
INTRODUCTION
Pancreatoblastoma is a rare malignant epithelial neoplasm of the pancreas with multiple lines of differentiation, including acinar differentiation and squamoid nests. Less frequently, this tumor may exhibit endocrine and ductal differentiation and may contain a distinct mesenchymal component [1]. Pancreatoblastoma usually occurs in childhood and is the most prevalent malignant pancreatic tumor in the first decade of life [2]. Exceptionally, it can also be encountered in adults, although such cases are so rare that fewer than fifty cases have been reported to date [3]. No effective therapy has been established, especially in cases with unresectable or metastatic lesions. There have been only a few reports on the molecular features of pancreatoblastoma.
Recently, we encountered an adult patient with pancreatoblastoma. Using a next generation sequencer (NGS), loss of heterozygosity (LOH) of the adenomatous polyposis coli (APC) gene was detected in the tumor. In the remaining APC allele, we observed a novel missense mutation, derived from the germline APC gene, which was not reported in the Universal Mutation Database (http://www.umd.be/) or Tohoku Medical Megabank Organization, a Japanese-specific genome cohort (http://www.megabank.tohoku.ac.jp/). In this report, we characterized the function of the novel APC mutant.
CASE PRESENTATION
A 37-year-old woman visited our hospital complaining of abdominal pain. She had no past or familial history of neoplasms. A computed tomography (CT) scan showed a 5 cm abnormal mass in the pancreas head. She was suspected for pancreatoblastoma by CT-guided biopsy (Figure 1A) and was sent for a pancreaticoduodenectomy.
The postoperative pathological findings showed squamoid nests, some with keratinization (Figure 1D). Immunohistological analysis indicated an abnormal nuclear accumulation of β-catenin proteins and the expression of Bcl-10, a pancreatic acinar cell marker, in the tumor tissues (Figures 1E and 1F). A pathological diagnosis of pancreatoblastoma depends on its acinar differentiation and on the existence of squamoid nests; therefore, this tumor was finally diagnosed as pancreatoblastoma according to these findings.
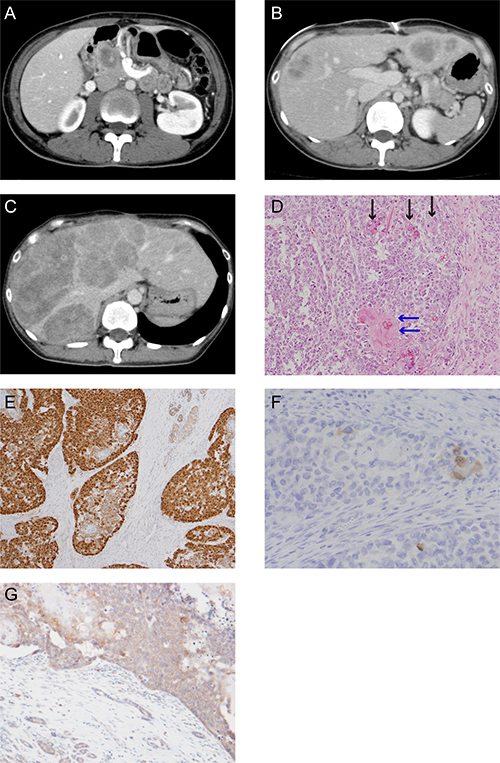
Figure 1: CT scan images and histological appearance of this case. (A) CT scan image at first visit. (B) CT scan image at postsurgical recurrence of liver metastasis. (C) The last CT scan image before the patient died. (D) Hematoxylin and eosin staining (magnification, ×200). The black arrow shows the squamoid nest and the blue arrow shows the squamoid nest with keratinization. (E) Immunohistochemical staining for β-catenin (magnification, ×100). (F) Scattered tumor cell shows positive for Bcl-10 (magnification, ×200). (G) Immunohistochemical staining for APC (magnification, ×50).
A CT scan taken three months postoperatively showed metastatic recurrence in the liver (Figure 1B). She received one course of adriamycin and gemcitabine chemotherapy, with no response. We treated her with cisplatin and S-1 as a second line chemotherapy. Although the tumor temporarily exhibited no change in size, after six cycles of cisplatin and S-1 chemotherapy her liver metastatic pancreatoblastoma increased in size (Figure 1C). We were unable to achieve any response via salvage treatment, and she ultimately died of pancreatoblastoma progression 13 months after diagnosis.
