
INTRODUCTION
The mechanistic target of rapamycin (mTOR) kinase is a highly conserved and fundamental regulator of growth, metabolism, and aging in all eukaryotes [1, 2]. mTOR is present in two structurally and functionally distinct complexes, mTORC1 and mTORC2, which are defined by their association with Raptor and Rictor, respectively. mTORC1 promotes growth-related cellular processes such as protein synthesis and ribosome biogenesis, and inhibits autophagy. Diverse extra- and intracellular stimuli activate mTORC1 signaling including growth factors (e.g. insulin), amino acids, and energy sufficiency. A key upstream activator of mTORC1 is the small GTPase Rheb (Ras homolog enriched in brain) which directly associates with mTORC1 and is able to stimulate its kinase activity [3, 4]. Activation of mTORC1 is inhibited by the mammalian tuberous sclerosis complex (comprised of Tsc1 and Tsc2) which acts as a GTPase-activating protein (GAP) on Rheb [5, 6]. Growth factors are important stimuli of the Akt kinase that directly phosphorylate and inactivate the Tsc1/Tsc2 complex, thereby de-repressing Rheb and promoting mTORC1 signaling. Recent studies indicate that Rheb-mediated activation of mTORC1 takes place at the lysosomal membrane [7–9].
mTORC1 balances anabolic and catabolic cellular processes. Under favorable conditions rich in nutrients and energy mTORC1 stimulates anabolic processes including the synthesis of proteins and lipids [10]. Upon activation mTORC1 phosphorylates a set of well-characterized targets, most notably the ribosomal S6 kinase (S6K) and the translational initiation factor 4E-binding protein (4E-BP1) to promote protein synthesis. While stimulating the synthesis of macromolecules, mTORC1 inhibits catabolic processes like autophagy, a cellular recycling mechanism [10].
mTORC1 signaling plays an important role in the regulation of cellular senescence and organismal aging [1]. Genetic suppression or pharmacological inhibition of mTOR by rapamycin extends lifespan in diverse model organisms ranging from yeast and C. elegans to mammals [11]. Notably, in C. elegans mTOR signaling plays a fundamental role in the regulation of growth and longevity [12–15]. Importantly, post-developmental inhibition of mTORC1 or rapamycin treatment cause lifespan extension in adult C. elegans and mice [16, 17] suggesting that inactivation of mTORC1 signaling might be a promising anti-aging strategy. Knockdown of mTORC1 also enhances tolerance against environmental stress consistent with a shift towards tissue maintenance. Previous work has identified cellular processes through which mTORC1 affects lifespan. Reduction of protein synthesis has emerged an important mTORC1-regulated mechanism for longevity. A low level of translation under unfavorable stress conditions might be beneficial because of notable energy savings, but further evidence indicates that protective mechanisms are also mobilized [18]. Genetic studies in diverse model organisms have demonstrated that inhibition of genes involved in translation initiation or ribosomal biogenesis promote longevity [19–21]. In addition to reducing mRNA translation, C. elegans mTORC1 inhibition exerts a positive effect on lifespan in part through activation of the transcription factors DAF-16/FoxO and SKN-1/Nrf2 [16]. DAF-16 and SKN-1 regulate genes that protect against environmental, metabolic, and proteotoxic stress, and contribute to longevity [20]. Moreover, longevity effects conferred by mTORC1 inhibition require autophagy [22, 23].
Aberrant mTOR signaling has been implicated in a large number of age-related diseases including cancer, neurodegenerative syndromes, and cardiovascular diseases [1]. Although considerable insights into the regulation of mTORC1 have been made, many unresolved and poorly understood issues remain. In this study, we identify the guanine nucleotide exchange factor CGEF-1 as a novel binding partner of C. elegans RHEB-1. CGEF-1 facilitates mTORC1 signaling to control aging, stress response, protein synthesis, and autophagy in C. elegans. DAF-16/FoxO and SKN-1/Nrf exert protective gene expression and increase stress tolerance and longevity when cgef-1 is genetically inhibited. Dbl, the homolog of CGEF-1 in mammals, associates with Rheb and promotes mTORC1 downstream signaling suggesting an evolutionary conserved function of Dbl/CGEF-1 in the mTOR pathway.
RESULTS
CGEF-1 interacts and functions in the same pathway with RHEB-1 to regulate longevity and stress response in C. elegans
Rheb is indispensable for the activation of mTORC1 signaling by amino acids and growth factors. To identify new regulators of RHEB-1 in C. elegans, we performed a yeast split-ubiquitin two-hybrid screen using full length RHEB-1 as bait. In this screen, CGEF-1 was isolated as a potential binding partner of C. elegans RHEB-1. cgef-1 belongs to the family of Dbl proteins and encodes a guanine nucleotide exchange factor (GEF). Interestingly, alterations of mammalian Dbl function lead to tumor formation and metastasis. We first aimed to confirm the interaction between RHEB-1 and CGEF-1 by co-immunoprecipitation from HEK 293T cells. Indeed, V5-tagged CGEF-1 co-precipitated with Flag-tagged C. elegans RHEB-1 but not with control proteins (Figure 1A) supporting that C. elegans RHEB-1 and CGEF-1 physically interact.
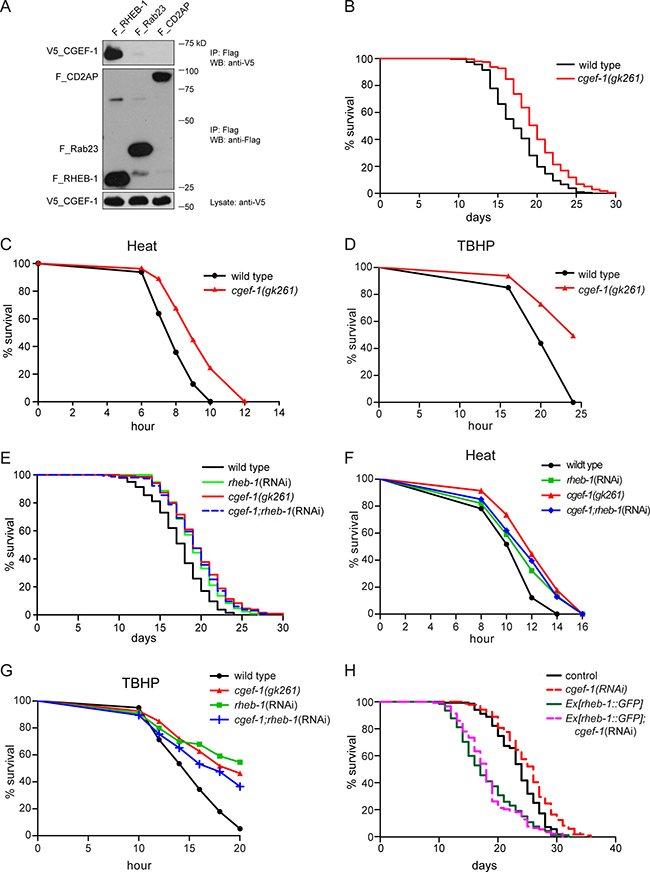
Figure 1: CGEF-1 is a novel RHEB-1 binding partner and functions in the same pathway with RHEB-1 to regulate longevity and stress response in C. elegans. (A) C. elegans CGEF-1 interacts with RHEB-1 in HEK 293T cells. After immunoprecipitation (IP) with anti-Flag antibody, the immobilized CGEF-1 was detected by western blot (WB) analysis using anti-V5 antibody (upper panel). The control proteins Rab23 and CD2AP failed to bind CGEF-1. Middle part shows immunoprecipitation of Flag-tagged proteins, the lower panel shows the expression of V5-tagged CGEF-1 in cell lysates. kD, kiloDalton. (B) Lifespan of cgef-1(gk261) mutants is increased relative to wild type N2. Survival analysis represents the combined data from three experiments. See Table 1 for statistical analyses. (C) Heat stress resistance is increased in cgef-1(gk261) mutants compared to wild type N2. (D) cgef-1 mutants show enhanced tolerance of oxidative stress from tert-butyl hydroperoxide (TBHP). (E) cgef-1 functions in the same pathway with rheb-1. Longevity of cgef-1(gk261) mutants is not altered by rheb-1(RNAi) knockdown. Combined data from two experiments are shown. Statistics are available in Table 1. (F) Inhibition of rheb-1 by RNAi does not affect heat tolerance of cgef-1(gk261) mutants. (G) The oxidative stress resistance conferred by mutation of cgef-1 is not enhanced by rheb-1(RNAi) knockdown. Heat survival assays (C and F) were performed at 35°C. Data from a representative experiment are shown. Replicates and statistics are available in Supplementary Table 2. In the oxidative stress experiments (D and G) worms were exposed to 7.5 mM TBHP. A representative experiment is shown. See Supplementary Table 3 for replicates and statistics. (H) Overexpression of rheb-1 suppresses cgef-1(RNAi) longevity. Combined data from two experiments are shown. Statistics are available in Table 1. The effectiveness of knockdown of cgef-1 and rheb-1 by RNAi was analyzed by feeding the RNAi plasmid to transgenic strains expressing a translational fusion of GFP to cgef-1 or rheb-1 respectively (Supplementary Figure 3).
