
INTRODUCTION
Lung cancer is the leading cause of cancer-related death worldwide [1, 2]. Non-small-cell lung cancer (NSCLC) is the dominant subtype in all clinical cases [3]. Patients with NSCLC are usually diagnosed in advanced stages, leaving the surgical operation frequently as an off-the-table option [4]. Chemotherapy and radiotherapy are the common treatments for NSCLC patients [5]. Despite significance advances in NSCLC pathogenesis and development of novel therapeutics such as targeted therapy and immunotherapy, the overall 5-year survival rate of NSCLC patients is still far less from satisfied [6–8].
If doable, surgery is the best choice for NSCLC because it provides the patients with most likelihood of cure and long-term survival [9]. Surgical operation is the primary treatment for patients with stage I and II lung cancer and an important approach in multimodality treatment for patients with stages III or with stage IV oligo-metastatic cases [10]. However, postoperative complications are critical challenges in clinical practice for NSCLC patients [11]. The most frequently observed postoperative complication is pulmonary infection that portends poor NSCLC prognosis [12]. Consequently, the long-term survival after surgery of lung cancer patients remains less than 50%, mostly due to the tumor recurrence [13].
Gram-negative bacteria are the most common pathogens in pulmonary infection of NSCLC patients, either in those with postoperative complications or in those with advanced disease, raising a possibility that gram-negative bacteria might facilitate NSCLC progression [14, 15]. Indeed, recent studies showed that gram-negative bacteria promoted the growth and metastasis of human lung cancer cell lines and primary lung cancer cells from clinical patients [16, 17]. Activation of Toll-like receptor 4 (TLR4) signaling conferred the function of gram-negative bacteria in lung cancer progression [16, 17]. However, the precise molecular mechanisms about how gram-negative bacteria affect the NSCLC progression and recurrence are still obscure.
Interleukin 33 (IL-33) is one new member of IL-1 superfamily and expressed in various tissues, especially in those with barrier functions [18, 19]. By binding to its receptor ST2, IL-33 drives production of Th2 cytokines and plays a critical role in asthma, inflammation and autoimmunity [20–23]. Recently, IL-33 in circulation was defined as a biomarker for evaluating lung cancer progression [24]. IL-33 was identified as a pro-cancer cytokine and fueled the glycolysis of NSCLC cells [25]. But whether IL-33 is involved in the crosstalk of gram-negative bacterial infection and NSCLC progression, still remains unknown.
In current study, we explored the function of gram-negative bacteria in tumor progression and recurrence in NSCLC patients. NSCLC cells isolated from surgical tissues of clinical patients would avoid the defect of established human cancer cell lines that usually do not represent the heterogeneity and characteristics of cancer cells in patients [25]. We determined an important role of IL-33 in gram-negative bacteria induced NSCLC progression and recurrence. Mechanistically, IL-33 conferred gram-negative bacteria in facilitating cancer metabolic reprograming and cancer stem cell properties. These findings could further our current knowledge in NSCLC immunopathology and provide clues for therapeutic explorations against lung cancer, especially in those with gram-negative bacterial infections.
RESULTS
Gram-negative bacterial infection portends advanced disease and reduced time interval to recurrence in NSCLC patients
To explore the effect of pulmonary infection with gram-negative bacteria on NSCLC progression, a cohort of 52 patients diagnosed with NSCLC were detected for the presence of pulmonary gram-negative bacterial infection. While 13 patients were negative for pulmonary infection, 25 patients were positive for gram-negative bacterial infections (Figure 1A). Of interest, high burden of gram-negative bacteria predicted advanced disease stages (stage III-IV) in NSCLC patients (Figure 1B).