RESULTS
Analysis of oncodriver mutations in a patient with pancreatoblastoma
We conducted genetic analysis to investigate genetic alterations in the tumor using a cancer panel (OncoDEEP®, Supplementary Table 1). We identified a previously unreported missense APC mutation of c.5503A > G (p.R1835G). We also confirmed the mutation of the APC gene by Sanger sequencing (Figure 2A) and the expression of APC protein by immunohistochemical staining (Figure 1G) in tumor tissue samples from the patient. Allele frequency of this mutated allele was 99% in the cancer panel analysis. In addition, we did not find any other alterations of the genes involving the Wnt signal pathway, including CTNNB1. After obtaining written informed consent from the patient, we analyzed her germ line APC gene from her saliva and found a heterogeneous mutation in the 1835 codon of the APC gene (Figure 2A). These data suggest that a LOH of the APC gene occurred in the tumor, and the products of the APC missense mutation took part in tumorigenesis.
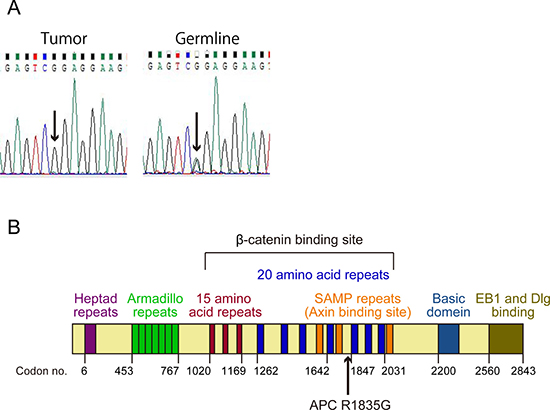
Figure 2: Status and position of the APC mutation. (A) The results of direct sequencing of the PCR product from a part of APC exon 15 in the tumor and the germline. The arrows show a point mutation of c.5503A > G (p.R1835G). (B) Structure of APC protein. The arrow shows position of the mutation (p.R1835G).
Evaluation of the suppression ability of the Wnt/β-catenin signaling pathway by R1835G mutant APC
Immunohistochemical analysis showed an abnormal nuclear accumulation of β-catenin protein (Figure 1E), indicating activation of the Wnt/β-catenin signaling pathway in the pancreatoblastoma. Therefore, to examine whether R1835G mutant APC affects Wnt/β-catenin signaling, we established cell models expressing APC R1835G mutant protein stably (Figure 3A). We performed quantitative RT-PCR (q-RT-PCR) to measure the endogenous Wnt/β-catenin transcriptional target, AXIN2, with three genotypes in Caco2 (APC-WT-Caco2, APC-R1835G-Caco2, Cntl-Caco2) and SW480 (APC-WT-SW480, APC-R1835G-SW480, Cntl-SW480). The q-RT-PCR analysis indicated that expression of AXIN2 in APC R1835G was reduced compared to Control (Cntl), but its expression was not downregulated as much as in APC-WT (Figure 3B and 3C). To examine how APC R1835G affects the Wnt/β-catenin signaling pathway via the TCF/LEF binding site, we carried out a TOPFlash assay with three genotypes in Caco2 and SW480 (Figure 3D and 3E). Similar results were also obtained from the TOPFlash assay. These results showed that a R1835G mutation in the APC gene partially inhibits Wnt/β-catenin signaling activity due to the APC mutant directly affecting the β-catenin/TCF transcriptional complex. In addition, we performed an immunofluorescence analysis for β-catenin, comparing the nuclear accumulation of β-catenin among the three genotypes in Caco2 and SW480. As can be seen in Figure 3F and 3G, there was greater accumulation of β-catenin in the nucleus with APC R1835G expressing cell lines than with APC-WT expressing cell lines. It is suggested that the product of APC R1835G has weak ability to bind β-catenin compared with WT.
