
INTRODUCTION
Papillary renal cell carcinoma (PRCC) is the second most common type of kidney cancer representing approximately 15–20% of all cases. PRCC is heterogeneous and categorized into two main histologic subtypes, type 1 PRCC and type 2 PRCC [1, 2]. PRCC can present as both sporadic disease or as a component of a hereditary syndrome, such as hereditary papillary renal cell carcinoma (HPRC) or hereditary leiomyomatosis and renal cell carcinoma (HLRCC) [3]. HPRC patients carry germline activating mutations within the tyrosine kinase domain of the MET oncogene and presents with bilateral, multifocal type 1 PRCC [4, 5], while HLRCC patients inherit germline inactivating mutation of the FH tumor suppressor gene and can have an aggressive variant of type 2 PRCC [6, 7]. Although numerous studies have been done on familial forms of PRCC, little was known about the genes responsible for the sporadic forms of PRCC until a recent report from the Cancer Genome Atlas (TCGA) in which 161 PRCC tumors were sequenced and analyzed [2]. This study highlighted that amplification of chromosome 7 and 17 and activating mutations of the MET gene, on chromosome 7, are associated with type 1 PRCC, while type 2 PRCC was more genetically diverse including tumors that had either germline or somatic FH mutation, chromosomal translocations producing TFE3-fusions, or NRF2 pathway mutations in NFE2L2 (NRF2) or CUL3 [2]. In addition, whole genome sequencing of all PRCC types identified five frequently mutated genes, MET, SETD2, NF2, KDM6A, and SMARCB1, suggesting that alterations to the MET signaling pathway, Hippo signaling pathway, and the chromatin modifier pathways may play an important role in PRCC carcinogenesis [2]. Currently, there are no standard of care therapies for most patients with advanced PRCC and clinical trials have been limited to targeting the MET signaling pathway [8].
Neurofibromatosis type 2 (NF2) is a tumor suppressor gene that regulates the Hippo signaling pathway, which normally controls organ growth by suppressing cell division and proliferation and activating programmed cell death/apoptosis [9]. Heterozygous germline inactivating mutations of NF2 are responsible for the hereditary tumor predisposition syndrome neurofibromatosis type 2, which is characterized by the development of multiple tumors in the nervous system [10, 11]. Somatic mutation or loss of the NF2 gene have been previously reported at a low frequency in a breast cancer, prostate cancer, clear cell renal cell carcinoma, and PRCC [2, 12, 13]. The Hippo signaling pathway is evolutionary conserved and regulates tissue growth and survival by transmitting extracellular signals such as contact inhibition to the cells. Cell-to-cell contact activates the Hippo signaling cascade through proteins such as Merlin (encoded by NF2) that induces the MST1/2 complex, with the regulatory protein Salvador, to phosphorylate the LATS1/2/MOB1 complex that leads to the phosphorylation dependent inhibition of the transcriptional activity of Yes-associated protein 1 (YAP1) and transcriptional co-activator with PDZ-binding motif (TAZ) [9]. Thus, the NF2 gene product Merlin suppresses the transcriptional activity of YAP1 and controls contact inhibition of normal cells. Complete loss of the NF2 gene would lead to an inability to suppress the activity of YAP/TAZ, driving the expression of multiple downstream transcription targets, including BIRC5 (survivin), CTGF (connective tissue growth factor), and CCND1 (cyclin D1), that all promote cell growth and survival [9]. Aberrant YAP1 transcriptional activity can also be driven in β-catenin active tumors via the activation of the Src family tyrosine-protein kinase Yes. Yes activates the YAP1-β-catenin-TBX5 complex transcriptional activity and promotes the survival of the cells by increasing expression of multiple genes, such as BIRC5 [14]. Yes inhibitors, such as dasatinib and saracatinib [15], have been shown to be active in preclinical studies of cancer cell lines with increased YAP1 transcription target expression due to increased β-catenin [14, 16] and therefore, could be an attractive potential therapeutic target in cancer cell lines with a deficiency in the Hippo signaling pathway.
In this report, we investigated the effect on the growth and survival of NF2-deficient PRCC cell lines of targeting YAP1 transcription activation resulting from the Hippo signaling pathway loss. We investigated three PRCC cell lines bearing NF2 mutations and assessed the effect of Yes inhibitors on their proliferation and survival in vitro and in vivo with the goal of developing a novel potential therapeutic approach for PRCC patients whose tumors are characterized by an NF2 deficiency.
RESULTS
The Hippo signaling pathway is altered in 22.5% of PRCC
The Cancer Genome Atlas (TCGA) data has recently shown that NF2 is among the five most commonly mutated genes in PRCC and was present in 3.6% of cases (10/280–cBioPortal; Figure 1A–1B) [17, 18]. Furthermore, copy number loss of the NF2 gene was observed in 21.8% of cases with most mutation positive cases also demonstrating copy number loss (Figure 1B). While the presence of a NF2 mutation did not correlate with survival, the 22.5% of PRCC cases that demonstrated either NF2 mutation or copy number loss demonstrated a strong correlation with poorer survival (p = 0.00189; Figure 1C). These data suggest that dysregulation of the Hippo signaling pathway might be a critical driver for PRCC growth.