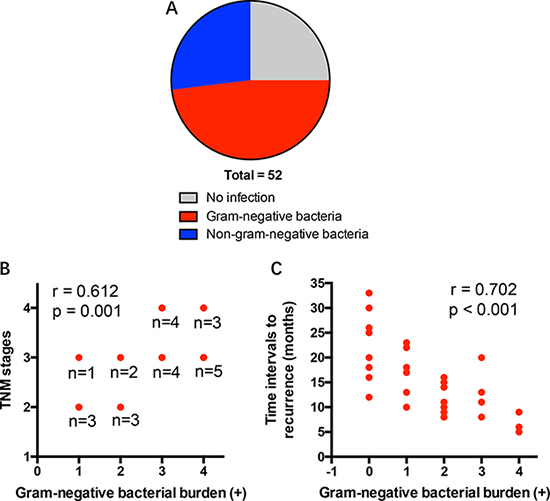
Figure 1: Gram-negative bacterial infection portends advanced disease and decreased tumor interval to recurrence in NSCLC patients. (A) A cohort of 52 NSCLC patients were screened for pulmonary infection of gram-negative bacteria. (B) The burden of gram-negative bacteria from NSCLC patients were associated with disease progression. (C) The burden of gram-negative bacteria in postoperative infection were correlated with time intervals to recurrence of NSCLC patients. Each dot represents the data from one patient.
To evaluate the involvement of gram-negative bacteria in tumor recurrence, a cohort of 28 NSCLC patients with or without postoperative pulmonary gram-negative bacterial infection were enrolled in a follow-up study. NSCLC patients suffered from strong postoperative complication of gram-negative bacterial infection have shorter time intervals to recurrence (Figure 1C).
Gram-negative bacteria drive outgrowth and metastasis of NSCLC through TLR4/MyD88 signaling
To understand the association of gram-negative bacterial infection and NSCLC progression, NSCLC cells were cultured with inactivated gram-negative bacteria [16, 17]. As expected, gram-negative bacteria promoted the growth capacity and invasion of NSCLC cells (Figure 2A–2B). Gram-negative bacteria enhanced tumor progression in a dose dependent manner and doubled the proliferative expansion rate and the number of invading NSCLC cells. The in vivo conformation was performed with NOD-SCID mice challenged with NSCLC cells that were pretreated with gram-negative bacteria. Prior treatment with gram-negative bacteria promoted the growth and metastasis of NSCLC cells in immune-deficient mice (Figure 2C–2D). Further, genetic knockdown of TLR4 expression in NSCLC cells efficiently abrogated the gram-negative bacteria mediated tumor progression both in vitro and in vivo (Supplementary Figure 1, Figure 2E–2H). These findings are consistent with previous studies [16, 17], pinpointing the requirement of TLR4 receptor in gram-negative bacteria mediated lung cancer progression. Accordingly, blocking MyD88 signaling by administration of MyD88 inhibitory peptide significantly inhibited gram-negative bacteria mediated NSCLC progression (Figure 2I–2L).
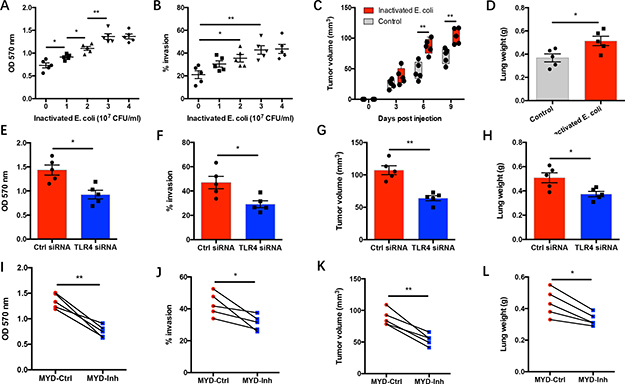
Figure 2: Gram-negative bacteria drive NSCLC progression via TLR4/MyD88 signaling. (A–B) NSCLC cells from clinical patients (n = 5) were cultured with an increasing dose of heat-inactivated E. coli. Proliferative expansion of NSCLC cells was detected after 72 hours (A). Invasion of NSCLC cells was analyzed after 24 hours (B). (C–D) NSCLC cells from 5 patients were pretreated with heat-inactivated E. coli (3x107 CFU/ml) for 24 hours and adoptively transferred into NOD-SCID mice. Tumor size was measured at the indicated time post NSCLC injection (C). Two weeks later, tumor metastasis was determined by analyzing lung weight to reflect tumor burden in lung (D). (E–H) NSCLC cells from 5 patients were transfected with TLR4 siRNA or control siRNA and incubated with heat-inactivated E. coli (3x107 CFU/ml). NSCLC growth capacity was analyzed after 72 hours (E). NSCLC invasion was detected after 24 hours (F). (G–H) 24 hours later, NSCLC were injected into NOD-SCID mice and assayed for tumor growth on day 7 (G) and tumor metastasis on day 14 (H). (I–L) NSCLC cells from 5 patients were cultured with heat-inactivated E. coli (3x107 CFU/ml) plus MyD88 inhibitory peptide (MYD-Inh, 50 μM) or control peptide (MYD-Ctrl, 50 μM). Tumor progression in vitro and in vivo were analyzed as described above. Each dot represents the data from one individual. *p < 0.05. **p < 0.01.