Table 1: Lifespan analyses
Strain |
RNAi |
mean lifespan ± SEM |
median |
75 %ile |
P value vs N2 |
P value vs control |
% change |
N |
No of exp |
Figure |
|---|---|---|---|---|---|---|---|---|---|---|
N2 |
17.6 ± 0.2 |
17 |
20 |
216/235 |
3 |
1B |
||||
cgef-1(gk261) |
19.9 ± 0.2 |
20 |
22 |
< 0.0001 |
+13 |
185/227 |
3 |
1B |
||
N2 |
control |
18.5 ± 0.2 |
18 |
21 |
216/231 |
3 |
S2A |
|||
N2 |
cgef-1 |
21.0 ± 0.3 |
21 |
23 |
< 0.0001 |
+14 |
198/219 |
3 |
S2A |
|
N2 |
control |
17.3 ± 0.3 |
18 |
20 |
136/142 |
2 |
1E |
|||
cgef-1(gk261) |
control |
19.5 ± 0.3 |
19 |
22 |
< 0.0001 |
+13 |
133/144 |
2 |
1E |
|
N2 |
rheb-1 |
19.2 ± 0.3 |
19 |
21 |
< 0.0001 |
+11 |
133/152 |
2 |
1E |
|
cgef-1(gk261) |
rheb-1 |
19.2 ± 0.3 |
19 |
22 |
< 0.0001 |
nsa; nsb |
+11 |
135/157 |
2 |
1E |
Ex[unc-119] |
control |
23.4 ± 0.3 |
24 |
26 |
130/138 |
2 |
1H |
|||
Ex[rheb-1::GFP, unc-119(+)] |
control |
18.1 ± 0.5 |
17 |
22 |
< 0.0001 |
-23 |
110/126 |
2 |
1H |
|
Ex[unc-119] |
cgef-1 |
25.2 ± 0.4 |
26 |
28 |
< 0.001 |
+7 |
128/142 |
2 |
1H |
|
Ex[rheb-1::GFP, unc-119(+)] |
cgef-1 |
18.3 ± 0.4 |
18 |
20 |
< 0.0001 |
nsi |
-22 |
105/126 |
2 |
1H |
N2 |
control |
17.8 ± 0.2 |
18 |
20 |
216/224 |
3 |
2A, B |
|||
cgef-1(gk261) |
control |
20.2 ± 0.2 |
20 |
22 |
< 0.0001 |
+13 |
205/219 |
3 |
2A, B |
|
N2 |
daf-15 |
21.1 ± 0.2 |
21 |
23 |
< 0.0001 |
+19 |
211/237 |
3 |
2B |
|
cgef-1(gk261) |
daf-15 |
21.7 ± 0.3 |
22 |
25 |
< 0.0001 |
nsc |
+22 |
205/230 |
3 |
2B |
N2 |
let-363 |
21.9 ± 0.3 |
22 |
25 |
< 0.0001 |
+23 |
210/238 |
3 |
2A |
|
cgef-1(gk261) |
let-363 |
21.7 ± 0.3 |
22 |
25 |
< 0.0001 |
nsd |
+22 |
208/233 |
3 |
2A |
N2 |
control |
20.1 ± 0.2 |
20 |
23 |
195/219 |
3 |
3A, B, C |
|||
N2 |
cgef-1 |
23.5 ± 0.3 |
24 |
26 |
< 0.0001 |
+17 |
186/217 |
3 |
3A, B, C |
|
skn-1(zu67) |
control |
15.5 ± 0.2 |
15 |
17 |
< 0.0001 |
−23 |
160/199 |
3 |
3B |
|
skn-1(zu67) |
cgef-1 |
17.9 ± 0.2 |
18 |
20 |
< 0.0001 |
< 0.0001e; < 0.0001h |
−11 |
163/204 |
3 |
3B |
daf-16(mgDf47) |
control |
18.0 ± 0.3 |
18 |
21 |
< 0.0001 |
−10 |
186/218 |
3 |
3A |
|
daf-16(mgDf47) |
cgef-1 |
17.8 ± 0.3 |
18 |
21 |
< 0.0001 |
nsf |
−11 |
175/214 |
3 |
3A |
daf-16;skn-1 |
control |
14.3 ± 0.2 |
14 |
16 |
< 0.0001 |
−29 |
162/205 |
3 |
3C |
|
daf-16,skn-1 |
cgef-1 |
14.4 ± 0.2 |
15 |
17 |
< 0.0001 |
nsg |
−28 |
183/207 |
3 |
3C |
These combined results were derived from individual experiments described in Supplementary Table 1. Lifespan extensions correspond to parallel N2 control experiments. N represents total number of animals dying of old age versus those in total experiment. SEM = standard error of the mean. ns = not significant.
p-values (log-rank test) refer to the following control experiments:
a cgef-1(gk261);control(RNAi),
b rheb-1(RNAi),
c daf-15/Raptor(RNAi),
d let-363/CeTOR(RNAi),
e skn-1(zu67);control(RNAi),
f daf-16(mgDf47);control(RNAi),
g daf-16(mgDf47);skn-1(zu67);control(RNAi),
h cgef-1(RNAi),
i Ex[rheb-1::GFP;unc-119(+)];control(RNAi).
cgef-1 exists in three isoforms with a common C-terminus and different N-termini (Supplementary Figure1A). To analyze the expression pattern of these different cgef-1 isoforms we used GFP reporter fusions. Pcgef-1::GFP was broadly expressed in many tissues including the intestine, the vulva area, and some of the head and tail neurons (Supplementary Figure 1B). This expression pattern resembled the expression of rheb-1::GFP (Supplementary Figure 1C) and let-363/CeTOR::GFP (Supplementary Figure 1D) supporting the idea that RHEB-1 and CGEF-1 might interact to regulate mTORC1 in C. elegans. We also tested whether cgef-1 is critical for the expression of rheb-1. No obvious alteration of RHEB-1::GFP was observed when cgef-1 was knocked down by RNAi suggesting that cgef-1 does not influence the expression of rheb-1 (Supplementary Figure 1C).
We hypothesized that CGEF-1 may function in RHEB-1 signaling. If CGEF-1 acts with RHEB-1 in a common pathway one would expect to observe similar phenotypes when inhibiting either cgef-1 or rheb-1 function. As knockdown of rheb-1 results in extension of lifespan and improved stress tolerance [16], we investigated the behavior of a cgef-1 mutant in lifespan and stress assays. The cgef-1 mutant gk261 is a reported null allele that bears a deletion of 318 bp disrupting the GEF domain [24] (Supplementary Figure 1A). Mutation of cgef-1 led to a moderate but significant increase of lifespan compared to wild type (+13 % lifespan extension) (Figure 1B and Table 1). This effect was similar to what we observed when rheb-1 was knocked down by RNAi (+11 %; Table 1). cgef-1 mutants also displayed enhanced resistance towards heat (Figure 1C) and the oxidizing agent tert-butyl hydroperoxide (TBHP) (Figure 1D). To exclude developmental effects of cgef-1 inactivation on lifespan and stress phenotypes, we tested whether inhibition of cgef-1 function by RNAi only during adulthood affects longevity. Indeed, adulthood cgef-1(RNAi) resulted in extended lifespan (Supplementary Figure 2A and Table 1) and enhanced heat and oxidative stress tolerance compared to control (Supplementary Figure 2B and 2C).