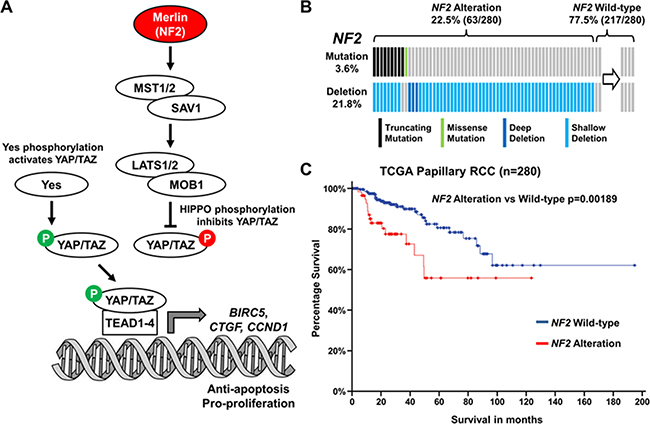
Figure 1: Hippo signaling pathway gene NF2 is altered in 22.5% of PRCC. (A) Cartoon depicting the Hippo signaling pathway and how Merlin (encoded by NF2) and tyrosine-protein kinase Yes play a role in this pathway. (B) Mutation and deletion of NF2 in the TCGA cohort of 280 PRCC tumors (Data downloaded for cBioPortal [17, 18]). (C) Survival of patients with NF2 altered PRCC tumors compared to NF2 unaltered PRCC tumors.
To provide a cell line model of Hippo signaling pathway loss, the patient-derived cell lines generated at the Urologic Oncology Branch from PRCC patient material were screened for complete mutational loss of the NF2 gene. This identified one cell line derived from a patient with type 1 PRCC, UOK342, with a homozygous NF2 insertion/deletion alteration that resulted in frameshift mutation and the loss of a splice site (Figure 2A) and one cell line derived from a patient with type 2 PRCC, UOK275, with a homozygous NF2 single nucleotide insertion that resulted in frameshift mutation (Figure 2B). These two newly identified NF2-deficient cell lines were combined with the existing ACHN cell line, known to have a homozygous NF2 stop codon mutation (https://cansar.icr.ac.uk/cansar/cell-lines/ACHN/mutations/) (Figure 2C), for further study.
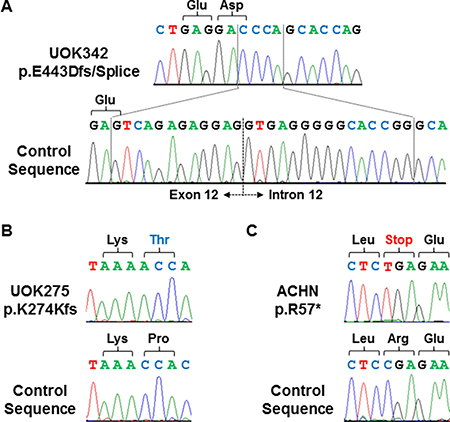
Figure 2: Homozygous mutations of NF2 in 3 PRCC cell lines. Homozygous frameshift and truncation mutations of NF2 were demonstrated by Sanger sequencing in UOK342 (A), UOK275 (B), and ACHN (C). DNA extracted from normal blood samples were used to generate control sequences.
Hippo signaling pathway loss in NF2-deficient PRCC cells supports their survival through YAP1 activation
NF2 alteration leads to the activation of the transcription factor YAP1 and the expression of multiple downstream targets promoting tumor cell survival and proliferation [9, 19]. Since homozygous mutation of NF2 was found in UOK275, UOK342 and ACHN, we investigated whether these alterations led to the expression of YAP1 downstream targets. As shown on Figure 3A–3B, survivin, cyclin D1 and CTGF were all overexpressed at the protein and mRNA levels compared to HEK293. Since all three genes can be transcribed by multiple transcription factors, we next measured YAP1 nuclear localization in NF2-deficient PRCC cells and HEK293 cells using a high content imaging system (InCell 2000 Analyzer). As shown in Figure 3C, YAP1 had a significantly higher nuclear localization in NF2-deficient PRCC cell lines compared to HEK293 cells suggesting that YAP1 is more activated in NF2-deficient PRCC cell lines (representative images are shown in Supplementary Figure 1A). Taken together, these data demonstrate that the Hippo pathway is activated in UOK275, UOK342 and ACHN.
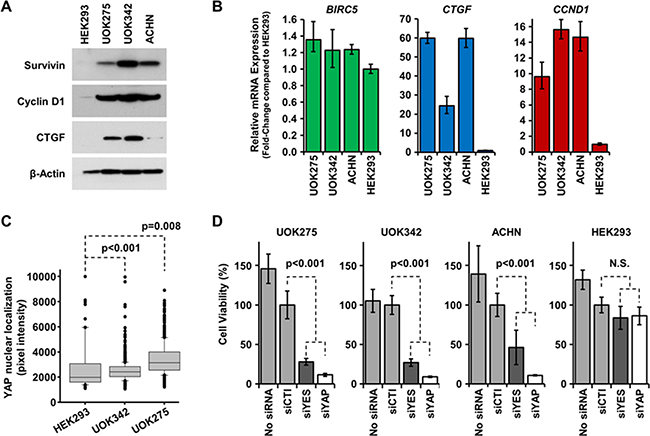
Figure 3: Inhibition of the Hippo pathway is cytotoxic to NF2-deficient tumor cells. (A) Basal protein expression level of YAP1 transcription targets survivin, cyclin D1 and CTGF were assessed by immunoblotting in UOK275, UOK342 and ACHN with HEK293 used to represent normal levels of expression. Β-actin expression levels were used as a loading control. (B) Gene expression of BIRC5 (survivin), CTGF (CTGF) and CCND1 (cyclin D1) in UOK275, UOK342 and ACHN. HEK293 were used as controls. (C) YAP1 nuclear localization in HEK293, UOK275, and UOK342 was quantified using an InCell Analyzer. (D) The effect of transient silencing of YES1 and YAP1 by small interference RNA or mock siRNA on cell viability was assessed in UOK275, UOK342, ACHN and HEK293 72 hours post-transfection by Cell-Titer Glo assay. N.S.: non-significant; siCTL: mock siRNA; siYES: siRNA against YES1; siYAP: siRNA against YAP1.