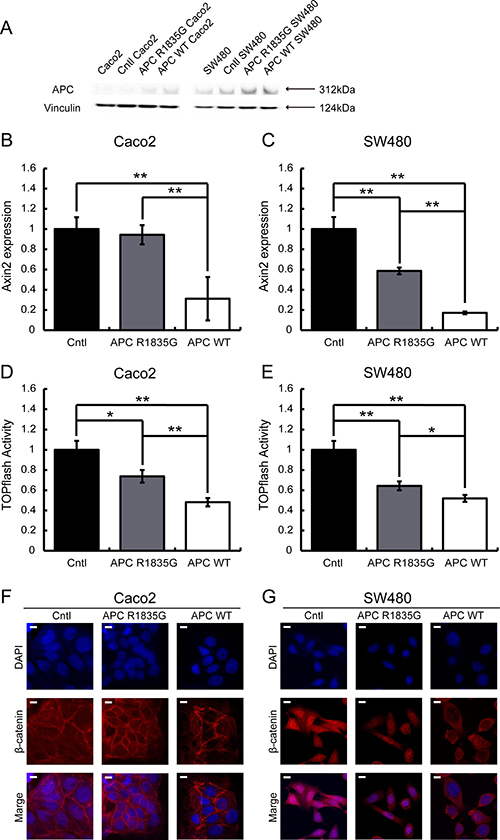
Figure 3: Evaluation of the suppression ability of the Wnt/β-catenin signaling pathway using a stable transformant. Western blotting analysis of APC in Caco2 and SW480 cells with three genotypes (APC-WT, APC-R1835G and Cntl) were shown (A). Relative expression level of AXIN2 mRNA with three genotypes in Caco2 (B) and SW480 (C) cells and activity of the TOPFlash assay with three genotypes in Caco2 (D) and SW480 (E) cells were indicated. Vertical axis of the result (B) and (C) shows relative AXIN2 expression and (D) and (E) show relative TOPflash activity. Data are shown as the mean ± SD, *p < 0.05 **p < 0.01 (Student’s t-test), and all assays were performed in triplicate. Immunofluorescence of β-catenin with three genotypes in Caco2 (F) and SW480 (G) cells were shown. Scale bar shows 10 μm.
Evaluation of the suppression ability of the Wnt/β-catenin signal by R1835G mutant APC with external Wnt stimuli
Next, we evaluated whether the R1835G mutant APC had the ability to suppress activated Wnt/β-catenin signaling in HEK293T cells, which originally have a wild-type APC allele. The activation of the Wnt/β-catenin pathway was mimicked by the inhibition of GSK-3β using the pharmacological inhibitor LiCl (lithium chloride). LiCl was added 6 h before the assay. As shown in Figure 4, activation of Wnt/β-catenin signaling was suppressed under the stably overexpressed APC WT. However, the stably overexpressed APC R1835G could not completely suppress activation of Wnt/β-catenin signaling. These results show that the R1835G mutant APC can partially, but not fully, inhibit Wnt/β-catenin signaling activity.
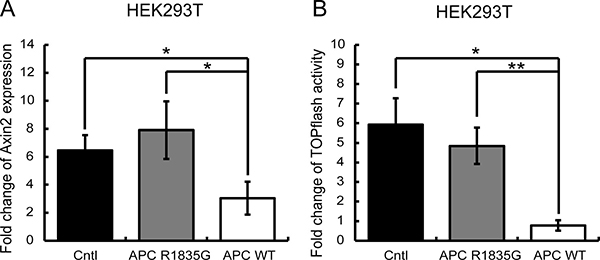
Figure 4: Evaluation of the suppressive ability of the Wnt/β-catenin signaling pathway under the condition of external Wnt signaling stimuli. (A) Relative expression levels of AXIN2 mRNA with three genotypes in HEK293T (APC-WT-HEK293T, APC-R1835G-HEK293T, and Cntl-HEK293T) cells are shown. (B) Activities of the TOPFlash assay with three genotypes in HEK293T were indicated. Vertical axis of result (A) shows fold change of AXIN2 expression and the result (B) shows fold change of TOPflash activity before and after LiCl stimulation. Data are shown as the mean ± SD, *p < 0.05 **p < 0.01 (Student’s t-test), and all assays were performed in triplicate.
Dose-dependent assessment of suppression ability of the Wnt/β-catenin signaling pathway by R1835G mutant APC
To determine if the suppression ability of Wnt/β-catenin signaling by APC R1835G was dose-dependent, we conducted a TOPFlash assay using ectopically expressed APC R1835G in SW480. Expression of increasing amounts of APC R1835G reduced TOPFlash reporter activity in a dose-dependent manner. We observed enhanced TOPFlash reporter activity of APC R1835G, compared to APC WT (Figure 5). These results show that R1835G mutant APC attenuate repression of the Wnt/β-catenin signaling pathway activity.