To test for a genetic interaction between cgef-1 and rheb-1, we analyzed the effect of rheb-1 knockdown in cgef-1 mutants. We found that rheb-1(RNAi) did not further increase the longevity of cgef-1(gk261) mutants (Figure 1E and Table 1). Moreover, heat as well as oxidative stress resistance of cgef-1 mutants was not further enhanced by rheb-1(RNAi) (Figure 1F and 1G). These findings indicate that cgef-1 and rheb-1 are epistatic. Additionally, we found that the lifespan extending effect associated with inhibition of cgef-1 could be suppressed by overexpression of rheb-1 (Figure 1H). Together, our data suggest that cgef-1 functions in the same pathway with rheb-1 to regulate lifespan and stress response in C. elegans.
CGEF-1 promotes mTORC1 signaling
Next, we assessed the genetic interaction between cgef-1 and mTORC1 signaling. We used RNAi to knock down the mTOR kinase let-363 and the mTORC1-specific gene daf-15/Raptor in cgef-1 mutants and wild-type animals. Knockdown of each mTORC1 gene that we examined strongly increased lifespan in wild type (Figure 2A and 2B, Table 1). Mutation of cgef-1 did not further enhance the longevity. Notably, the mean lifespan associated with let-363/CeTOR(RNAi) and daf-15/Raptor(RNAi) was similar in wild-type animals and cgef-1 mutants (Figure 2A and 2B). Thus, we conclude that cgef-1 acts in the same pathway with mTORC1 to regulate longevity. mTORC1 is also important for growth and development. While mutation of cgef-1 had no effect on body size (Supplementary Figure 4A), inactivation of cgef-1 led to a reduced brood size (Supplementary Figure 4B).
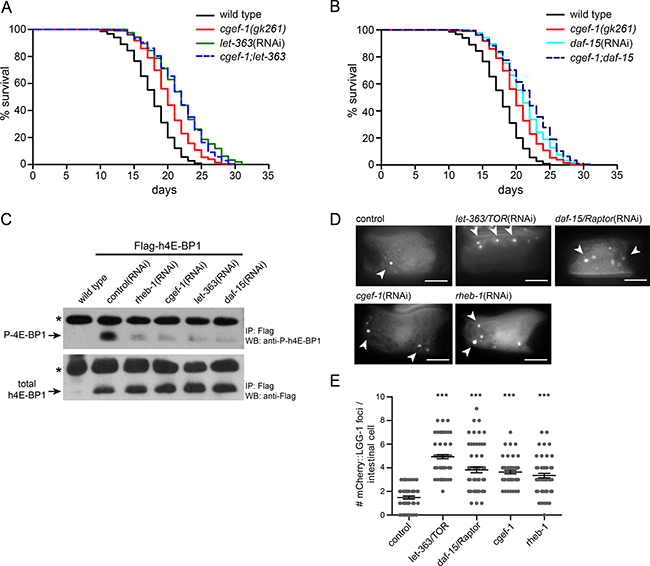
Figure 2: cgef-1 functions in the mTORC1 signaling pathway.(A) Longevity of let-363/CeTOR(RNAi) was not affected by the cgef-1(gk261) mutation. (B) Survival of daf-15/Raptor(RNAi) was not altered by cgef-1(gk261) mutation. For lifespan analysis (in A and B) cgef-1(gk261) and wild-type N2 worms were fed with let-363/CeTOR, daf-15/Raptor, or control(RNAi). Survival plots represent the combined data from three experiments. See Table 1 for statistical analyses. (C) CGEF-1 activates mTORC1 to phosphorylate the elongation factor 4E-BP1. Phosphorylation of transgenically expressed human 4E-BP1 was used to assess the activity of mTORC1 signaling. Worms carrying the Pges-1::Flag-h4E-BP1 transgene were subjected to RNAi treatment as indicated. After immunoprecipitation (IP) with anti-Flag antibody the Flag-tagged h4E-BP1 was detected by western blot (WB) analysis (lower panel). Phosphorylation of h4E-BP1 was analyzed using anti-P-4E-BP antibody (upper panel). Star indicates antibody light chains. (D) Autophagy is increased after cgef-1 and mTORC1-pathway gene knockdown. Representative pictures of L4 animals expressing mCherry::LGG-1 in the intestine. In control animals mCherry::LGG-1 is diffusely distributed in the cytosol. After inhibition of cgef-1, rheb-1, let-363/CeTOR, and daf-15/Raptor by RNAi an induction of mCherry::LGG-1-positive foci was observed (arrowheads). Scale bar represents 10 μm. (E) Quantification of mCherry::LGG-1 foci in intestinal cells in L4 larvae. Individual data points are plotted to illustrate variability. Bars indicate the mean and SEM for each genotype. n > 25 animals per each condition. ***p < 0.001 versus control, ANOVA.
In mammals, mTORC1 signaling promotes growth by increasing protein synthesis through phosphorylation of 4E-BP and S6K and by inhibiting catabolic processes such as autophagy [10]. Monitoring the phosphorylation state of the mTORC1 substrate 4E-BP has been previously utilized to characterize mTORC1 activity in C. elegans [25]. Here, we took advantage of this readout and expressed human 4E-BP1 (h4E-BP1) tagged with Flag under control of the ges-1 promoter in the intestine of C. elegans. We observed that let-363/CeTOR(RNAi) and daf-15/Raptor(RNAi) knockdown almost completely abolished the phosphorylation of h4E-BP1 (Figure 2C) indicating that human 4E-BP1 serves as substrate for C. elegans mTORC1. Interestingly, RNAi against cgef-1 and rheb-1 mimicked these effects and blocked h4E-BP1 phosphorylation to the same level as caused by let-363(RNAi) or daf-15(RNAi) (Figure 2C). These results indicate that CGEF-1 serves as an upstream activator for mTORC1 signaling.
We next wondered whether cgef-1 also regulates autophagy, another mTORC1 downstream process. LGG-1, the worm ortholog of the yeast Atg8 and mammalian MAP1-LC3, localizes to autophagosomal membranes and has been widely deployed as an indicator of autophagy in C. elegans [22, 26]. Here, we used a reporter strain expressing mCherry-tagged LGG-1 in the intestine [27]. Upon induction of autophagy, mCherry::LGG-1 changes its diffuse cytoplasmic distribution pattern to punctate structures reflecting LGG-1 sequestration to autophagosomal membranes. In agreement with published data [16, 22], we observed a significant increase of mCherry::LGG-1 positive foci in intestinal cells in let-363/CeTOR(RNAi) and daf-15/Raptor(RNAi) treated animals compared to wild type (Figure 2D and 2E). An induction of mCherry::LGG-1 foci was also observed upon rheb-1 and cgef-1(RNAi) knockdown indicating an increase in autophagy levels (Figure 2D and 2E). Taken together, we conclude that CGEF-1 associates with RHEB-1 and activates mTORC1 signaling to inhibit autophagy in C. elegans.
Lifespan extension and enhanced stress resistance of cgef-1 require the function of the transcription factors DAF-16/FoxO and SKN-1/Nrf2
Two transcription factors, DAF-16 and SKN-1, have been implicated in the prolonged lifespan that results from genetic inhibition of mTORC1 [16]. To test the roles of daf-16 and skn-1 in cgef-1-mediated longevity we analyzed the lifespan of skn-1 and daf-16 mutants upon cgef-1(RNAi) knockdown. We found that mutation of daf-16 strongly suppressed the lifespan extension associated with inhibition of cgef-1 (Figure 3A and Table 1). Inactivation of cgef-1 by RNAi, however, still increased the lifespan in skn-1(zu67) mutants (Figure 3B and Table 1). A daf-16;skn-1 double mutant completely eliminated the increase in lifespan deriving from cgef-1(RNAi) (Figure 3C and Table1). We conclude that DAF-16 profoundly contributes to the increases in longevity that are associated with reductions in cgef-1 activity, while SKN-1 is not required.