Tyrosine-protein kinase Yes, encoded by the YES1 gene, is one of the main regulators of YAP1, in addition to the Hippo signaling pathway, and drives transcriptional activity in β-catenin active tumors [14]. To assess whether loss of the Hippo signaling pathway due to NF2 mutation may provide a therapeutic target, either YAP1 or its regulator Yes were transiently silenced using small interference RNAs (Supplementary Figure 1B). Transient silencing of either YAP1 or YES1 proved to be cytotoxic to UOK275, UOK342 and ACHN cells, while there was no effect on the survival of HEK293 (Figure 3D). This suggests that inhibition of the Hippo pathway in NF2-deficient PRCC may have a therapeutic value and may selectively target the tumor cells.
Inhibition of Yes-mediated YAP1 activation is cytotoxic to NF2-deficient PRCC cells
Yes is one of nine members of the Src kinase family along with Src, Fyn, Lyn, ABL, and Lck. Despite the role of Yes in regulating the Hippo signaling pathway through phosphorylation of YAP and supporting tumor growth, there are currently only a small proportion of the clinically approved Src kinase family inhibitors that effectively target Yes, such as dasatinib and saracatinib. Dasatinib is a Yes, Bcr-ABL, Src, and Kit inhibitor, and has been approved by the Food and Drug Administration for the treatment of chronic phase Philadelphia chromosome-positive chronic myelogenous leukemia (CP-CML) and Philadelphia chromosome-positive acute lymphoblastic leukemia (ALL) [20–22]. Saracatinib is a known inhibitor of Fyn, Yes, Src, Bcr-ABL and Lck, and has recently been repurposed for the treatment of Alzheimer’s disease [23]. Since silencing of YES1 was lethal to NF2-deficient PRCC tumor cells, their survival was assessed following dasatinib and saracatinib treatment. Both decreased the survival of NF2-deficient PRCC (Figure 4A and Supplementary Figure 2A) as well as the expression of YAP1 downstream targets (Figure 4B–4C, Supplementary Figure 2B). The embryonic kidney cell line HEK293 used as control was unaffected by the treatments (Figure 4A and 4C). To distinguish the specific effects of Yes inhibition from addition effects due to inhibition of other Src kinase family members, two alternative Src kinase family inhibitors, PP2 and WH-4-023, that do not inhibit Yes were used. They did not affect cell viability or alter the expression of the YAP1 downstream target genes (Supplementary Figure 2A–2B). The levels of Yes-dependent phosphorylation on YAP1 (Tyr357) were also assessed by immunoblotting. As shown in Figure 4B, dasatinib decreased Yes-dependent phosphorylation of YAP1 in all 3 NF2-deficient cell lines.
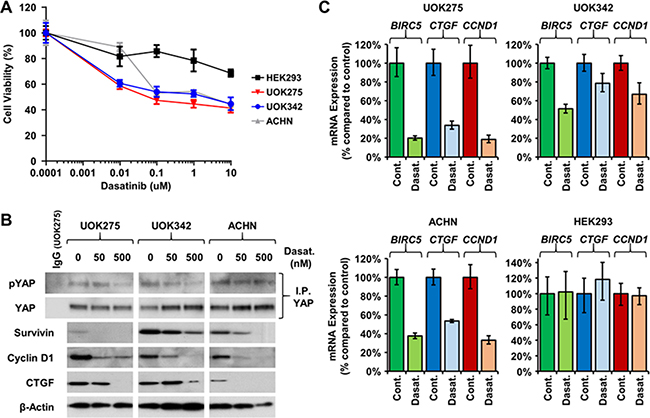
Figure 4: Yes inhibitors lower YAP1-mediated transcriptional network. (A) Dose concentration treatment of dasatinib (μM) on the viability of UOK275, UOK342 and ACHN. HEK293 were used as controls. Cells were treated as indicated for 48 hours then viability was assessed by Cell-Titer Glo assay. (B) Yes-dependent phosphorylation of YAP1 and protein expression of survivin, cyclin D1 and CTGF was evaluated following 24 hours of dasatinib treatment by immunoblotting. (C) Expression of BIRC5 (survivin), CTGF (CTGF) and CCND1 (cyclin D1) was assessed by TaqMan assay 24 hours after dasatinib treatment (100nM) in HEK293, UOK275, UOK342 and ACHN.