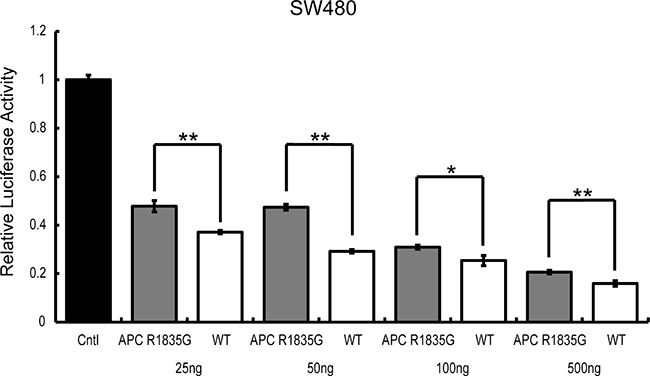
Figure 5: TOPFlash reporter activity by APC-R1835G expression levels in SW480. TOPflash activity was assessed after transfection of pCMV-Neo-Bam APC-R1835G and pCMV-Neo-Bam APC in SW480 cells. Each plasmid is transfected at 25 ng, 50 ng, 100 ng, and 500 ng. Data are shown as the mean ± SD, *p < 0.05 **p < 0.01 (Student’s t-test), and all assays were performed in triplicate.
DISCUSSION
This is a case report of pancreatoblastoma in an adult patient. Pancreatoblastoma is a very uncommon neoplasm in adults, and its molecular pathogenesis is largely unknown. We found a novel missense mutation in the APC gene. Although the tumor cells had only novel missense mutation alleles of the APC gene, germ cell lines had wild-type and mutant alleles of APC. Moreover, neoplasms showed abnormal nuclear accumulation of β-catenin protein by immunohistological analysis. Therefore, this case of pancreatoblastoma was likely caused by activation of the Wnt/β-catenin signal by LOH of the APC gene.
According to the cBioPortal for Cancer Genomes (http://www.cbioportal.org/), somatic mutations in the APC and CTNNB1 loci have rarely been identified in pancreatic cancer, but a few CTNNB1 gene mutations have been reported in cases of pancreatoblastoma [4–7]. Because of the rarity of pancreatoblastoma, the population of APC gene mutations is not known. Only one case has been reported of a germline truncating mutation of the APC gene, a 5-bp deletion at the β-catenin binding site in an individual with familial adenomatous polyposis (FAP) [6]; LOH of the APC gene in pancreatoblastoma was also observed in this case (Table 1). Both pancreatoblastoma and acinar cell carcinomas consistently exhibit acinar differentiation [1], as well as alterations in the genes for Wnt/β-catenin signaling, including inactivating mutations in APC and activating mutations in CTNNB1; however, they lack alterations in the genes commonly mutated in pancreatic duct adenocarcinoma, including KRAS, TP53, P16/CDKN2A, and SMAD4 [8]. We have therefore discussed the activation of Wnt/β-catenin signaling in the pancreas cause not ductal cancer but acinar cell carcinoma, including pancreatoblastoma. This is consistent with previous reports of alterations in the genes for Wnt/β-catenin signaling, such as APC and CTNNB1, in pancreatoblastoma (Table 1).
Table 1: Mutation profiles of CTNNB1 and APC in pancreatoblastoma reported in the past
Case |
Age/Sex |
CTNNB1 alternations |
APC alternations |
Author |
|---|---|---|---|---|
1 |
5/Male |
S37P |
Not evaluated |
Tanaka et al.(4) |
2 |
3/Male |
S33P |
Not evaluated |
|
3 |
5/Male |
N32H |
Not evaluated |
Abraham et al.(6) |
4 |
3/Male |
Wild type |
Not evaluated |
|
5 |
4/Male |
S33F |
Not evaluated |
|
6 |
45/Female |
S37F |
Not evaluated |
|
7 |
51/Female |
Wild type |
Germline |
|
8 |
6/Male |
Wild type |
Wild type |
|
9 |
13/Female |
Wild type |
Wilde type |
|
10 |
5/Female |
G34R |
Not evaluated |
|
11 |
3.5/Female |
N32H |
Not evaluated |
|
12 |
3 day/Male |
T44A |
Not evaluated |
Ismale et al.(7) |
APC regulates turnover of cytosolic β-catenin and is the key effector of the canonical Wnt signal pathway. The nucleotide sequence of the APC gene is conserved across species, and APC gene mutations exhibit closely related oncogenesis. The position of the mutation on the APC gene is also important because this gene carries a variety of functional domains, including a heptad repeat, an Arm repeat, a β-catenin binding site (three 15-amino acid repeats and seven 20-amino acid repeats), an Axin binding site (SAMP repeats), a basic region, and an EB1 binding site [9]. The mutation we report here is located between the fourth and fifth 20-amino acid repeat in the β-catenin binding site (Figure 2B). The mutation, which caused an amino acid to change from hydrophobic arginine to hydrophilic glycine, was located very close to the β-catenin binding domain. Given this, we hypothesized that this mutation may reduce the ability to bind to β-catenin. This hypothesis was supported by the result of the immunofluorescence analysis of β-catenin (Figures 3F and 3G).