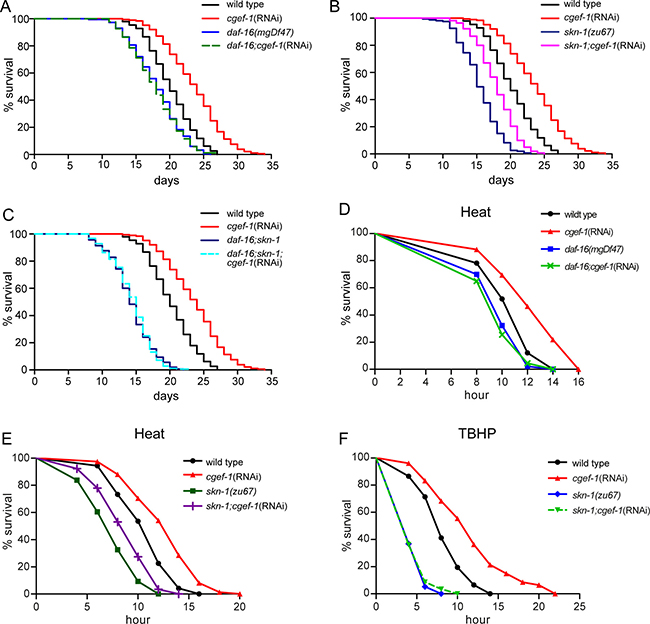
Figure 3: cgef-1 regulates lifespan and stress response through the transcription factors DAF-16/FoxO and SKN-1/Nrf2.(A) Lifespan extension deriving from loss of cgef-1 function requires daf-16 function. (B) cgef-1(RNAi) extends lifespan in skn-1 mutants. (C) Lifespan of daf-16;skn-1 double mutants is unaffected by cgef-1(RNAi). In the lifespan experiments shown in (A–C), wild type N2, daf-16(mgDf47), skn-1(zu67), or daf-16(mgDf47);skn-1(zu67) mutants were fed with cgef-1(RNAi) or control(RNAi). Survival plots show composites from three experiments. See also Table 1 for statistics. (D) cgef-1(RNAi) increases resistance to heat (35°C) dependent upon daf-16 function. (E) skn-1 is not required to protect against heat stress. (D-E) Wild type N2, daf-16(mgDf47), or skn-1(zu67) mutants worms were fed with cgef-1(RNAi) or control(RNAi). Data from a representative experiment are shown in which animals were exposed to 35°C. Replicates and statistics are presented in Supplementary Table 2. (F) The oxidative stress resistance phenotype (TBHP) of cgef-1(RNAi) requires skn-1 function. Wild type N2 and skn-1(zu67) mutants fed with cgef-1(RNAi) or control(RNAi) were exposed to 7.5 mM TBHP. A representative experiment is shown. See Supplementary Table 3 for replicates and statistics.
Next, we tested whether the cgef-1 phenotype of elevated stress resistance require daf-16 and skn-1. We observed that the increased resistance of cgef-1(RNAi) to heat stress was completely abolished by mutation of daf-16 (Figure 3D), while mutation of skn-1 only partially prevented the thermo-tolerance (Figure 3E). Consistent with the role of skn-1 in phase 2 detoxification, mutation of skn-1 dramatically suppressed the increases in oxidative stress resistance seen with cgef-1(RNAi) (Figure 3F). These data suggest that the increases in heat stress resistance resulting from cgef-1 knockdown are predominantly mediated by DAF-16, while SKN-1 is essential under TBHP oxidative stress conditions.
We wondered whether the transcription activities of SKN-1 and DAF-16 are actually controlled by cgef-1. DAF-16 and SKN-1 orchestrate a well described gene expression program [28, 29]. When mTORC1 components are knocked down by RNAi, several DAF-16 and SKN-1 targets are upregulated [16]. Accordingly, we observed that the transcriptional reporter Psod-3::GFP, in which the promoter of the conserved DAF-16/FoxO target superoxide dismutase (sod-3) is fused to GFP, was activated in the intestine through cgef-1(RNAi) (Figure 4A). We also analyzed the effects on endogenous DAF-16 regulated gene expression in cgef-1 mutants using quantitative PCR. We assayed mRNA production from the well-characterized DAF-16 targets sod-3, mtl-1, and hsp-12.6 [29]. cgef-1 mutation increased the expression of sod-3 and mtl-1, but not the small heat shock gene hsp-12.6 (Figure 4B). We conclude that, when cgef-1 is inactivated, DAF-16 induces a protective transcriptional response.
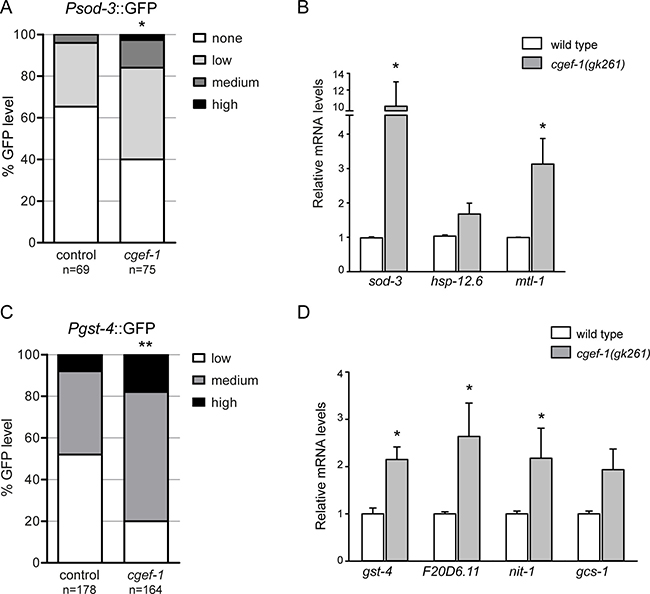
Figure 4: Inhibition of cgef-1 induces DAF-16 and SKN-1-mediated transcription.(A) Analysis of Psod-3::GFP expression after exposure to cgef-1(RNAi) or control(RNAi) in adult worms. Induction of sod-3 expression was quantified as described (see Experimental Procedures). p-values were calculated by the Chi-square-test. *p < 0.05. n, number of animals analyzed. Pooled data from three experiments. (B) Induction of endogenous DAF-16 target gene expression in cgef-1(gk261) mutants analyzed by qPCR. Data are presented as fold change compared to wild type averaged from three independent experiments, error bars represent SEM. p-values were derived from a Student’s t-test. *p < 0.05. (C) Pgst-4::GFP expression is induced in response to cgef-1(RNAi) knockdown. Induction of the Pgst-4::GFP reporter was quantified as described (see Experimental Procedures). p-values were calculated by the Chi-square-test. **p < 0.01. n, number of animals analyzed. Pooled data from three experiments. (D) Activation of endogenous SKN-1 target genes in cgef-1(gk261) mutants analyzed by qPCR. Data are mean ± SEM. p-values were derived from a Student’s t-test. *p < 0.05.
We further investigated whether cgef-1 also regulates the transcriptional activity of SKN-1. We examined how cgef-1(RNAi) affected the expression of the conserved SKN-1 target glutathione S-transferase gst-4 using a transcriptional Pgst-4::GFP reporter. In the intestine, Pgst-4::GFP was expressed at low levels under normal conditions, but was strongly upregulated by inhibition of cgef-1 through RNAi (Figure 4C). We also analyzed effects on endogenous SKN-1 regulated gene expression focusing on a set of target genes involved in various stress processes, such as gst-4, gcs-1, nit-1, and oxidoreductase F20D6.11 [28]. We observed that the expression of all four genes was increased in cgef-1 mutants (Figure 4D). Taken together, these data indicate that both SKN-1 and DAF-16 induce protective gene expression when cgef-1 is inactivated.
Dbl interacts with human Rheb and activates mTORC1
C. elegans CGEF-1 displays the N- to C-terminal Dbl-homology (DH) and pleckstrin-homology (PH) domain structure shared by the Dbl family of GEFs (Figure 5A) [30]. A database search identified human Dbl (also called MCF2) as potential ortholog of C. elegans CGEF-1. C. elegans CGEF-1 showed 30 % identity to the human Dbl protein. Dbl is a prototype member of GEFs that stimulate the activation of GTPases from the Rho family [31]. Mutations of the dbl gene leading to deregulated GEF activity and persistent stimulation of the GTPase substrate are associated with cancer. Oncogenic activation of the dbl proto-oncogene involves the loss of the amino-terminal half of the protein giving rise to a constitutively active GEF with potent transforming potential (Figure 5A) [32]. Given the homology and structural similarity, we analyzed whether Dbl can also interact with human Rheb (hRheb) by co-immunoprecipitation from HEK 293T cells. hRheb specifically precipitated both, the full-length proto-oncogenic form of Dbl (proto-Dbl) and the truncated oncogenic form of Dbl (onco-Dbl) (Figure 5B).