Inhibition of Yes arrests the cell cycle of NF2-deficient PRCC cells
In normal cells, the balance between cell cycle arrest and cell proliferation is tightly regulated by extracellular signals and contact inhibition. The Hippo signaling pathway is critical in mediating those signals, thus alteration of NF2 and YAP1 activation lead to aberrant cell proliferation and loss of contact inhibition [24]. To investigate how dasatinib and saracatinib affect cell cycle, NF2-deficient cells were treated with either 50 nM or 500 nM of dasatinib or 100 nM of saracatinib, stained with BrdU and propidium iodide and analyzed by flow cytometry. As shown in Figure 5A–5C, inhibition of YAP1 with dasatinib led to a G0/G1 arrest in NF2-deficient cell lines but did not affect the cell cycle of HEK293 (see also Supplementary Table 1). Similar results, although less pronounced, were observed with saracatinib (Supplementary Table 2). Src inhibitors PP2 and WH-4-023 did not affect the cell cycle of NF2-deficient cells suggesting that the previous observed effect is likely to be Yes-mediated (Supplementary Tables 3 and 4 respectively).
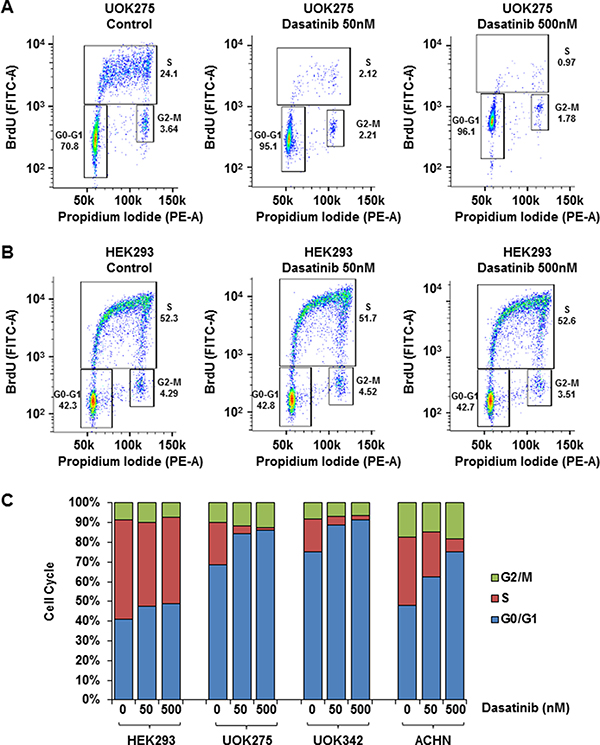
Figure 5: Yes inhibitors block NF2-deficient PRCC cell cycle. Quantification of UOK275 (A) and HEK293 (B) cell cycle phases by BrdU and propidium iodide (PI) staining. The panels display the position of gates used to quantify cells in G0-G1, S or G2-M phases after 18 hours of treatment with DMSO (control), or dasatinib (50 nM or 500 nM). (C) Quantification of cells in the different cell cycle phases in HEK293, UOK275, UOK342 and ACHN after DMSO or dasatinib treatment (average of 3 distinct experiments).
Dasatinib inhibits tumor growth in two NF2-deficient xenograft animal models
Next, we evaluated the in vivo effect of dasatinib on two PRCC NF2-deficient tumor xenograft models (UOK275 and UOK342). Female nude mice bearing UOK275 xenografts or UOK342 xenografts were treated with dasatinib by oral gavage (25 mg/kg/week days) or vehicle (water/PEG 1:1; 10 mice per groups). Tumors were measured on a weekly basis and, after six to seven weeks, tumors were excised and the mice sacrificed. As shown in Figure 6A and 6B, dasatinib treatment significantly inhibited tumor growth in both UOK275 xenografts and UOK342 xenografts, suggesting that targeted inhibition of Yes in YAP-activated tumors with dasatinib may present a therapeutic potential for patients with NF2-deficient PRCC tumors that have lost regulation by the Hippo signaling pathway.
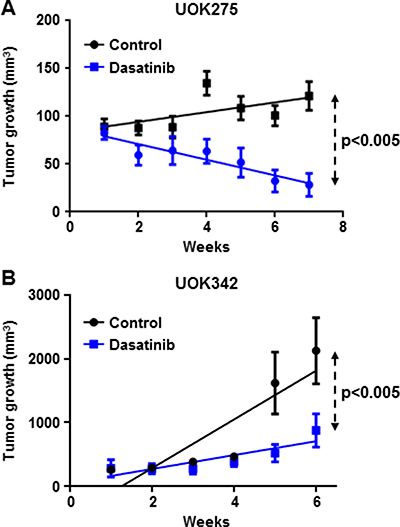
Figure 6: Dasatinib inhibits NF2-deficient tumor growth in two xenograft animal models. (A) Twenty female athymic nude mice were injected subcutaneously with 2 million of UOK275 cells. When the tumor volumes reached > 80 mm3, mice were randomized into 2 groups. One group was treated with dasatinib (25 mg/kg week days by oral gavage) while the other group received the vehicle (water/PEG 1:1) in a similar manner. Tumor volumes were measured once a week using calipers. Tumor volume was estimated using the formula v = lxL2. (B) Twenty female athymic nude mice were injected with 2 million of UOK342 cells. When the tumor volumes reached >100 mm3, mice were randomized into 2 groups treated either with dasatinib (25 mg/kg week days by oral gavage) or the vehicle (water/PEG 1:1). Tumor volumes were measured using calipers once a week. Tumor volume was estimated using the formula v = lxL2.