TLR4 activation by gram-negative bacteria induces NSCLC progression in IL-33 dependent manner
To detect whether IL-33 was involved in the effect of gram-negative bacteria on NSCLC progression, NSCLC cells were incubated with inactivated gram-negative bacteria and analyzed for IL-33 expressions. We found that gram-negative bacteria efficiently induced mRNA and protein expressions of IL-33 in NSCLC cells (Figure 3A–3C). Genetic knockdown of TLR4 expression significantly reduced IL-33 expression in response to gram-negative bacteria (Figure 3D–3E).
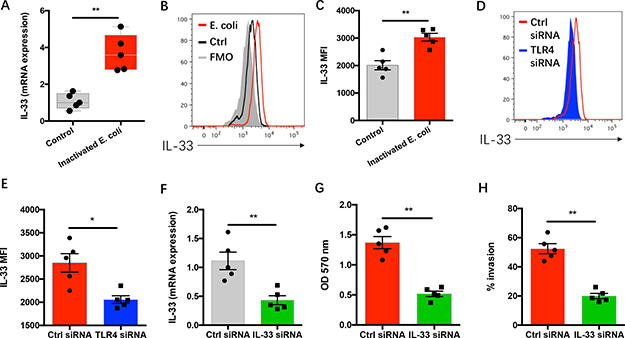
Figure 3: Gram-negative bacteria-induced NSCLC progression relies on TLR4/IL-33 pathway. (A) NSCLC cells from 5 patients were incubated with or without heat-inactivated E. coli (3x107 CFU/ml) for 12 hours and analyzed for IL-33 mRNA expressions. (B–C) NSCLC cells from clinical patients (n = 5) were incubated with or without heat-inactivated E. coli (3x107 CFU/ml) for 24 hours and analyzed for IL-33 protein expressions. (D–E) NSCLC cells from 5 clinical patients were transfected with TLR4 siRNA or control siRNA and incubated with heat-inactivated E. coli (3x107 CFU/ml) for 24 hours. IL-33 protein expressions were detected by flow cytometry. (F) NSCLC cells from 5 patients were transfected with IL-33 siRNA or control siRNA and analyzed for IL-33 mRNA expressions after 12 hours. (G–H) NSCLC cells from 5 patients were transfected with IL-33 siRNA or control siRNA and incubated with heat-inactivated E. coli (3x107 CFU/ml). NSCLC growth capacity was detected after 72 hours (G) and the invasion was determined after 24 hours (H). Each dot represents the data from one patient. *p < 0.05. **p < 0.01.
To evaluate the potential role of IL-33 in gram-negative bacteria mediated NSCLC progression, NSCLC cells were transfected with IL-33 siRNA and cultured with inactivated gram-negative bacteria. Knockdown of IL-33 expression abrogated gram-negative bacteria mediated NSCLC progression (Supplementary Figure 2 and Figure 3F–3H).
IL-33 confers gram-negative bacteria-enhanced cancer metabolism
High rates of glycolysis and lipogenesis are two hallmarks of cancer metabolic reprograming to support their uncontrolled outgrowth and metastasis [16, 25, 26]. To explore how IL-33 regulates gram-negative bacteria induced NSCLC progression, NSCLC cells with or without IL-33 knockdown were incubated with inactivated gram-negative bacteria and analyzed for expressions of cancer metabolism reflecting genes GLUT1, PKM2, FASN and ACC1. While gram-negative bacteria increased their mRNA expressions (Figure 4A), this process could be abrogated by IL-33 knockdown (Figure 4B). IL-33 is essential for gram-negative bacteria to enhance the membrane GLUT1 protein levels (Figure 4C–4D). Accordingly, gram-negative bacteria promoted lactate production in NSCLC cells, which again could be impeded by IL-33 knockdown (Figure 4E–4F).