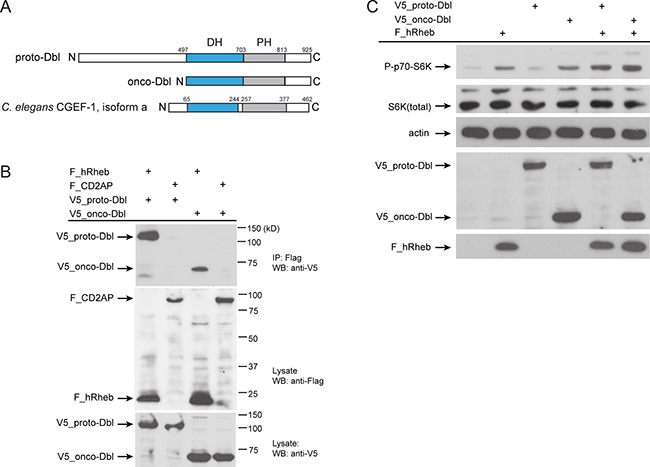
Figure 5: Human Dbl interacts with Rheb-1.(A) Schematics of the proto-Dbl, onco-Dbl and the CGEF-1 protein domain structure. The Dbl homology (DH) domain is shaded blue, the PH pleckstrin homology (PH) domain grey. Oncogenic activation of proto-Dbl occurs through truncation of the N-terminal 497 residues. The C-terminal half of Dbl includes the DH domain and PH domain which constitutes the minimum module essential for cell transformation. (B) Dbl proteins interact with human Rheb (hRheb) in HEK293T cells. HEK293T cells expressing V5-tagged proto-Dbl or onco-Dbl and Flag-tagged hRheb-1 or CD2AP as indicated were subjected to immunoprecipitation (IP) with anti-Flag antibody. Association of V5-Dbl with Flag-hRheb was determined by western blot (WB) analysis with anti-V5 antibody (upper panel). The control protein CD2AP failed to bind Dbl. Middle part shows expression of Flag-tagged proteins, the lower panel shows the expression of V5-tagged proto- and onco-Dbl in cell lysates. kD, kiloDalton. (C) Onco-Dbl but not proto-Dbl induces S6K phosphorylation in HEK293T cells. V5-tagged proto-Dbl or onco-Dbl and Flag-tagged hRheb were co-expressed in transiently transfected HEK 293T cells. Cell lysates were analyzed by immunoblotting with the indicated antibodies. Anti-actin was used to verify equivalent input of total cellular protein.
Next, we tested whether Dbl can promote mTORC1 signaling. Therefore epitope-tagged hRheb, proto-Dbl, and onco-Dbl were expressed in HEK cells and the phosphorylation level of the mTORC1 substrate S6 Kinase was evaluated. Previous studies have shown an increase in the phosphorylation of S6 Kinase when Rheb was overexpressed in HEK293 cells [3, 4]. Accordingly, we observed increased S6 kinase phosphorylation by overexpression of hRheb (Figure 5C). Intriguingly, overexpression of onco-Dbl, but not the full length proto-Dbl resulted in enhanced phosphorylation of S6 kinase (Figure 5C). Co-expression of hRheb together with onco-Dbl further increased S6K phosphorylation levels. These observations suggest that Dbl can associate with Rheb to activate the mTORC1 pathway and this interaction is conserved between C. elegans and humans.
DISCUSSION
Our C. elegans and biochemical experiments have identified the guanine nucleotide exchange factor CGEF-1 as a novel regulator of the mTORC1 pathway. CGEF-1 directly binds to RHEB-1 and stimulates mTORC1 signaling activity to mediate stress responses and aging. Absence of cgef-1 results in longevity similar to deficiency of mTORC1. Previous studies uncovered effects of mTORC1 inhibition that may enhance lifespan and stress tolerance including decreased protein synthesis, increased autophagy, and induction of a protective gene expression program by the transcription factors DAF-16/FoxO and SKN-1/Nrf2 [16, 19, 20, 23]. Intriguingly, we found that cgef-1 involves these mTORC1-associated mechanisms. The finding that human Dbl also associates with Rheb and regulates well-established mTORC1 targets indicates a conserved function of CGEF-1/Dbl proteins in invertebrates and in mammals.
mTOR signaling is evolutionarily highly conserved and has a fundamental role in metabolism, cell growth, and aging [1, 2]. Genetic evidence from several model organisms established Rheb as an indispensable upstream activator of mTORC1 signaling by growth factors and amino acids [33]. Thus C. elegans RHEB-1 served as an attractive bait protein in our yeast-two hybrid screen for mTORC1 regulators. Thereby, we identified and verified CGEF-1 as a novel binding partner of RHEB-1 (Figure 1A). Dbl, the mammalian ortholog of C. elegans CGEF-1, is also associated with human Rheb (Figure 5B), indicating that the interaction is conserved across species. cgef-1 encodes a guanine nucleotide exchange factor and has previously been involved in the activation of the GTPase CDC-42 during cell polarization and asymmetric division in the C. elegans embryo [24, 30]. Dbl was initially identified as an oncogene in mammalian cells causing tumorgenic growth. Dbl proteins function as guanine nucleotide exchange factors for Rho family GTPases facilitating the exchange of GDP to GTP and thus stimulating the active GTP-bound state. Dbl family proteins share a structural tandem module of DH and PH domains responsible for activation of Rho [31]. Dbl proteins activate Rho GTPases in diverse biological processes, including actin cytoskeletal dynamics, cell growth, proliferation, and survival. Aberrant function of Dbl proteins has been implicated in malignant transformation, invasion and metastasis [31].
Based on the initial finding that CGEF-1 directly binds to RHEB-1 we have been able to show that cgef-1 affects mTORC1 signaling in vivo. The expression pattern of cgef-1 in C. elegans substantially overlaps with rheb-1 and let-363/CeTOR (Supplementary Figure 1B–1D). Interestingly, cgef-1 is strongly expressed in the intestine where mTOR activity is required for adaptation to environmental changes [16]. cgef-1 mutants and also cgef-1 knockdown by RNAi during adulthood shared the longevity and stress phenotypes associated with loss of rheb-1, let-363/CeTOR kinase and daf-15/Raptor function. Moreover, the longevity produced by inhibition of rheb-1 and mTORC1 was not further increased by cgef-1 mutation (Figure 1E–1G, and Figure 2A–2B), and overexpression of rheb-1 completely suppressed cgef-1 associated longevity (Figure 1H). Together, our observations are consistent with a role of CGEF-1 upstream in the Rheb/mTORC1 pathway. Of note, cgef-1 mutants exhibit only mild developmental phenotypes compared to loss of rheb-1, let-363/CeTOR, and daf-15/Raptor function (Supplementary Figure 4). Absence of rheb-1 or mTORC1 results in arrest at the L3 larval stage [12, 13, 15] while cgef-1 mutants display no severe embryonic lethality or larval developmental defects. The cgef-1(gk261) deletion destructs the DH domain essential for GEF function and therefore was previously characterized as strong loss of function or null allele [24]. However, cgef-1(gk261) does not completely eliminate CDC-42 activity during asymmetric cell division in the embryo indicating the presence of alternative factors [24, 30]. Indeed, a large number of GEFs is present in C. elegans [34] and other animals, and redundant activation by multiple GEFs may be a common theme [35]. Thus, a more complex and partially redundant GEF network might control mTORC1 activity.
We further implicated CGEF-1 in the regulation of mTORC1 and showed that suppression of cgef-1 caused inactivation of mTORC1 downstream signaling. Active mTORC1 regulates a set of well-characterized targets including S6 kinase and the translational repressor 4E-BP1 to stimulate protein synthesis, and inhibits catabolic processes like autophagy [10]. Knockdown of cgef-1 in C. elegans, similarly to inhibition of rheb-1 and mTORC1 gene function, reduced the phosphorylation of human 4E-BP1 (Figure 2C). In mammals, 4E-BP1 is phosphorylated in response to various signals resulting in its dissociation from the translation initiation factor 4E (eIF4E) and activation of cap-dependent mRNA translation. Consistent with mTORC1 signaling being downregulated by loss of cgef-1 function, we also observed increased LGG-1 positive autophagic vesicles in the intestine (Figure 2D, 2E). Together, our data show that CGEF-1 engages mTORC1-associated mechanisms and therefore we link CGEF-1 with RHEB-1 as regulator of mTORC1 signaling.
