of human cancer cells. Cancer Res. 2010; 70:7710–22. https://doi.org/10.1158/0008-5472.CAN-10-1887.
79. Pan LX, Chen ZP, Liu YS, Zhao JH. Magnetic resonance imaging and biological markers in pituitary adenomas with invasion of the cavernous sinus space. J Neurooncol. 2005; 74:71–6. https://doi.org/10.1007/s11060-004-6150-9.
80. Barry S, Gadaleta E, Chelala C, Korbonits M. Gene expression profiling of familial and sporadic pituitary adenomas. Endocrine Abstracts.15th European Congress of Endocrinology, Copenhagen, Denmark. 2013; 32:823.
81. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009; 119:1420–8. https://doi.org/39104.
82. Lekva T, Berg JP, Fougner SL, Olstad OK, Ueland T, Bollerslev J. Gene Expression Profiling Identifies ESRP1 as a Potential Regulator of Epithelial Mesenchymal Transition in Somatotroph Adenomas from a Large Cohort of Patients with Acromegaly. J Clin Endocrinol Metab. 2012; 97:E1506–14. https://doi.org/10.1210/jc.2012-1760.
83. Papa L, Manfredi G, Germain D. SOD1, an unexpected novel target for cancer therapy. Genes Cancer. 2014; 5:15–21. https://doi.org/10.18632/genesandcancer.4.
84. Hubina E, Nanzer AM, Hanson MR, Ciccarelli E, Losa M, Gaia D, Papotti M, Terreni MR, Khalaf S, Jordan S, Czirjak S, Hanzely Z, Nagy GM, et al. Somatostatin analogues stimulate p27 expression and inhibit the MAP kinase pathway in pituitary tumours. Eur J Endocrinol. 2006; 155:371–9. https://doi.org/10.1530/eje.1.02213.
85. Secondo A, De MM, Zirpoli L, Santillo M, Mondola P. The Cu-Zn superoxide dismutase (SOD1) inhibits ERK phosphorylation by muscarinic receptor modulation in rat pituitary GH3 cells. Biochem Biophys Res Commun. 2008; 376:143–7. https://doi.org/10.1016/j.bbrc.2008.08.110.
86. Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci. 2005; 6:11–22. https://doi.org/10.1038/nrn1587.
87. Thompson A, Schafer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Johnstone R, Mohammed AK, Hamon C. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem. 2003; 75:1895–904. https://doi.org/10.1021/ac0262560.
88. Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999; 20:3551–67.
89. QIAGEN. Ingenuity Pathways Analysis. www.qiagen.com/ingenuity. 2015. Access date: 12/17/2013.
90. Leontiou CA, Gueorguiev M, van der Spuy J, Quinton R, Lolli F, Hassan S, Chahal HS, Igreja SC, Jordan S, Rowe J, Stolbrink M, Christian HC, Wray J, et al. The role of the aryl hydrocarbon receptor-interacting protein gene in familial and sporadic pituitary adenomas. J Clin Endocrinol Metab. 2008; 93:2390–401. https://doi.org/10.1210/jc.2007-2611.
91. Schindelin J, Rueden CT, Hiner MC, Eliceiri KW. The ImageJ ecosystem: An open platform for biomedical image analysis. Mol Reprod Dev. 2015; 82:518–29. https://doi.org/10.1002/mrd.22489.
92. Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc. 2006; 224:213–32. https://doi.org/10.1111/j.1365-2818.2006.01706.x.
93. Dunn KW, Kamocka MM, McDonald JH. A practical guide to evaluating colocalization in biological microscopy. Am J Physiol Cell Physiol. 2011; 300:C723-C42. https://doi.org/10.1152/ajpcell.00462.2010.
94. White DJ, Kazimiers T, Schindelin J. Coloc 2 - Standardized, modular, pixel intensity correlation over space based colocalization analysis. http://imagej.net/Coloc_2. 2015. Access date: 21-10-2016.
95. Li S, Wang L, Fu B, Berman MA, Diallo A, Dorf ME. TRIM65 regulates microRNA activity by ubiquitination of TNRC6. Proc Natl Acad Sci USA. 2014; 111:6970–5. https://doi.org/10.1073/pnas.1322545111.
96. Petrulis JR, Hord NG, Perdew GH. Subcellular localization of the aryl hydrocarbon receptor is modulated by the immunophilin homolog hepatitis B virus X-associated protein 2. J Biol Chem. 2000; 275:37448–53. https://doi.org/10.1074/jbc.M006873200.
97. Kazlauskas A, Poellinger L, Pongratz I. The immunophilin-like protein XAP2 regulates ubiquitination and subcellular localization of the dioxin receptor. J Biol Chem. 2000; 275:41317–24. https://doi.org/10.1074/jbc.M007765200.