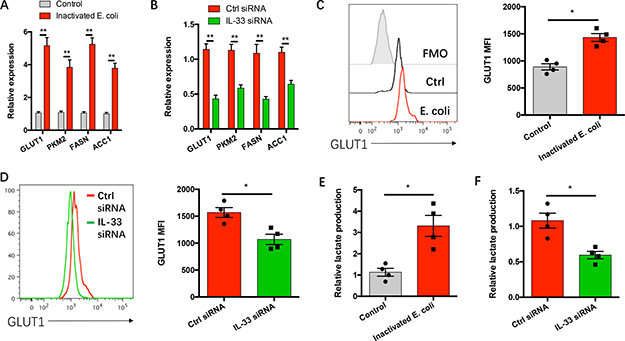
Figure 4: IL-33 confers gram-negative bacteria-induced cancer metabolism. (A) NSCLC cells from 5 clinical patients were incubated with or without heat-inactivated E. coli (3x107 CFU/ml) for 12 hours and detected the mRNA expressions of the indicated genes. (B) NSCLC cells from 5 clinical patients were transfected with IL-33 siRNA or the control siRNA, and incubated with heat-inactivated E. coli (3x107 CFU/ml) for 12 hours. (C, E) NSCLC cells from 4 clinical patients were incubated with or without heat-inactivated E. coli (3x107 CFU/ml) for 36 hours. (D, F) NSCLC cells from 4 clinical patients were transfected with IL-33 siRNA or the control siRNA, and incubated with heat-inactivated E. coli (3x107 CFU/ml) for 36 hours. Each dot represents the data from one patient. *p < 0.05. **p < 0.01.
IL-33 licenses gram-negative bacteria-induced cancer stem cell properties
Cancer stem cells (CSCs) are well-acknowledged as a niche of cancer cells that are endowed with stem cell properties, chemotherapy resistant, hardly eradicated by clinical management and thus lead to tumor recurrence [27, 28]. Incubation with gram-negative bacteria increased the expressions of CSC-related genes including CD133, CD44, CD166 and ALDH1A1 (Figure 5A). This process was dependent on TLR4 signaling and IL-33 expression in NSCLC cells and could be abrogated by knockdown of TLR4 and IL-33 expressions (Figure 5B–5C). In support, gram-negative bacteria significantly enhanced the mammosphere formation through TLR4 activation and IL-33 upregulation in NSCLC cells (Figure 5D–5F).
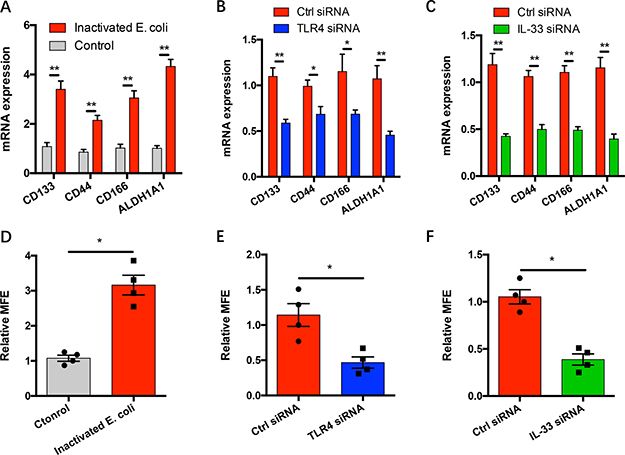
Figure 5: IL-33 licenses gram-negative bacteria-induced stem cell properties. (A) NSCLC cells from 5 patients were incubated with heat-inactivated E. coli (3x107 CFU/ml) for 12 hours and detected for mRNA expressions of the indicated CSC-related genes. (B–C) NSCLC cells from 5 patients were transfected with TLR4 siRNA, IL-33 siRNA or the control respectively, incubated with heat-inactivated E. coli (3x107 CFU/ml) for 12 hours. (D) NSCLC cells from 4 patients were incubated with or without heat-inactivated E. coli (3x107 CFU/ml) and detected for MFE. (E–F) NSCLC cells from 4 patients were transfected with TLR4 siRNA, IL-33 siRNA or the control respectively, incubated with heat-inactivated E. coli (3x107 CFU/ml) and analyzed for MFE. Each dot represents the data from one patient. *p < 0.05. **p < 0.01.