Studies in several organisms demonstrate that autophagy and translational processes are central effector mechanisms of mTORC1 on aging [19, 23, 36]. Moreover, mTORC1 signaling influences longevity by opposing the transcription factors DAF-16/FoxO and SKN-1/Nrf2 in C. elegans [14, 16]. When the mTORC1 pathway is inhibited, DAF-16 and SKN-1 activate a protective gene expression response, and increase stress resistance and longevity. We observed that mutation of daf-16 completely suppressed the extended lifespan of cgef-1(RNAi) (Figure 3A). cgef-1(RNAi), however, increased lifespan modestly, but reproducibly, in wild-type animals and skn-1 mutants (Figure 3B). These observations indicate that longevity upon loss of cgef-1 involves DAF-16, but not SKN-1. Along that line, DAF-16 was also critical for thermo-tolerance associated with inactivation of cgef-1 while SKN-1 played a less important role (Figure 3D, E). Notably, it was shown in a previous study that delays in aging resulting from mTORC1 inhibition by knockdown of the RAG GTPases ragc-1 and raga-1 required both DAF-16 and SKN-1 [16]. Perhaps DAF-16 and SKN-1 are affected differently by these regimens. The enhanced oxidative stress resistance derived from cgef-1(RNAi), however was completely abolished in skn-1 mutants suggesting that SKN-1 is the central player for phase 2 detoxification and adaptation to oxidative stress (Figure 3F). Inactivation of cgef-1 resulted in upregulation of a set of genes (Figure 4) for which the expression has been shown to strongly depend on DAF-16 and SKN-1 [29, 37]. Together, these findings support the notion that the transcriptional responses of DAF-16 and SKN-1 are critical for cgef-1 mediated longevity and protection from environmental stress, as was observed previously for genetic mTORC1 inhibition [16].
Our observations would provide a model whereby CGEF-1 stimulates mTORC1 signaling to regulate key substrates for protein synthesis, autophagy and longevity in C. elegans. How could CGEF-1 promote mTORC1 signaling? It is well established that Rheb plays a crucial role in the activation of mTORC1 upon growth factor or amino acid stimulation [33]. One might speculate that CGEF-1 straightly activates RHEB-1 to promote mTORC1 signaling. Rheb is negatively controlled by the TSC complex that functions as GAP promoting conversion of active Rheb to the inactive form. This leads to decreased mTORC1 activity [5, 38]. No obvious homologs of the TSC complex have been identified in the nematode. Interestingly, it was described that RalGAP (HGAP-1 and HGAP-2) shares similarity with TSC and loss of hgap-1/hgap-2 phenocopied mTORC1 activation and decreased lifespan in C. elegans, consistent with a TSC-like function [39]. The role of GEF molecules that counter the activity of the TSC in the control of Rheb remains so far ill-defined. Studies in Drosophila identified the translationally controlled tumor protein (TCTP) functioning as GEF to activate Rheb and implicated TCTP in the mTORC1 pathway [40]. Yet, following studies reported that TCTP does not affect mTORC1 signaling in mammals [41, 42]. Hence, so far, no positive regulator of Rheb activity has been identified in the mammalian system. Our study now proposes CGEF-1 as a new activator of RHEB-1/mTORC1 signaling. Genetic analyses establish that cgef-1 is epistatic to rheb-1 and functions upstream of mTORC1 in the control of aging. Biochemical experiments show that CGEF-1 and RHEB-1 form a complex together (Figure 1A). Unfortunately, we have not detected direct in vitro GEF activity towards RHEB-1 due to technical problems of purifying functional CGEF-1 protein. Notably, CGEF-1 has been described as GEF and activator of the Rho GTPase CDC-42 [24, 30]. Yet, multiple lines of evidence indicate that some GEFs can target and activate multiple GTPases [reviewed in 31]. An alternative model is that the ability of CGEF-1 to regulate mTORC1 activity is rather indirect. CGEF-1 might facilitate RHEB-1/mTORC1 complex formation and thereby promote mTORC1 signaling.
Strikingly, the regulation of mTORC1 signaling by CGEF-1/Dbl proteins appears to be conserved in evolution. First, we observed that human Dbl can engage complexes with Rheb (Figure 5B). The oncogenic form of Dbl bearing the DH-PH domains that promote nucleotide exchange in vivo was sufficient to bind hRheb (Figure 5B). Next, overexpression of onco-Dbl, just as Rheb in cell culture, strongly increased the phosphorylation level of S6K (Figure 5C) supporting the hypothesis that Dbl stimulates mTORC1 activity. It will be of great importance to elucidate how Dbl/CGEF-1 influences mTORC1-mediated mechanisms and longevity across species.
MATERIALS AND METHODS
C. elegans growth conditions
Unless otherwise indicated C. elegans were cultured at 20°C on standard NGM plates seeded with E. coli OP50. Nematode strains used in this study are described in Supplementary Table 4.
RNA interference
RNAi experiments were carried out as described [43]. HT115 bacteria transformed with RNAi clones or empty vector pL4440 were grown over night in 12.5 μg/ml tetracycline and 50 μg/ml ampicillin. The following day, cultures were diluted 1:10 and grown to an OD600 of 0.8-1.0 and induced with 0.7 mM IPTG. This culture was used to seed NGM plates containing tetracycline, ampicillin, and 1 mM IPTG. RNAi plasmids for knockdown of cgef-1 and rheb-1 were derived from the ORF-RNAi library. skn-1(RNAi) plasmid was obtained from K. Blackwell [37] and let-363(RNAi) plasmid was previously described [13]. For daf-15(RNAi) partial cDNA was cloned into pPD129.36. All clones were confirmed by sequencing.
Lifespan analysis
Lifespan assays were performed at 20°C and measured from L4. At L4 stage, worms were transferred to NGM plates containing 50 μM FUdR or RNAi plates with FUdR. Animals were scored daily for surviving. Animals that crawled off the plate, ruptured, or showed internally hatched larvae were censored. Data were analyzed using a log-rank test with Holm-Sidak method performed with SigmaStat 3.5.
Plasmids and transgenic strains
We generated a rheb-1::gfp translational fusion by inserting a SalI/BamHI fragment containing the complete genomic rheb-1 locus and 2.8 kb upstream regulatory sequence into pPD95.75 (A. Fire, Carnegie Institute of Washington, Baltimore, MD). To generate animals overexpressing rheb-1, the rheb-1::gfp plasmid (ID8424) was injected at 10 ng/μL into unc-119(ed4) mutants along with unc-119 wild-type rescuing plasmid (50 ng/μL).
For the analysis of 4E-BP phosphorylation in C. elegans, Flag-tagged human 4E-BP1 cDNA was cloned under control of 2.5 kb ges-1 promoter into pPD30.38 (ID8132). Transgenic strains expressing h4E-BP1 were created by injection of the Pges-1::Flag_h4E-BP1 plasmid at 10 ng/μl along with 20 ng/μl pRF4 (rol-6) in N2. Extrachromosomal arrays of Flag_h4E-BP1 were integrated into the genome by γ-irradiation. The integrated lines were backcrossed at least four times with wild type N2.
We generated cgef-1::gfp translational fusion by inserting a 1386 bp fragment of cgef-1a cDNA under control of 2280 bp ges-1 promotor region into pPD95.75. Pges-1::cgef-1::GFP plasmid (ID9096) was injected at 20 ng/μl into wild-type N2 animals along with the coinjection marker 20 ng/μl pRF4 (rol-6) to generate animals expressing cgef-1::GFP.
Analysis of brood size
Animals were synchronized by timed egg laying on OP50 or RNAi and grown to the L4 stage at 20°C. To assess the brood size, individual animals were transferred every other day onto plates until egg-laying ceased and their total number of progeny was counted.
Analysis of body size
Animals were raised at 20°C and fed with RNAi for two generation. F1 generation was synchronized by egg laying and after 72 hours, worms were anaesthetized with Tetramisolhydrochloride (2mM) and images were taken at 5x magnification. To measure the body size, the perimeter was traced and the area was calculated using the software application AxioVision4 (Zeiss).