DISCUSSION
Papillary renal cell carcinoma (PRCC) is a genetically and histologically heterogeneous disease. The Cancer Genome Atlas Research Network’s recent comprehensive analysis of the molecular and genetic features of PRCC has greatly improved our understanding of the pathways involved in PRCC carcinogenesis and is opening novel targeted therapeutic opportunities. Mutation of NF2, a key regulator of the Hippo signaling pathway was observed in 3.6% of PRCC and, in combination with copy number loss, NF2 alterations were present in 22.5% PRCC, including both type 1 and type 2 PRCC histologies [2].
We identified 2 PRCC cell lines bearing NF2 mutations; one derived from a patient with type 2 PRCC, UOK275, and one derived from a patient with type 1 PRCC, UOK342. In addition, a known, commonly used type 1 PRCC cell line, ACHN, had been reported to have a NF2 mutation and this was confirmed within this study. A series of siRNA experiments demonstrated that all 3 NF2-deficient PRCC cell lines were dependent on YAP1 transcriptional activity to support their survival and growth. Similar to that observed within β-catenin active tumors [14], Yes activation and expression were also critical for the survival of these NF2-deficient PRCC cell lines and for YAP1 transcriptional activity. YAP1 transcriptional activity is regulated at the posttranslational level by phosphorylation by multiple kinases, including both the downstream members of the Hippo signaling pathway and several members of the src kinase family, including Yes [25–27]. Dasatinib and saracatinib are src inhibitors that have a broad inhibitory effect on most of the src family members, including Yes. While alterative src inhibitors, such as WH-4-023 and PP2, effect similar members of the src kinase family, but are not reported to inhibit Yes. Comparative analysis using these two different sets of src inhibitors demonstrated a specific effect viability and expression of YAP1 downstream targets from the Yes kinase inhibitors and no effect from the src inhibitors that lacked Yes kinase inhibition. This strengthened our hypothesis that these NF2-deficient PRCC cell lines could be specifically targeted with dasatinib and saracatinib in a predominantly Yes-dependent manner. These data were further complemented by showing that Yes kinase inhibitors induced a G0/G1 arrest in all 3 NF2-deficient PRCC cell lines, but had no effect on the HEK293 control cell lines, while the alternative src inhibitors had no effect on cell cycle in any cell line.
The Hippo pathway controls organ size and cell proliferation by regulation of YAP1 cellular localization and transcriptional activity. In NF2-deficient PRCC, the disruption of the Hippo pathway and aberrant activation of YAP1 are likely to promote cell proliferation. The YAP1 downstream targets cyclin D1 (CCDN1) and survivin (BIRC5) could both regulate the cell cycle. Cyclin D1 expression is required and maintained during the G1 phase and for the initiation of S phase, after which it is degraded. Survivin is part of the inhibitor of apoptosis (IAP) family and is only expressed in G2/M phase. It is thought that survivin affects the M phase by binding to mitotic spindles. The G0/G1 arrest observed in the treated cell lines is most likely due, at least in part, to the decrease in cyclin D1 expression.
The in vitro analysis had demonstrated the effectiveness of Yes inhibitors on cell lines, so we proceeded to assess the effect of dasatinib in vivo on the growth of two NF2-deficient PRCC xenograft models using the type 2 PRCC cell line UOK275 and the type 1 PRCC cell line UOK342. Both models presented a tumor growth inhibition in the dasatinib-treated groups compared with the vehicle-treated groups. Taken together our data identify both Yes kinase activity and YAP1 transcriptional activity as attractive therapeutic targets in NF2-deficient PRCC.
The recent uncovering of a subgroup of PRCC patients with NF2 mutation has paved the way for the development of novel targeted therapies. This preclinical study is providing some scientific rationale as to why to target Yes kinase activity and identifies Yes inhibitors (such as dasatinib) as promising potential therapeutic agents for patients with NF2-deficient PRCC tumors.
MATERIALS AND METHODS
Cell lines and cell culture
UOK275 and UOK342 cell lines were established in the Urologic Oncology Branch from tumor specimens (National Cancer Institute, Bethesda, MD). A full characterization of the cell lines will be published elsewhere. Briefly, UOK275 was established from a 10 cm retroperitoneal mass that proved to be metastatic RCC in a 55-year-old male who had undergone a right radical nephrectomy the year before for multifocal renal masses involving a 9 cm area of the right kidney and invading the renal vein. UOK342 was established from ascites fluid taken from a 63-year-old male with a history of type 1 PRCC. The immortalized human embryonal kidney cell line HEK293 and the papillary kidney tumor cell line ACHN were both purchased from ATCC (Manassas, VA, USA). Cells were cultured in high glucose DMEM containing pyruvate and glutamine and supplemented with 10%FBS. The cells were harvested or treated when they reached 70–80% confluence.