TLR4/IL-33 pathway correlates with tumor growth and expression of CSC-related gene in NSCLC patients with gram-negative bacterial infection
To investigate the in vivo relevance of TLR4 signaling with IL-33 expression and NSCLC progression in clinical patients, NSCLC tissues with or without gram-negative bacterial infection were analyzed for TLR4, IL-33 and CD133 expressions. We observed higher TLR4, IL-33 and CD133 expressions in NSCLC patients with gram-negative bacterial infection (Figure 6A–6C). TLR4 expressions were closely correlated with IL-33 expressions in gram-negative bacterial infected patients (Figure 6D). Of note, TLR4 and IL-33 expressions were positively associated with Ki-67 proliferative index (PI) and expressions of CSC-related gene CD133 in NSCLC patients with gram-negative bacterial infection (Figure 6E–6H).
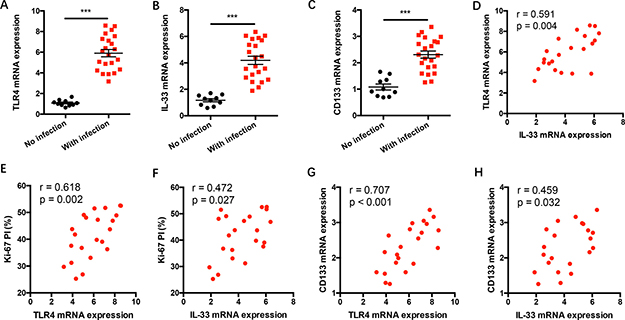
Figure 6: TLR4/IL-33 signaling correlates with tumor progression in NSCLC patients with gram-negative bacterial infection. (A–C) NSCLC patients with or without gram-negative bacterial infection were detected for mRNA expressions of TLR4, IL-33 and CD133 in tumor tissues. (D–H) TLR4 and IL-33 expressions were analyzed for correlations with tumor Ki-67 proliferation index and CSC-related gene CD133 expression in gram-negative bacteria affected NSCLC tissues. Each dot represents the data from one patient. ***p < 0.001.
DISCUSSION
The relationship between pathogenic infection and cancer progression, especially the pulmonary infection and lung cancer, has long been a subject of active investigation [16, 17]. Gram-negative bacterial infection is the most common pathogen that affects patients with NSCLC, either in patients with postoperative complications or in patients with advanced disease [16, 17, 29]. Herein, we identified a novel mechanism underlies gram-negative bacteria mediated NSCLC progression. Specifically, by stimulating TLR4 signaling, gram-negative bacteria induced robust IL-33 expression in NSCLC cells, facilitating cancer metabolic reprograming and development of cancer stem cell properties.
Toll-like receptors are pattern recognition receptors that recognize pathogen-associated molecular patterns, a crucial protective mechanism in innate immunity and human diseases [30, 31]. Accumulating evidence have demonstrated an important function of TLR signaling in lung cancer pathogenesis [32, 33]. As such, activation of TLR4 or TLR9 enhanced growth and metastasis of lung cancer [34–37]. Triggering of TLR7 and TLR8 expressed by human lung cancer cells induced cell survival and chemo-resistance [38]. In current study, we determined that blockade of TLR4 signaling efficiently abrogated gram-negative bacteria mediated NSCLC progression. In consistent, gram-negative bacteria activated TLR4 in NSCLC cells, promoting tumor growth and metastasis [16, 17]. It is reasonable because lipopolysaccharides, the major component of the outer membrane of gram-negative bacteria, is a well-known ligand for TLR4 [35, 39].