Microscopy and transgenic reporter scoring
Light microscopy was performed using a Zeiss Axioplan 2-microscope equipped with Nomarski differential interference contrast (DIC), an AxioCam-camera and the AxioVision-Software Rel.4.8. GFP was detected using appropriate EGFP-filter sets (480/20 nm excitation, 510/20 nm emission). Images were processed and figures compiled with Adobe Illustrator CS5.
Intestinal Pgst-4::GFP expression was scored as essentially published [16, 21]. Worms were raised on cgef-1(RNAi) or control(RNAi) until L4 stage, then placed on slides on 2% agarose pads and stunned by levamisole. “High” refers to high GFP levels anteriorly and throughout most of the intestine, “medium” refers to intense GFP levels only anteriorly or posteriorly, and “low” refers to no or only barely visible GFP expression. Chi-square tests were performed with SigmaStat 3.5.
Intestinal Psod-3::GFP expression was scored based on the intensity of GFP expression as described [44]. Worms were raised on cgef-1 (RNAi) or control (RNAi) until young adult. “High” refers to bright GFP expression throughout most of the intestine, “medium” refers to GFP expression only anteriorly or posteriorly, “low” refers to weak visible GFP expression, and “no” indicates barely detectable GFP expression. Chi-square tests were performed with SigmaStat 3.5.
GFP intensity of animals expressing cgef-1::GFP and rheb-1::GFP was analyzed with a Zeiss Axiovert 200 M microscope and AxioCam camera. GFP fluorescence intensity of the images was measured with ZenBlue2011. Background intensity was subtracted. Fluorescence intensity of the wild type was set to 100%.
Analysis of autophagy in C. elegans
The level of autophagy in intestinal cells was assessed using a Pnhx-2::mCherry::lgg-1 translational reporter characterized previously [27]. Animals were raised at 20°C and fed with RNAi until L4. L4 larvae were then scored for mCherry-positive puncta using an Axioplan 2 microscope at high magnification (630x). Foci were counted based on the intensity of mCherry expression in the proximal region of the intestine as described [27, 45]. Three independent biological samples were analyzed for autophagic events. ANOVA test was performed with SigmaStat 3.5.
Heat and oxidative stress resistance assays
To assess heat resistance, L4 stage worms were fed with RNAi bacteria for four days at 20°C. Worms were then shifted to 35°C and tested for survival. For oxidative stress resistance, worms that had been treated as described above were transferred to NGM plates that contained 7.5mM tert-Butyl hydroperoxide (TBHP) (Sigma-Aldrich) and were seeded with OP50. Animals were scored for surviving and worms that did not respond to gentle prodding with a platinum wire were scored as dead. Worms that crawled off the plate, ruptured, or died from internal hatching of progeny were excluded from the analysis. Survival plots and p-values (log-rank) were determined using SigmaStat software.
RNA Isolation and quantitative PCR
Total RNA was isolated from four day adult worms using TRI Reagent (Sigma-Aldrich) and a RNA clean and concentrator kit (Zymo Research Corp.). DNase treatment was performed using the on-column DNase digestion (Quiagen). To generate cDNA 1 μg of RNA was reverse transcribed with oligo-dT primer and the Superscript III Kit (Invitrogen).
Quantitative PCR reactions were performed with Roche LightCycler 480 using Takyon No Rox SYBR MasterMix dTTP Blue (Eurogentec). qPCR reactions were performed in at least three independent samples in triplicates. The quantification of changes in mRNA expression levels was based on the 2–ΔCt method [46]. Results were normalized against endogenous cdc-42 and Y45F10D.4 expression. Significant differences were assessed by Student’s unpaired t-test. Primer sequences are listed in Supplementary Table 6.
Analysis of h4E-BP phosphorylation in C. elegans
Worms carrying the Is[Pges-1::Flag-h4E-BP1] transgene (ENH441) were collected at young adult stage in IP lysis buffer containing 40 mM HEPES (pH7.5), 120 mM NaCl, 1 mM EDTA and 0.3 % CHAPS supplemented with 1 mM PMSF and protease inhibitor cocktail (Sigma), and dissolved by sonication (10-s sonication with a 10-s break, 10 cycles) and incubation with constant rotation for two hours at 4°C to fully lyse the worms. FLAG-tagged h4E-BP1 was immunoprecipitated with anti-FLAG M2 affinity beads (Sigma). Immunoprecipitates were washed with CHAPS-free IP lysis buffer three times at 4°C, and eluted by boiling with 2× SDS sample buffer at 95°C for 3 min. Immunoprecipitates were subjected to subsequent western blot analysis using anti-FLAG antibody (Sigma) and anti-phospho-4E-BP1 antibody (Cell Signaling).
Yeast split-ubiquitin two-hybrid interaction screen
The RHEB-1 protein interaction screen was performed using the yeast two-hybrid (Y2H) split-ubiquitin system as described before [47]. As bait the coding sequence of C. elegans rheb-1 was fused to the C-terminus of ubiquitin (Cub) in pPcup1-ubc9-CRU and used for transformation of the yeast strain JD53. A C. elegans Nub I-split ubiquitin cDNA library was used as prey. Positive clones were identified by sequencing and BLAST with the C. elegans genome.
Cell culture, immunoprecipitation, and immunoblotting
HEK293T cells were grown in DMEM with 10% FBS. Cells were transfected with plasmid DNA applying the calcium phosphate method and incubated for 24h. Cells were lysed in IP buffer (1% Triton-X 100, 20 mM Tris pH 7.5, 50 mM NaCl, 50 mM NaF, 15 mM Na4P2O7, 0.1 mM EDTA pH 8.0) supplemented with protease inhibitors. After centrifugation (43,000 rpm, 30 min, 4°C) the supernatants containing equal amounts of total protein were incubated with anti-FLAG M2 affinity beads (Sigma) for at least 1 h at 4°C. The precipitates were washed extensively with IP buffer and subjected to SDS-PAGE and immunoblotting analysis with anti-Flag and anti-V5 antibodies (Sigma).
For the analysis of S6K phosphorylation HEK293T cells were transfected with epitope-tagged Rheb and Dbl protein. 24h after transfection, cells were starved 1h in MEM, Hank’s Balanced Salts (from life technologies) and then lysed in IP buffer. Lysates were centrifuged (43,000 rpm, 30 min, 4°C). Equal amounts of protein samples were analyzed by immunoblotting. Anti-p70-S6K and anti-phospho-p70-S6K antibodies (Thr389) were obtained from Cell signaling, anti-actin antibodies from Sigma.
Expression constructs are described in detail in the Supplementary Table 5.
Author contributions
Y.L., S.F., A.G., A.T. and J.G. planned and conducted experiments, analyzed the results, and contributed to manuscript preparation. M.K. analyzed autophagy and helped with microscopy recordings. M.G. and J.K. planned and conducted experiments. T.J. contributed to experimentation. G.B. and K.T. supported manuscript preparation. G.W. and R.B. contributed to project guidance and manuscript preparation. E.N.-H. planned and guided the project, planned and analyzed the experiments, and wrote the manuscript.
ACKNOWLEDGMENTS
We thank M. Dottermusch and A. Schwierzok for expert technical assistance and R. Nitschke from the Life Imaging Center (University Freiburg) for support in image recording and analysis. We thank M. Messerschmid for help with the split-ubiquitin interaction screen. We would like to thank S. Takagi (Division of Biological Science, Nagoya University Graduate School of Science, Japan) for h4E-BP1 plasmids and A. Eva (Laboratorio di Biologia Molecolare, Istituto G Gaslini, Genova, Italy) for the DBL constructs. We are also grateful to the Caenorhabditis Genetic Center (University of Minnesota, Minneapolis) and the Japanese knockout consortium National BioResource Project for providing C. elegans strains.
CONFLICTS OF INTEREST
None.
FUNDING
This study was supported by grants from the Deutsche Forschungsgemeinschaft DFG KFO 201 (to E.N.-H., R.B., G.W.), DFG CRC1140 (to E.N.-H.), DFG CRC746 and BIOSS- The Centre for Biological Signaling Studies (to R.B.). E.N.-H. is supported by the European Social Fund and the Ministry of Science, Research, and Arts Baden-Württemberg (Margarete von Wrangell Programm). K.T. is supported by the BMBF e:Med projects MAPTor-NET (031A426B) and GlioPATH (01ZX1402B), a Rosalind-Franklin-Fellowship of the University of Groningen, the Ubbo Emmius Fund, Stichting TSC Fonds (calls 2015 and 2017) and the German Research Foundation (TH 1358/3-1). K.T. is recipient of the TSC Research Award 2017 of the German TS Foundation.