98. Dunham EE, Stevens EA, Glover E, Bradfield CA. The aryl hydrocarbon receptor signaling pathway is modified through interactions with a Kelch protein. Mol Pharmacol. 2006; 70:8–15. https://doi.org/10.1124/mol.106.024380.
99. Schimmack G, Eitelhuber AC, Vincendeau M, Demski K, Shinohara H, Kurosaki T, Krappmann D. AIP augments CARMA1-BCL10-MALT1 complex formation to facilitate NF-kappaB signaling upon T cell activation. Cell Commun Signal. 2014; 12:49. https://doi.org/10.1186/s12964-014-0049-7.
100. Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K, Dong R, Guarani V, Vaites LP, et al. The BioPlex network: a systematic exploration of the human interactome. Cell. 2015; 162:425–40. https://doi.org/10.1016/j.cell.2015.06.043.
101. Taipale M, Tucker G, Peng J, Krykbaeva I, Lin ZY, Larsen B, Choi H, Berger B, Gingras AC, Lindquist S. A quantitative chaperone interaction network reveals the architecture of cellular protein homeostasis pathways. Cell. 2014; 158:434–48. https://doi.org/10.1016/j.cell.2014.05.039.
102. Wang J, Huo K, Ma L, Tang L, Li D, Huang X, Yuan Y, Li C, Wang W, Guan W, Chen H, Jin C, Wei J, et al. Toward an understanding of the protein interaction network of the human liver. Mol Syst Biol. 2011; 7:536. https://doi.org/10.1038/msb.2011.67.
103. Zhang M, Han G, Wang C, Cheng K, Li R, Liu H, Wei X, Ye M, Zou H. A bead-based approach for large-scale identification of in vitro kinase substrates. Proteomics. 2011; 11:4632–7. https://doi.org/10.1002/pmic.201100339.
104. Deribe YL, Wild P, Chandrashaker A, Curak J, Schmidt MH, Kalaidzidis Y, Milutinovic N, Kratchmarova I, Buerkle L, Fetchko MJ, Schmidt P, Kittanakom S, Brown KR, et al. Regulation of epidermal growth factor receptor trafficking by lysine deacetylase HDAC6. Sci Signal. 2009; 2:ra84. https://doi.org/10.1126/scisignal.2000576.
105. Kristensen AR, Gsponer J, Foster LJ. A high-throughput approach for measuring temporal changes in the interactome. Nat Methods. 2012; 9:907–9. https://doi.org/10.1038/nmeth.2131.
106. Cai W, Kramarova TV, Berg P, Korbonits M, Pongratz I. The immunophilin-like protein XAP2 is a negative regulator of estrogen signaling through interaction with estrogen receptor alpha. PLoS One. 2011; 6:e25201. https://doi.org/10.1371/journal.pone.0025201.
107. Havugimana PC, Hart GT, Nepusz T, Yang H, Turinsky AL, Li Z, Wang PI, Boutz DR, Fong V, Phanse S, Babu M, Craig SA, Hu P, et al. A census of human soluble protein complexes. Cell. 2012; 150:1068–81. https://doi.org/10.1016/j.cell.2012.08.011.
108. Nakata A, Urano D, Fujii-Kuriyama Y, Mizuno N, Tago K, Itoh H. G-protein signalling negatively regulates the stability of aryl hydrocarbon receptor. EMBO Rep. 2009; 10:622–8. https://doi.org/10.1038/embor.2009.35.
109. Meyer BK, Perdew GH. Characterization of the AhR-hsp90-XAP2 core complex and the role of the immunophilin-related protein XAP2 in AhR stabilization. Biochemistry. 1999; 38:8907–17. https://doi.org/10.1021/bi982223w.
110. Laenger A, Lang-Rollin I, Kozany C, Zschocke J, Zimmermann N, Ruegg J, Holsboer F, Hausch F, Rein T. XAP2 inhibits glucocorticoid receptor activity in mammalian cells. FEBS Lett. 2009; 583:1493–8. https://doi.org/10.1016/j.febslet.2009.03.072.
111. Schülke JP, Wochnik GM, Lang-Rollin I, Gassen NC, Knapp RT, Berning B, Yassouridis A, Rein T. Differential impact of tetratricopeptide repeat proteins on the steroid hormone receptors. PLoS One. 2010; 5:e11717. https://doi.org/10.1371/journal.pone.0011717.
112. Ravasi T, Suzuki H, Cannistraci CV, Katayama S, Bajic VB, Tan K, Akalin A, Schmeier S, Kanamori-Katayama M, Bertin N, Carninci P, Daub CO, Forrest AR, et al. An atlas of combinatorial transcriptional regulation in mouse and man. Cell. 2010; 140:744–52. https://doi.org/10.1016/j.cell.2010.01.044.