IL-33 is recognized as a dual-functional factor in maintenance of barrier functions and in driving Th2 response [19, 23]. Recent studies identified IL-33 as a critical factor involved in cancer pathogenies [40, 41]. IL-33 signaling contributed to intestinal tumorigenesis in humans and mice [42]. IL-33/ST2 pathway facilitated metastasis of human colorectal cancer [43]. IL-33 promoted growth and liver metastasis of colorectal cancer in mice by remodeling the tumor microenvironment and inducing angiogenesis [44]. Besides, IL33/ST2 axis could promote epithelial cell transformation and breast tumorigenesis via upregulation of COT activity [45]. IL-33/ST2 axis promoted breast cancer growth and metastases by facilitating intratumoral accumulation of immunosuppressive and innate lymphoid cells [46]. In addition, IL-33 expression predicted poor prognosis and promoted ovarian cancer cell growth and metastasis through regulating ERK and JNK signaling pathways [47]. IL-33 also promoted gastric cancer cell metastasis through ST2-ERK1/2 pathway [48]. Similarly, higher decline extent of serum IL-33 level was a predictor of shorter progression-free survival after chemotherapy in gastric cancer patients [49]. Mechanistically, IL-33 activated tumor stroma to promote intestinal polyposis [50]. IL-33 in tumor microenvironment was crucial for the accumulation and function of myeloid-derived suppressor cells [51]. Herein, we extended previous studies by demonstrating that gram-negative bacterial infection led to high expression of IL-33 in NSCLC cells through TLR4 activation. IL-33 knockdown decreased the enhanced cancer glycolysis, lipogenesis and stemness by gram-negative bacteria, suggesting IL-33 as an effective regulator of cancer metabolism and cancer stem cell properties. In consistent with these findings, IL-33 signaling promoted membrane GLUT1 localization and glycolysis of NSCLC cells [25]. IL-33 facilitated endocrine resistance of breast cancer by inducing cancer stem cell properties [52]. Of important, blockade of IL-33 restricted tumor progression of human NSCLC xenograft in immune-deficient mice [53].
IL-33 production could be induced by TLR agonists in IRF3 dependent manner [54]. TLR4 activation upregulated IL-33 expression in bone marrow-derived dendritic cells, alveolar macrophages, and respiratory dendritic cells [55]. TLR4 activation by short ragweed pollen led to IL-33 mRNA and protein expression in MyD88 and NF-kB dependent manner in ocular surface, cervical lymph nodes and isolated CD4 T cells of BALB/c mice [56]. Cyclic stretch activated TLR4 signaling to induce IL-33 production through HMGB1 in murine respiratory epithelial cells [57]. On the other hand, IL-33 increased the expression of the LPS receptor components MD2 and TLR4, the soluble form of CD14 and the MyD88 adaptor molecule, enhancing the LPS-induced pro-inflammatory response in macrophage [58]. Combing these findings indicate a positive feedback loop for TLR4/IL-33 pathway. Herein, we showed that gram-negative bacteria stimulation resulted in up-regulation of IL-33 expression in NSCLC cells and this process could be inhibited by TLR4 knockdown. Considering the aberrant expression of IRF3 in human lung cancer and the crucial role of NF-kB activity in lung cancer carcinogenesis [59, 60], the mechanisms underlie TLR4-induced IL-33 expression in NSCLC cells might involve IRF3 and NF-kB pathway, as well as HMGB1 since it binds to LPS to facilitate LPS transfer and initiate TLR4 signaling [61].
It should be pointed out that plasma IL-33 levels could be inversely associated with progression of lung cancer [62]. IL-33 was reported to enhance programmed oncosis of ST2L-positive low-metastatic Lewis lung carcinoma [63]. Tumoral expression of IL-33 could inhibit tumor growth and modify tumor microenvironment through CD8 T and NK cells in mice bearing B16 cells and 4T1 cells [64]. IL-33 could restrict tumor growth and inhibit pulmonary metastasis in melanoma-bearing mice through eosinophils [65]. Systemic administration of recombinant IL-33 protein could inhibit growth of established tumors in transplant and de novo melanoma tumorigenesis models through inducing CD8 T cell expansion and IFN-γ production, as well as through restoring dendritic cell activation and maturation [66]. These studies suggest an anti-cancer activity of IL-33 in tumor immunity, reflecting a complexity of IL-33 in cancer pathogenesis. These conflicting phenomena might due to distinct types of cancer and different cohorts of clinical patients. Clinical therapeutics such as radiotherapy and chemotherapy could induce IL-33 expressions in tumor tissue of clinical patients [67, 68], suggesting clinical managements as critical factors in determining IL-33 expressions. Besides, research findings from established cancer cell lines could be different with those from primary cancer cells in clinical patients. We propose tumoral IL-33 expression and its function in tumor progression as an individualized element to be considered in lung cancer patients.