Cell line mutation identification and gene sequencing
DNA was extracted from cell pellets using a Promega Maxwell 16 Cell DNA Purification Kit (Promega, WI, USA). UOK275 and UOK342 cell line DNA was assessed using the OncoVar assay as previously described [28, 29]. Mutations in NF2 were validated by PCR using a Qiagen Taq PCR Core Kit (Qiagen, MD, USA) per the manufacturer’s specifications, followed by bidirectional sequencing using the BigDye Terminator v.1.1 Cycle Sequencing Kit (Applied Biosystems, CA, USA) per the manufacturer’s specifications. Sequencing was performed on an ABI 3730/ABI 3130xl Genetic Analyzer automated sequencing machine (Applied Biosystems, CA, USA). Sanger Sequencing was conducted at the CCR Genomics Core at the National Cancer Institute. Forward and reverse sequences were evaluated using Sequencher 5.4.6 (Genecodes, MI, USA). NF2-specific primers used were exon 2F: GAGAGTGGAGAGTGCAGAGA, exon 2R: CTTCTCTACTATACAGCTACAG, exon 9F: GGTTTAGTGCCTGGATACTG, exon 9R: CTTCACAAGATGTCACTCTGA, exon 12F: ACAGCACATGATCCCACTTC, exon 12R: GACAACTGCTGTAGAGCTCA.
Reagents
Dasatinib, sarcatinib, PP2 and WH-4-023 were purchased from Selleck Chemicals (Houston, TX, USA). Smartpool ON-TARGETplus siRNAs targeting YAP and YES were purchased from GE Healthcare (Pittsburgh, PA, USA).
Immunoblotting
Ten to twenty micrograms of protein were loaded in 4–20% polyacrylamide gels (Biorad, Hercules, CA, USA). After electrophoresis, proteins were transferred to PVDF membranes, blocked with 5% fat-free milk for 1h, and incubated with primary antibodies overnight at 4°C (dilution 1:1000). After 3 × 10 min washes with TBS-Tween, blots were incubated with horseradish peroxidase-linked secondary antibodies (1:3000 dilution in 2% fat-free milk; Biorad) for 2 h before development with the ECL protein detection system (Thermo Fisher Scientific, Rockford, IL). Rabbit antibodies against survivin (#2808), YAP (#4912), and cyclin D1 (#2922), and mouse antibodies against β-actin (#3700) and α-tubulin (#3873) were from Cell Signaling Technology, Inc (Danvers, MA, USA). Rabbit antibody against CTGF (#ab5097) and phospho-YAP (Tyr357) (#ab62751) was from Abcam (Cambridge, MA, USA).
Cell viability and siRNA transfections
Cell viability was measured using a Cell-Titer Glo purchased from Promega Biosciences, Inc. (San Luis Obispo, CA, USA) following the manufacturer’s protocol and as previously described [30].
RNA extraction and Real-Time PCR analysis
Cell lines were grown to approximately 80–90% confluency, washed with 5 mL of sterile PBS, and RNA was extracted using the RNeasy mini kit (Qiagen, MD) in accordance with the manufacturer’s protocol. RNA was eluted in 50 μL of RNase-free water and the measured using a NanoDrop 2000 UV-Vis Spectrophotometer (Thermo Fisher Scientific Inc., MA, USA). Complementary DNA was generated from 2 μg of RNA using the SuperScript® VILO™ cDNA Synthesis Kit (Invitrogen, CA, USA) in a 20 μL volume and diluted 10-fold with RNase-free water for downstream applications. RT-PCR amplification was performed using an ABI ViiA7 real-time PCR system (Thermo Fisher Scientific Inc., MA, USA) and TaqMan® Gene Expression Assays (Thermo Fisher Scientific Inc., MA) using 2 μL of dilute cDNA for each reaction. All reactions were performed in triplicate using the standard ViiA7 reaction template and expression was normalized to the 18S control gene (Hs99999901_m1) and calculated using the ViiA7 software as comparative CT (ΔΔCT) values. The following set probes were used: BIRC5 (Hs03043574_m1), CTGF (Hs01026927_g1), and CCND1 (Hs00765553_m1).
YAP nuclear/cytoplasmic localization analyses
Two thousand cells were seeded in 96-black well plates (Corning). The following day, cells were fixed with 4% paraformaldehyde for 1 hour and permeabilized with 1% triton for 10 minutes. After blocking with 1% bovine serum albumin (BSA)/PBS for 1 hour, the cells were incubated overnight with FITC-conjugated rabbit YAP antibody (Cell Signaling #14729) in 0.1% BSA/PBS at 4°C. After being washed with PBS, cells were incubated 10 mg/mL Hoechst 33342 and Texas Red phalloidin (Invitrogen) in 0.1% BSA/PBS for 1 hour. Cells were washed 3 times with PBS prior to image acquisition. Image acquisition for each well was performed on an IN Cell Analyzer 2000 (GE Healthcare) by using a 10x objective lens. YAP nuclear/cytoplasmic localization was determined with the IN Cell Developer software (GE Healthcare) by using Hoechst staining to define nuclear regions and phalloidin staining to define cells cytoplasm.