APC germline mutations are often seen in patients with FAP, with germline truncating mutations frequently observed at the β-catenin binding site, such as in Case 7 in Table 1. In contrast, our patient had no family history of FAP or past history of colon polyposis. This indicated that the mutation of the APC gene in this case caused pancreatoblastoma, not adenomatous polyposis. Another report described gastric polyposis caused by a missense mutation in the 5′ terminal side of APC [10]; and Ikenoue et al. reported that a truncated mutation at the 3′ terminal side of the APC gene caused attenuated polyposis [11]. In addition, there has been a report of APC missense mutations in cases of hepatoblastoma; however, these cases all featured childhood disease onset, and we were unable to determine whether the patients developed adenomatous polyposis [12].
As described above, in this case the mutation seemed to result in partial loss of biding ability to β-catenin. Differences in the ability to suppress Wnt/β-catenin signaling may account for the phenotypic differences that determine whether adenomatous polyposis occurs. In future studies, we need to investigate the relationship between phenotypes and mutations under in vivo conditions.
Comprehensive analysis by NGS gave us a lot of information. On the other hand, we found many variant of uncertain significance (VUS) mutations. There is no established method to confirm if mutations found by clinical sequencing have pathological significance or not. This case report suggests a desperate need for an established evaluation method of VUS mutations in the NGS era.
MATERIALS AND METHODS
Mutation analysis by cancer panel and Sanger sequence
The gene analysis of tumor cells was performed using the OncoDEEP Clinical® (OncoDNA, Belgium), a cancer panel for 409 kinds of genes by NGS. Supplementary Table 1 shows the 409 gene varieties that are detected by OncoDEEP Clinical®. We submitted five tumor tissue sections with 5 μm thickness and one hematoxylin and eosin staining section to the OncoDNA Company. Almost all exons of the APC and CTNNB1 genes were covered: for APC, exons 1–10, 12, 15, and 16 were 100% covered, exons 13 and 14 were more than 90% covered, and exon 11 was more than 30% covered; for CTNNB1, exons 1–3, 6–9, and 11–15 were 100% covered, and exons 4, 5, and 10 were more than 90% covered.
To confirm the mutation detected by OncoDEEP Clinical®, we performed Sanger sequencing of the APC gene using a primer, 5′-TCGTCTTCTGCACCCAACAA–3′. For Sanger sequencing of the APC gene from the germline, genomic DNA was extracted from saliva using the Oragene DNA® (DNA genotek, ON, Canada).
Immunohistochemical staining
Five micron sections of paired neoplastic and normal formalin-fixed, paraffin-embedded tissues were used for immunohistochemical staining with anti-β-catenin (clone β-catenin-1), anti-Bcl10 (clone sc-5273) and anti-APC (clone GTX116009) listed in Supplementary Table 2, according to the manufacturers’ instructions. For β-catenin, immunohistochemical labeling was evaluated for the presence of nuclear, cytoplasmic, and membranous β-catenin accumulation in both the neoplasms and the normal surrounding tissues.
Plasmid construction
We used pCMV-Neo-Bam APC, which was a gift from Bert Vogelstein (Addgene plasmid # 16507)] as a template of APC [13]. DNA fragments of APC were amplified by PCR using a set of primers, 5′-CGAGGTTAACGAATTATGGCTGCAGCTTCATATG-3′ and 5′-CTACCCGGTAGAATTTTAAACAGATGTCACAAGGTA-3′. Amplified APC fragments were subcloned into EcoRI-digested pMSCV PIG, which was a gift from David Bartel (Addgene plasmid # 21654) [14], using an In-Fusion® HD cloning kit (Takara). The constructed plasmid containing APC WT was named pMSCV-APC-WT.
Plasmids containing APC R1835G (c.5503a > g) were generated from pMSCV-APC-WT using site-directed mutagenesis and named pMSCV-APC-R1835G. The primers used for site-directed mutagenesis were 5′-GAAGATAGAGTCGGAGGAAGTTTTG-3′ and 5′-CAAAACTTCCTCCGACTCTATCTTC-3′.