113. Li S, Wang L, Berman M, Kong YY, Dorf ME. Mapping a dynamic innate immunity protein interaction network regulating type I interferon production. Immunity. 2011; 35:426–40. https://doi.org/10.1016/j.immuni.2011.06.014.
114. Zhou Q, Lavorgna A, Bowman M, Hiscott J, Harhaj EW. Aryl hydrocarbon receptor interacting protein targets IRF7 to suppress antiviral signaling and the induction of type I interferon. J Biol Chem. 2015; 290:14729–39. https://doi.org/10.1074/jbc.M114.633065.
115. Marschang P, Brich J, Weeber EJ, Sweatt JD, Shelton JM, Richardson JA, Hammer RE, Herz J. Normal development and fertility of knockout mice lacking the tumor suppressor gene LRP1b suggest functional compensation by LRP1. Mol Cell Biol. 2004; 24:3782–93. https://doi.org/10.1128/MCB.24.9.3782-3793.2004.
116. Gupta GD, Coyaud E, Goncalves J, Mojarad BA, Liu Y, Wu Q, Gheiratmand L, Comartin D, Tkach JM, Cheung SW, Bashkurov M, Hasegan M, Knight JD, et al. A dynamic protein interaction landscape of the human centrosome-cilium interface. Cell. 2015; 163:1484–99. https://doi.org/10.1016/j.cell.2015.10.065.
117. Bolger GB, Peden AH, Steele MR, MacKenzie C, McEwan DG, Wallace DA, Huston E, Baillie GS, Houslay MD. Attenuation of the activity of the cAMP-specific phosphodiesterase PDE4A5 by interaction with the immunophilin XAP2. J Biol Chem. 2003; 278:33351–63. https://doi.org/10.1074/jbc.M303269200.
118. de Oliveira SK, Hoffmeister M, Gambaryan S, Muller-Esterl W, Guimaraes JA, Smolenski AP. Phosphodiesterase 2A forms a complex with the co-chaperone XAP2 and regulates nuclear translocation of the aryl hydrocarbon receptor. J Biol Chem. 2007; 282:13656–63. https://doi.org/10.1074/jbc.M610942200.
119. Wan C, Borgeson B, Phanse S, Tu F, Drew K, Clark G, Xiong X, Kagan O, Kwan J, Berzginov A, Chessman K, Pal S, Cromar G, et al. Panorama of ancient metazoan macromolecular complexes (pre-publication dataset). http://thebiogrid.org/185267/publication/panorama-of-ancient-metazoan-macromolecular-complexes.html. 2015. Access date: 19/08/2016.
120. Sumanasekera WK, Tien ES, Turpey R, Vanden Heuvel JP, Perdew GH. Evidence that peroxisome proliferator-activated receptor alpha is complexed with the 90-kDa heat shock protein and the hepatitis virus B X-associated protein 2. J Biol Chem. 2003; 278:4467–73. https://doi.org/10.1074/jbc.M211261200.
121. Couzens AL, Knight JD, Kean MJ, Teo G, Weiss A, Dunham WH, Lin ZY, Bagshaw RD, Sicheri F, Pawson T, Wrana JL, Choi H, Gingras AC. Protein interaction network of the mammalian Hippo pathway reveals mechanisms of kinase-phosphatase interactions. Sci Signal. 2013; 6:rs15. https://doi.org/10.1126/scisignal.2004712.
122. Vargiolu M, Fusco D, Kurelac I, Dirnberger D, Baumeister R, Morra I, Melcarne A, Rimondini R, Romeo G, Bonora E. The tyrosine kinase receptor RET interacts in vivo with aryl hydrocarbon receptor-interacting protein to alter survivin availability. J Clin Endocrinol Metab. 2009; 94:2571–8. https://doi.org/10.1210/jc.2008-1980.
123. Zhao Y, Meng XM, Wei YJ, Zhao XW, Liu DQ, Cao HQ, Liew CC, Ding JF. Cloning and characterization of a novel cardiac-specific kinase that interacts specifically with cardiac troponin I. J Mol Med (Berl). 2003; 81:297–304. https://doi.org/10.1007/s00109-003-0427-x.
124. Froidevaux MS, Berg P, Seugnet I, Decherf S, Becker N, Sachs LM, Bilesimo P, Nygard M, Pongratz I, Demeneix BA. The co-chaperone XAP2 is required for activation of hypothalamic thyrotropin-releasing hormone transcription in vivo. EMBO Rep. 2006; 7:1035–9. https://doi.org/10.1038/sj.embor.7400778.