Flow cytometry analysis of cell cycle
Cells were incubated with 10 μM BrdU for 2 hours prior harvest. Cells were then trypsinized and fixed overnight at 4°C in 70% ethanol in calcium- and magnesium-free phosphate buffered saline (PBS). Ethanol solution was removed and cells were incubated in 3 mL of 0.08% pepsin in 0.1 N HCl at 37°C for 20 minutes. Pepsin was removed and nuclei were incubated in 1.5 mL of 2 N HCl at 37°C for 20 minutes. The nuclei containing acid solution was neutralized with 3 mL of 0.1 M sodium borate. Nuclei were spun out of neutralized acid and washed with 2 mL IFA buffer (10 mM HEPES pH 7.4, 150 mM NaCl, 4% FBS and 0.1% sodium azide with 0.5% Tween-20 added on the day of use) and then incubated overnight at 4°C with anti-BrdU clone MoBU-1 conjugated to AlexaFluor488 (Invitrogen B35130) in IFA buffer. DNA was stained for 30 minutes with IFA buffer with of 50 μg/mL propidium iodide (PI) (Sigma-Aldrich #P4864) and 5 μg/mL RNase A (Sigma #R4642). Cell cycle analysis (bivariate plots of BrdU incorporation and DNA content) was performed on a FACSCanto II (Becton Dickinson). Data were collected and analyzed using FlowJo software (FlowJo, Ashland, OR, USA).
Animal study
Animal experiments were performed in accordance with the guidelines of the Animal Care and Use Committee of the National Institutes of Health. Forty female athymic nude mice (Taconic, Germantown, NY, USA) were injected on the right flank with 2 million of UOK342 or UOK275 cells diluted in matrigel (100%; BD Bioscience, San Jose, CA, USA); 20 mice per cell lines). Two weeks after injection, tumor volume reached > 100 mm3. Mice were randomized into four groups of ten mice bearing either UOK342 or UOK275. One group of UOK342 and one group of UOK275 were treated with dasatinib (25 mg/kg week days by oral gavage) while the other groups received the vehicle (water/PEG 1:1) in a similar manner. After 6-7 weeks treatment, animals were sacrificed.
Statistics
All values are expressed as mean ± standard error. All experiments have been performed three times, with exception of the animal study. Values were compared using the Student-Newman-Keul’s test. P < 0.05 was considered significant.
ACKNOWLEDGMENTS
The authors would like to thank Mrs Catherine Wells for help with the animal studies.
CONFLICTS OF INTEREST
The authors have no conflicts of interest.
FUNDING
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
REFERENCES
1. Delahunt B, Eble JN. Papillary renal cell carcinoma: a clinicopathologic and immunohistochemical study of 105 tumors. Mod Pathol. 1997; 10:537–44.
2. Linehan WM, Spellman PT, Ricketts CJ, Creighton CJ, Fei SS, Davis C, Wheeler DA, Murray BA, Schmidt L, Vocke CD, Peto M, Al Mamun AA, Shinbrot E, et al, and Cancer Genome Atlas Research Network. Comprehensive Molecular Characterization of Papillary Renal-Cell Carcinoma. N Engl J Med. 2016; 374:135–45. https://doi.org/10.1056/NEJMoa1505917..
3. Linehan WM, Srinivasan R, Schmidt LS. The genetic basis of kidney cancer: a metabolic disease. Nat Rev Urol. 2010; 7:277–85. https://doi.org/10.1038/nrurol.2010.47.
4. Zbar B, Tory K, Merino M, Schmidt L, Glenn G, Choyke P, Walther MM, Lerman M, Linehan WM. Hereditary papillary renal cell carcinoma. J Urol. 1994; 151:561–66. https://doi.org/10.1016/S0022-5347(17)35015-2.
5. Schmidt L, Duh FM, Chen F, Kishida T, Glenn G, Choyke P, Scherer SW, Zhuang Z, Lubensky I, Dean M, Allikmets R, Chidambaram A, Bergerheim UR, et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet. 1997; 16:68–73. https://doi.org/10.1038/ng0597-68.
6. Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, Leigh I, Gorman P, Lamlum H, Rahman S, Roylance RR, Olpin S, Bevan S, et al, and Multiple Leiomyoma Consortium. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 2002; 30:406–10. https://doi.org/10.1038/ng849.
7. Grubb RL 3rd, Franks ME, Toro J, Middelton L, Choyke L, Fowler S, Torres-Cabala C, Glenn GM, Choyke P, Merino MJ, Zbar B, Pinto PA, Srinivasan R, et al. Hereditary leiomyomatosis and renal cell cancer: a syndrome associated with an aggressive form of inherited renal cancer. J Urol. 2007; 177:2074–79. https://doi.org/10.1016/j.juro.2007.01.155.
8. Srinivasan R, Ricketts CJ, Sourbier C, Linehan WM. New strategies in renal cell carcinoma: targeting the genetic and metabolic basis of disease. Clin Cancer Res. 2015; 21:10–17. https://doi.org/10.1158/1078-0432.CCR-13-2993.
9. Harvey KF, Zhang X, Thomas DM. The Hippo pathway and human cancer. Nat Rev Cancer. 2013; 13:246–57. https://doi.org/10.1038/nrc3458.
10. Rouleau GA, Merel P, Lutchman M, Sanson M, Zucman J, Marineau C, Hoang-Xuan K, Demczuk S, Desmaze C, Plougastel B, Pulst SM, Lenoir G, Bijlsma E, et al. Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature. 1993; 363:515–21. https://doi.org/10.1038/363515a0.
11. Striedinger K, VandenBerg SR, Baia GS, McDermott MW, Gutmann DH, Lal A. The neurofibromatosis 2 tumor suppressor gene product, merlin, regulates human meningioma cell growth by signaling through YAP. Neoplasia. 2008; 10:1204–12. https://doi.org/10.1593/neo.08642.