Cell lines and culture
Colorectal cancer cell lines Caco-2 and SW480 (APC-null) and embryonic kidney cell lines HEK293T (APC wild type) were cultured according to recommendations of the American Type Culture Collection (ATCC). PLAT-A cells [15] were maintained at 37°C in a 5% CO2 incubator in DMEM, 10% fetal calf serum (FCS), 1 μg/mL puromycin, 10 μg/mL blasticidin, and penicillin.
Retroviral transduction and establishment of a stable transformant
The day before transfection, PLAT-A cells, at 2 × 106 per dish, were seeded in a 60 mm culture dish and incubated overnight. Each of pMSCV-APC-WT, pMSCV-APC-R1835G and pMSCV PIG was transfected into PLAT-A cells at 3 μg using GeneJuice Transfection Reagent (Novagen). Retrovirus particles from PLAT-A cells were collected 48 h after transfection and added to Caco-2, SW480, and HEK293T cells with 8 μg/ml Polybren (Sigma). We generated APC-WT-Caco2, APC-R1835G-Caco2, Cntl-Caco2, APC-WT-SW480, APC-R1835G-SW480, Cntl-SW480, APC-WT-HEK293T, APC-R1835G-HEK293T, and Cntl-HEK293T cells. After infection, Caco-2, SW480 and HEK293T cells were incubated with the 8 ng/μl, 4 ng/μl, and 1.5 ng/μl puromycin selection agents, respectively, to obtain stable cell lines.
Western blotting
Cellular lysates were prepared and the protein concentrations were determined using a PierceTM BCA Protein Assay Kit (Thermo Fisher Scientific). Samples with 13 μg and 5 μg of total protein were used for western blots of Caco-2 and SW480, respectively. The samples were separated via electrophoresis on 5%–12% SDS-polyacrylamide gel and then transferred onto a nylon membrane for the western blot analysis. The membrane was probed with the polyclonal rabbit antibodies listed in Supplementary Table 2, anti-APC (clone ab15270), and anti-Vinculin (clone ab73412) as the loading control. Anti-rabbit IgG.
HRP-linked Antibody (Cell Signaling Technology) was used as the secondary antibody. The protein bands were detected using Pierce® ECL Plus Western Blotting Substrate (Thermo Fisher Scientific).
Transient transfection
We generated pCMV-Neo-Bam APC-R1835G from pCMV-Neo-Bam APC by site-directed mutagenesis using a set of primers, 5′- GAAGATAGAGTCGGAGGAAGTTTTG-3′ and 5′- CAAAACTTCCTCCGACTCTATCTTC-3′. The day before transfection, SW480 cells, at 2 × 105 cells per dish, were seeded in a 60 mm culture dish and incubated overnight. Each of pCMV-Neo-Bam APC-R1835G, pCMV-Neo-Bam APC, and pCMV were transfected into SW480 cells at 25 ng, 50 ng, 100 ng, and 500 ng using GeneJuice Transfection Reagent (Novagen).
Luciferase assay
In an assay of stable cell lines, the day before transfection for reporter gene assays, APC-WT-Caco2, APC-R1835G-Caco2, Cntl-Caco2, APC-WT-SW480, APC-R1835G-SW480, Cntl-SW480, APC-WT-HEK293T, APC-R1835G-HEK293T, and Cntl-HEK293T cells, at 2 × 105 per dish, were seeded in six-well plates. Each well was transfected with a total of 78 ng of plasmids, including 65 ng of M50 Super 8× TOPFlash, which was a gift from Randall Moon (Addgene plasmid # 12456) [16], or M51 Super 8× FOPFlash, also a gift from Randall Moon (Addgene plasmid # 12457) [16], and 13 ng of pRL-SV40P, which was a gift from Ron Prywes (Addgene plasmid # 27163) [17], with GeneJuice Transfection Reagent (Novagen). Luciferase activity was monitored by SpectraMax®i3 (MOLECULAR DEVICES) using the dual luciferase assay system (Promega). We controlled the experimental LEF-luciferase reporter activity for transfection efficiency and potential treatment toxicity using the constitutively expressed pRL-SV40P luciferase. The specificity of APC-mediated effects on LEF reporters was confirmed using the M51 Super 8× FOPFlash, which harbors mutated LEF binding sites and an unrelated AP-1 reporter. Concerning to APC-WT-HEK293T, APC-R1835G-HEK293T, and Cntl-HEK293T cells, LiCl was added as a stimulant 6 h before the luciferase assay. In assay of transiently transfected SW480 cells, each of M50 Super 8× TOPFlash, M51 Super 8× FOPFlash, and pRL-SV40P were transfected in conjunction with transfection of pCMV-Neo-Bam APC-R1835G, pCMV-Neo-Bam APC, and pCMV.