125. Danielsen JM, Sylvestersen KB, Bekker-Jensen S, Szklarczyk D, Poulsen JW, Horn H, Jensen LJ, Mailand N, Nielsen ML. Mass spectrometric analysis of lysine ubiquitylation reveals promiscuity at site level. Mol Cell Proteomics. 2011; 10:M110. https://doi.org/10.1074/mcp.M110.003590.
126. Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, Sowa ME, Rad R, Rush J, Comb MJ, Harper JW, Gygi SP. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell. 2011; 44:325–40. https://doi.org/10.1016/j.molcel.2011.08.025.
127. Udeshi ND, Mani DR, Eisenhaure T, Mertins P, Jaffe JD, Clauser KR, Hacohen N, Carr SA. Methods for quantification of in vivo changes in protein ubiquitination following proteasome and deubiquitinase inhibition. Mol Cell Proteomics. 2012; 11:148–59. https://doi.org/10.1074/mcp.M111.016857.
128. Povlsen LK, Beli P, Wagner SA, Poulsen SL, Sylvestersen KB, Poulsen JW, Nielsen ML, Bekker-Jensen S, Mailand N, Choudhary C. Systems-wide analysis of ubiquitylation dynamics reveals a key role for PAF15 ubiquitylation in DNA-damage bypass. Nat Cell Biol. 2012; 14:1089–98. https://doi.org/10.1038/ncb2579.
129. Stes E, Laga M, Walton A, Samyn N, Timmerman E, De Smet I, Goormachtig S, Gevaert K. A COFRADIC protocol to study protein ubiquitination. J Proteome Res. 2014; 13:3107–13. https://doi.org/10.1021/pr4012443.
130. Gadelha MR, Prezant TR, Une KN, Glick RP, Moskal SF 2nd, Vaisman M, Melmed S, Kineman RD, Frohman LA. Loss of heterozygosity on chromosome 11q13 in two families with acromegaly/gigantism is independent of mutations of the multiple endocrine neoplasia type I gene. J Clin Endocrinol Metab. 1999; 84:249–56.
131. Igreja S, Chahal HS, King P, Bolger GB, Srirangalingam U, Guasti L, Chapp le JP, Trivellin G, Gueorguiev M, Guegan K, Stals K, Khoo B, Kumar AV, et al, and International FIPA Consortium. Characterization of aryl hydrocarbon receptor interacting protein (AIP) mutations in familial isolated pituitary adenoma families. Hum Mutat. 2010; 31:950–60. https://doi.org/10.1002/humu.21292.
132. Aflorei ED, Radian S, Chen C, Klapholz B, Brown N, Stanewsky R, Korbonits M. Functional homology between human and fruitfly AIP protein - an in vivo assay system to test the pathogenicity of AIP mutations. Endocr Rev. 2015; 36:SAT-440.
133. Chahal HS, Stals K, Unterlander M, Balding DJ, Thomas MG, Kumar AV, Besser GM, Atkinson AB, Morrison PJ, Howlett TA, Levy MJ, Orme SM, Akker SA, et al. AIP mutation in pituitary adenomas in the 18th century and today. N Engl J Med. 2011; 364:43–50. https://doi.org/10.1056/NEJMoa1008020.
134. Radian S, Diekmann Y, Gabrovska P, Holland B, Bradley L, Wallace H, Stals K, Bussell AM, McGurren K, Cuesta M, Ryan AW, Herincs M, Hernandez-Ramirez LC, et al. Increased Population Risk of AIP-Related Acromegaly and Gigantism in Ireland. Hum Mutat. 2017; 38:78–85. https://doi.org/10.1002/humu.23121.
135. Occhi G, Jaffrain-Rea ML, Trivellin G, Albiger N, Ceccato F, De Menis E, Angelini M, Ferasin S, Beckers A, Mantero F, Scaroni C. The R304X mutation of the aryl hydrocarbon receptor interacting protein gene in familial isolated pituitary adenomas: Mutational hot-spot or founder effect? J Endocrinol Invest. 2010; 33:800–5. https://doi.org/10.3275/6956.
136. Lecoq AL, Viengchareun S, Hage M, Bouligand J, Young J, Boutron A, Zizzari P, Lombes M, Chanson P, Kamenicky P. AIP mutations impair AhR signaling in pituitary adenoma patients fibroblasts and in GH3 cells. Endocr Relat Cancer. 2016; 23:433–43. https://doi.org/10.1530/ERC-16-0041.
137. Georgitsi M, Raitila A, Karhu A, Tuppurainen K, Makinen MJ, Vierimaa O, Paschke R, Saeger W, van der Luijt RB, Sane T, Robledo M, De ME, Weil RJ, et al. Molecular diagnosis of pituitary adenoma predisposition caused by aryl hydrocarbon receptor-interacting protein gene mutations. Proc Natl Acad Sci USA. 2007; 104:4101–5. https://doi.org/10.1073/pnas.0700004104.