REFERENCES
1. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011; 12:21–35.
2. Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006; 124:471–84.
3. Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J. Rheb binds and regulates the mTOR kinase. Curr Biol. 2005; 15:702–13.
4. Yang Q, Inoki K, Kim E, Guan KL. TSC1/TSC2 and Rheb have different effects on TORC1 and TORC2 activity. Proc Natl Acad Sci USA. 2006; 103:6811–16.
5. Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003; 17:1829–34.
6. Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol. 2003; 13:1259–68.
7. Menon S, Dibble CC, Talbott G, Hoxhaj G, Valvezan AJ, Takahashi H, Cantley LC, Manning BD. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell. 2014; 156:771–85.
8. Demetriades C, Doumpas N, Teleman AA. Regulation of TORC1 in response to amino acid starvation via lysosomal recruitment of TSC2. Cell. 2014; 156:786–99.
9. Bar-Peled L, Schweitzer LD, Zoncu R, Sabatini DM. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell. 2012; 150:1196–208.
10. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012; 149:274–93.
11. Cornu M, Albert V, Hall MN. mTOR in aging, metabolism, and cancer. Curr Opin Genet Dev. 2013; 23:53–62.
12. Long X, Spycher C, Han ZS, Rose AM, Müller F, Avruch J. TOR deficiency in C. elegans causes developmental arrest and intestinal atrophy by inhibition of mRNA translation. Curr Biol. 2002; 12:1448–61.
13. Vellai T, Takacs-Vellai K, Zhang Y, Kovacs AL, Orosz L, Müller F. Genetics: influence of TOR kinase on lifespan in C. elegans. Nature. 2003; 426:620.
14. Jia K, Chen D, Riddle DL. The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development. 2004; 131:3897–906.
15. Honjoh S, Yamamoto T, Uno M, Nishida E. Signalling through RHEB-1 mediates intermittent fasting-induced longevity in C. elegans. Nature. 2009; 457:726–30.
16. Robida-Stubbs S, Glover-Cutter K, Lamming DW, Mizunuma M, Narasimhan SD, Neumann-Haefelin E, Sabatini DM, Blackwell TK. TOR signaling and rapamycin influence longevity by regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab. 2012; 15:713–24.
17. Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009; 460:392–95.
18. Evans DS, Kapahi P, Hsueh WC, Kockel L. TOR signaling never gets old: aging, longevity and TORC1 activity. Ageing Res Rev. 2011; 10:225–37.
19. Hansen M, Taubert S, Crawford D, Libina N, Lee SJ, Kenyon C. Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell. 2007; 6:95–110.
20. Kenyon CJ. The genetics of ageing. Nature. 2010; 464:504–12.
21. Wang J, Robida-Stubbs S, Tullet JM, Rual JF, Vidal M, Blackwell TK. RNAi screening implicates a SKN-1-dependent transcriptional response in stress resistance and longevity deriving from translation inhibition. PLoS Genet. 2010; 6:6.
22. Hansen M, Chandra A, Mitic LL, Onken B, Driscoll M, Kenyon C. A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet. 2008; 4:e24.
23. Tóth ML, Sigmond T, Borsos E, Barna J, Erdélyi P, Takács-Vellai K, Orosz L, Kovács AL, Csikós G, Sass M, Vellai T. Longevity pathways converge on autophagy genes to regulate life span in Caenorhabditis elegans. Autophagy. 2008; 4:330–38.
24. Kumfer KT, Cook SJ, Squirrell JM, Eliceiri KW, Peel N, O’Connell KF, White JG. CGEF-1 and CHIN-1 regulate CDC-42 activity during asymmetric division in the Caenorhabditis elegans embryo. Mol Biol Cell. 2010; 21:266–77.
25. Nukazuka A, Tamaki S, Matsumoto K, Oda Y, Fujisawa H, Takagi S. A shift of the TOR adaptor from Rictor towards Raptor by semaphorin in C. elegans. Nat Commun. 2011; 2:484.
26. Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res. 2014; 24:24–41.
27. Gosai SJ, Kwak JH, Luke CJ, Long OS, King DE, Kovatch KJ, Johnston PA, Shun TY, Lazo JS, Perlmutter DH, Silverman GA, Pak SC. Automated high-content live animal drug screening using C. elegans expressing the aggregation prone serpin α1-antitrypsin Z. PLoS One. 2010; 5:e15460.
28. Oliveira RP, Porter Abate J, Dilks K, Landis J, Ashraf J, Murphy CT, Blackwell TK. Condition-adapted stress and longevity gene regulation by Caenorhabditis elegans SKN-1/Nrf. Aging Cell. 2009; 8:524–41.
29. Kwon ES, Narasimhan SD, Yen K, Tissenbaum HA. A new DAF-16 isoform regulates longevity. Nature. 2010; 466:498–502.
30. Chan E, Nance J. Mechanisms of CDC-42 activation during contact-induced cell polarization. J Cell Sci. 2013; 126:1692–702.
31. Cook DR, Rossman KL, Der CJ. Rho guanine nucleotide exchange factors: regulators of Rho GTPase activity in development and disease. Oncogene. 2014; 33:4021–35.
32. Bi F, Debreceni B, Zhu K, Salani B, Eva A, Zheng Y. Autoinhibition mechanism of proto-Dbl. Mol Cell Biol. 2001; 21:1463–74.
33. Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell. 2010; 40:310–22.
34. Lundquist EA. Small GTPases. WormBook : the online review of C elegans biology. 2006:1–18.
35. Rossman KL, Sondek J. Larger than Dbl: new structural insights into RhoA activation. Trends Biochem Sci. 2005; 30:163–65.
36. Kapahi P, Chen D, Rogers AN, Katewa SD, Li PW, Thomas EL, Kockel L. With TOR, less is more: a key role for the conserved nutrient-sensing TOR pathway in aging. Cell Metab. 2010; 11:453–65.
37. Tullet JM, Hertweck M, An JH, Baker J, Hwang JY, Liu S, Oliveira RP, Baumeister R, Blackwell TK. Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell. 2008; 132:1025–38.
38. Zhang Y, Gao X, Saucedo LJ, Ru B, Edgar BA, Pan D. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat Cell Biol. 2003; 5:578–81.
39. Martin TD, Chen XW, Kaplan RE, Saltiel AR, Walker CL, Reiner DJ, Der CJ. Ral and Rheb GTPase activating proteins integrate mTOR, GTPase signaling in aging, autophagy, and tumor cell invasion. Mol Cell. 2014; 53:209–20.
40. Hsu YC, Chern JJ, Cai Y, Liu M, Choi KW. Drosophila TCTP is essential for growth and proliferation through regulation of dRheb GTPase. Nature. 2007; 445:785–88.
41. Rehmann H, Brüning M, Berghaus C, Schwarten M, Köhler K, Stocker H, Stoll R, Zwartkruis FJ, Wittinghofer A. Biochemical characterisation of TCTP questions its function as a guanine nucleotide exchange factor for Rheb. FEBS Lett. 2008; 582:3005–10.
42. Wang X, Fonseca BD, Tang H, Liu R, Elia A, Clemens MJ, Bommer UA, Proud CG. Re-evaluating the roles of proposed modulators of mammalian target of rapamycin complex 1 (mTORC1) signaling. J Biol Chem. 2008; 283:30482–92.
43. Kamath RS, Martinez-Campos M, Zipperlen P, Fraser AG, Ahringer J. Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome biology. 2001; 2:RESEARCH0002.
44. Libina N, Berman JR, Kenyon C. Tissue-specific activities of C. elegans DAF-16 in the regulation of lifespan. Cell. 2003; 115:489–502.
45. Lapierre LR, Gelino S, Meléndez A, Hansen M. Autophagy and lipid metabolism coordinately modulate life span in germline-less C. elegans. Curr Biol. 2011; 21:1507–14.
46. Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008; 3:1101–08.
47. Dirnberger D, Messerschmid M, Baumeister R. An optimized split-ubiquitin cDNA-library screening system to identify novel interactors of the human Frizzled 1 receptor. Nucleic Acids Res. 2008; 36:e37.