Quantitative RT-PCR
The day before RNA extraction, APC-WT-Caco2, APC-R1835G-Caco2, Cntl-Caco2, APC-WT-SW480, APC-R1835G-SW480, Cntl-SW480, APC-WT-HEK293T, APC-R1835G-HEK293T, and Cntl-HEK293T cells, at 2 × 105 cells per dish, were seeded in six-well plates. Total RNAs were extracted from each well of these cells with NucleoSpin RNA (MACHEREY-NAGEL). Quantification of AXIN2 gene transcripts was performed by quantitative RT-PCR (q-RT-PCR) using the GoTaq 1-Step RT-qPCR System (Promega). HPRT1 was used as an internal control. The primer pairs used for the human AXIN2 gene were 5′-CTGGCTTTGGTGAACTGTTG-3′ and 5′-AGTTGCTCACAGCCAAGACA-3′. The primer pair used for the human HPRT1 gene was 5′-GC ACCACCAACTGCTTA-3′ and 5′-AGTAGAGGCAGG GATGAT-3′. Concerning APC-WT-HEK293T, APC-R183 5G-HEK293T, and Cntl-HEK293T cells, LiCl was added as stimulant 6 h before RNA extraction.
Immunofluorescence
Caco2 and SW480 cells grown and transfected onto 12-well chambers were fixed for 5 min in 10% methanol at –20° C. The cells were then washed three times with phosphate-buffered saline (PBS) and incubated with 1% bovine serum albumin (BSA) in PBS for 30 min at room temperature. Anti-β-catenin (clone 14/β-catenin) antibody in 1% BSA was then added to the samples, which were incubated overnight at 4° C. After washing three times with PBS, the cells were treated with Goat Anti-Mouse IgG H&L (Alexa Fluor® 594; Abcam) as the secondary antibody, diluted at 1:200 in 1% BSA for 1 h at room temperature. After washing three more times with PBS, the nuclei were counterstained with ProLongTM Gold antifade reagent with DAPI (Invitrogen). The samples were observed using a BZ-X710 (Keyence).
Abbreviations
NGS: next generation sequencer; LOH: loss of heterozygosity; APC: adenomatous polyposis coli; CT: computed tomography; q-RT-PCR: quantitative RT-PCR; LiCl: lithium chloride; FAP: familial adenomatous polyposis; VUS: variant of uncertain significance; ATCC: American type culture collection; FCS: fetal calf serum; PBS: phosphate-buffered saline; BSA: bovine serum albumin.
ACKNOWLEDGMENTS
The authors would like to thank Dr. T Kitamura for providing of the PLAT-A packaging cells, Dr. H Komiyama for providing of the SW480 cells, Enago (www.enago.jp) for the English language review.
CONFLICTS OF INTEREST
All authors have no conflicts of interest to disclose.
GRANT SUPPORT
This work was supported in part by JSPS <26860227> and grants from Chugai Pharmaceutical Co., Ltd. and Taiho Pharmaceutical Co., Ltd.
REFERENCES
1. Morohoshi T, Hruban RH, Klimstra DS, Ohike N, Terris B, Bosman F, Carmeiro F, Hruban RH. WHO classification of tumours of the digestive system, 4th ed. Int Agency Res Cancer. 2010; 319–21. https://doi.org/10.1016/j.annpat.2011.08.001.
2. Mylonas KS, Nasioudis D, Tsilimigras DI, Doulamis IP, Masiakos PT, Kelleher CM. A population-based analysis of a rare oncologic entity, malignant pancreatic tumors in children. J Pediatr Surg. 2017 Jun 30. https://doi.org/10.1016/j.jpedsurg.2017.06.024. [Epub ahead of print].
3. Omiyale AO. Clinicopathological review of pancreatoblastoma in adults. Gland Surg. 2015; 4:322–8. https://doi.org/10.3978/j.issn.2227-684X.2015.04.05.