12. Chen F, Zhang Y, Şenbabaoğlu Y, Ciriello G, Yang L, Reznik E, Shuch B, Micevic G, De Velasco G, Shinbrot E, Noble MS, Lu Y, Covington KR, et al. Multilevel Genomics-Based Taxonomy of Renal Cell Carcinoma. Cell Reports. 2016; 14:2476–89. https://doi.org/10.1016/j.celrep.2016.02.024.
13. Petrilli AM, Fernández-Valle C. Role of Merlin/NF2 inactivation in tumor biology. Oncogene. 2016; 35:537–48. https://doi.org/10.1038/onc.2015.125.
14. Rosenbluh J, Nijhawan D, Cox AG, Li X, Neal JT, Schafer EJ, Zack TI, Wang X, Tsherniak A, Schinzel AC, Shao DD, Schumacher SE, Weir BA, et al. β-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell. 2012; 151:1457–73. https://doi.org/10.1016/j.cell.2012.11.026.
15. Patel PR, Sun H, Li SQ, Shen M, Khan J, Thomas CJ, Davis MI. Identification of potent Yes1 kinase inhibitors using a library screening approach. Bioorg Med Chem Lett. 2013; 23:4398–403. https://doi.org/10.1016/j.bmcl.2013.05.072.
16. Taccioli C, Sorrentino G, Zannini A, Caroli J, Beneventano D, Anderlucci L, Lolli M, Bicciato S, Del Sal G. MDP, a database linking drug response data to genomic information, identifies dasatinib and statins as a combinatorial strategy to inhibit YAP/TAZ in cancer cells. Oncotarget. 2015; 6:38854–65. https://doi.org/10.18632/oncotarget.5749.
17. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013; 6:pl1.
18. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2:401–04.
19. Johnson R, Halder G. The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat Rev Drug Discov. 2014; 13:63–79.
20. Talpaz M, Shah NP, Kantarjian H, Donato N, Nicoll J, Paquette R, Cortes J, O’Brien S, Nicaise C, Bleickardt E, Blackwood-Chirchir MA, Iyer V, Chen TT, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med. 2006; 354:2531–41. https://doi.org/10.1056/NEJMoa055229.
21. Santos FP, Cortes J. Dasatinib for the treatment of Philadelphia chromosome-positive leukemias. Expert Opin Pharmacother. 2012; 13:2381–95. https://doi.org/10.1517/14656566.2012.725722.
22. Keating GM. Dasatinib: A Review in Chronic Myeloid Leukaemia and Ph+ Acute Lymphoblastic Leukaemia. Drugs. 2017; 77:85–96. https://doi.org/10.1007/s40265-016-0677-x.
23. Kaufman AC, Salazar SV, Haas LT, Yang J, Kostylev MA, Jeng AT, Robinson SA, Gunther EC, van Dyck CH, Nygaard HB, Strittmatter SM. Fyn inhibition rescues established memory and synapse loss in Alzheimer mice. Ann Neurol. 2015; 77:953–71. https://doi.org/10.1002/ana.24394.
24. Ehmer U, Sage J. Control of Proliferation and Cancer Growth by the Hippo Signaling Pathway. Mol Cancer Res. 2016; 14:127–40. https://doi.org/10.1158/1541-7786.MCR-15-0305.
25. Levy D, Adamovich Y, Reuven N, Shaul Y. Yap1 phosphorylation by c-Abl is a critical step in selective activation of proapoptotic genes in response to DNA damage. Mol Cell. 2008; 29:350–61. https://doi.org/10.1016/j.molcel.2007.12.022.
26. Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, Zheng P, Ye K, Chinnaiyan A, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007; 21:2747–61. https://doi.org/10.1101/gad.1602907.
27. Vassilev A, Kaneko KJ, Shu H, Zhao Y, DePamphilis ML. TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes-associated protein localized in the cytoplasm. Genes Dev. 2001; 15:1229–41. https://doi.org/10.1101/gad.888601.
28. Sorber R, Teper Y, Abisoye-Ogunniyan A, Waterfall JJ, Davis S, Killian JK, Pineda M, Ray S, McCord MR, Pflicke H, Burkett SS, Meltzer PS, Rudloff U. Whole Genome Sequencing of Newly Established Pancreatic Cancer Lines Identifies Novel Somatic Mutation (c.2587G>A) in Axon Guidance Receptor Plexin A1 as Enhancer of Proliferation and Invasion. PLoS One. 2016; 11:e0149833. https://doi.org/10.1371/journal.pone.0149833.
29. Killian JK, Miettinen M, Walker RL, Wang Y, Zhu YJ, Waterfall JJ, Noyes N, Retnakumar P, Yang Z, Smith WI Jr, Killian MS, Lau CC, Pineda M, et al. Recurrent epimutation of SDHC in gastrointestinal stromal tumors. Sci Transl Med. 2014; 6:268ra177.
30. Sourbier C, Ricketts CJ, Matsumoto S, Crooks DR, Liao PJ, Mannes PZ, Yang Y, Wei MH, Srivastava G, Ghosh S, Chen V, Vocke CD, Merino M, et al. Targeting ABL1-mediated oxidative stress adaptation in fumarate hydratase-deficient cancer. Cancer Cell. 2014; 26:840–50. https://doi.org/10.1016/j.ccell.2014.10.005.