4. Tanaka Y, Kato K, Notohara K, Nakatani Y, Miyake T, Ijiri R, Nishimata S, Ishida Y, Kigasawa H, Ohama Y, Tsukayama C, Kobayashi Y, Horie H. Significance of aberrant (cytoplasmic/nuclear) expression of beta-catenin in pancreatoblastoma. J Pathol. 2003; 199:185–90. https://doi.org/10.1002/path.1262.
5. Honda S, Okada T, Miyagi H, Minato M, Suzuki H, Taketomi A. Spontaneous rupture of an advanced pancreatoblastoma: Aberrant RASSF1A methylation and CTNNB1 mutation as molecular genetic markers. J Pediatr Surg. 2013; 48:e29–32. https://doi.org/10.1016/j.jpedsurg.2013.02.038.
6. Abraham SC, Wu TT, Klimstra DS, Finn LS, Lee JH, Yeo CJ, Cameron JL, Hruban RH. Distinctive molecular genetic alterations in sporadic and familial adenomatous polyposis-associated pancreatoblastomas: frequent alterations in the APC/beta-catenin pathway and chromosome 11p. Am J Pathol. 2001; 159:1619–27. https://doi.org/S0002-9440(10)63008-8.
7. Ismael O, Shimada A, Hama A, Takahashi Y, Sato Y, Hayakawa M, Tsuchiya H, Tainaka T, Ono Y, Kaneko K, Ando H, Sato K, Kojima S. Congenital pancreatoblastoma associated with β-catenin mutation. Pediatr Blood Cancer. 2012; 58:827–827. https://doi.org/10.1002/pbc.23337.
8. Jiao Y, Yonescu R, Offerhaus GJ, Klimstra DS, Maitra A, Eshleman JR, Herman JG, Poh W, Pelosof L, Wolfgang CL, Vogelstein B, Kinzler KW, Hruban RH, et al. Whole-exome sequencing of pancreatic neoplasms with acinar differentiation. J Pathol. 2014; 232:428–35. https://doi.org/10.1002/path.4310.
9. Aoki K, Taketo MM. Adenomatous polyposis coli (APC): a multi-functional tumor suppressor gene. J Cell Sci. 2007; 120:3327–35. https://doi.org/10.1242/jcs.03485.
10. Li J, Woods SL, Healey S, Beesley J, Chen X, Lee JS, Sivakumaran H, Wayte N, Nones K, Waterfall JJ, Pearson J, Patch AM, Senz J, et al. Point Mutations in Exon 1B of APC Reveal Gastric Adenocarcinoma and Proximal Polyposis of the Stomach as a Familial Adenomatous Polyposis Variant. Am J Hum Genet. 2016; 98:830–42. https://doi.org/10.1016/j.ajhg.2016.03.001.
11. Ikenoue T, Yamaguchi K, Komura M, Imoto S, Yamaguchi R, Shimizu E, Kasuya S, Shibuya T, Hatakeyama S, Miyano S, Furukawa Y. Attenuated familial adenomatous polyposis with desmoids caused by an APC mutation. Hum Genome Var. 2015; 2:15011. https://doi.org/10.1038/hgv.2015.11.
12. Oda H, Imai Y, Nakatsuru Y, Hata J, Ishikawa T. Somatic Mutations of the APC Gene in Sporadic Hepatoblastomas. Cancer Res. 1996; 56:3320–23. http://cancerres.aacrjournals.org/content/56/14/3320.abstract.
13. Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of β-Catenin-Tcf Signaling in Colon Cancer by Mutations in β-Catenin or APC. Science. 1997; 275:1787–90. http://science.sciencemag.org/content/275/5307/1787.abstract.
14. Mayr C, Bartel DP. Widespread Shortening of 3′UTRs by Alternative Cleavage and Polyadenylation Activates Oncogenes in Cancer Cells. Cell. 2009; 138:673–84. https://doi.org/10.1016/j.cell.2009.06.016.
15. Morita S, Kojima T, Kitamura T. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 2000; 7:1063–6. https://doi.org/10.1038/sj.gt.3301206.
16. Veeman MT, Slusarski DC, Kaykas A, Louie SH, Moon RT. Zebrafish prickle, a modulator of noncanonical Wnt/Fz signaling, regulates gastrulation movements. Curr Biol. 2003; 13:680–85. https://doi.org/10.1016/S0960-9822(03)00240-9.
17. Chen X, Prywes R. Serum-induced expression of the cdc25A gene by relief of E2F-mediated repression. Mol Cell Bi ol. 1999; 19:4695–702. https://doi.org/10.1128/MCB.19.7.4695.