In essence, pulmonary infection with gram-negative bacteria in NSCLC patients results in TLR4 activation and IL-33 overexpression, promoting tumor progression and recurrence by enhancing cancer metabolic reprograming and cancer stem cell properties. These findings derived from primary tumor cells reflect a realistic condition in clinical patients. Blocking TLR4/IL-33 pathway could be an optimistic strategy in controlling NSCLC progression and recurrence.
MATERIALS AND METHODS
Patients
A cohort of 121 patients diagnosed with NSCLC was enrolled in this study. Surgical tissues and clinical parameters were collected after written consent form was obtained. The NSCLC patients were forty-one to eighty-three years old. Fifty-eight patients were males and eighty-one cases were adenocarcinoma. The burden of infected gram-negative bacteria was from medical laboratory. Ki-67 proliferation index was from pathology division. Experiments were performed in accordance with the 1964 Helsinki declaration including its later amendments and approved by Jilin Institutional Ethics Committee.
Mice
Immuno-deficient NOD-SCID mice of 8–10 weeks old were from Beijing Vital River Laboratory Animal Technology Co., Ltd and housed under specific pathogen free animal facility. Experiments were approved by Jilin Institutional Ethics Committee.
Cells and reagents
Clinical surgical tissues were dissociated into a single-cell suspension with the human Tumor Dissociation Kit (Miltenyi Biotec). NSCLC cells were isolated using the human Tumor Cell Isolation Kit (Miltenyi Biotec). Experiments were performed according to the manufactures’ instructions. Cells were cultured in complete RPMI 1640 medium containing 10% FBS (Thermo Fisher Scientific) plus Penicillin-Streptomycin-Glutamine (1:100, Thermo Fisher Scientific) at 37°C under 5% CO2 as previously described [25].
Heat-inactivation of gram-negative bacteria Escherichia coli (ATCC® 11775™) was performed as previously described [16, 17]. Human TLR4 siRNA, IL-33 siRNA and the controls were from Santa Cruz Biotechnology. MyD88 inhibitory peptide and control peptide were from In vivo Gen. Cellular Nucleofector™ kit was from Lonza. All reagents were used according to the manufactures’ instructions.
Real-time PCR
Real-time qPCR was performed using SYBR Green qPCR Master Mix (Bimake) as previously described [69]. All qPCR primers were from Sino Biological. Beta-actin was used as the internal control.
Flow cytometry
Flow cytometric analysis with NSCLC cells were performed as previously described [25, 69]. Briefly, 0.2 million NSCLC cells were fixed with BD Fix Buffer I, washed and stained with APC labeled anti-human TLR4 antibody (BioLegend) or PE anti-human GLUT1 antibody (R&D Systems) at 4°C for 45 min. Otherwise, cells were fixed with BD Fix Buffer I, permeabilized with BD Perm Buffer III, washed and stained with PE labeled anti-human IL-33 antibody (R&D Systems) for 45 min under room temperature. Cells were acquired using BD LSR II flow cytometer and data analysis was performed with FlowJo software (Tree Star).
Lactate production
Lactate production was determined with Lactate Assay Kit (Sigma) as previously described [25].
Mammosphere (MS) formation assay
NSCLC cells were seeded at 1×105 cells/ml at ultralow attachment plates and cultured for 7 days. MS with a diameter ≥ 50μm were counted and MS-forming efficiency (MFE) was calculated by dividing the number of MS with original number of seeded cells [52].
MTT assay
In vitro growth capacity of NSCLC cells was determined with MTT assay [16, 25]. Briefly, 10 thousand cells were incubated with or without heat-inactivated E. coli for 72 hours and detected for proliferative expansion with MTT cell proliferation kit (Cayman Chemical) according to the manufacturer’s instructions.
Invasion assay
In vitro invasion of NSCLC cells was detected using Corning® BioCoat™ Matrigel® Invasion Chambers [16, 25]. 0.1 million NSCLC cells suspended in 0.5 mL of serum-free culture medium were seeded onto the upper chamber of the Matrigel inserts. Complete culture medium containing 10% FBS was placed into the lower chambers. 24 hours later, invading cells on the lower side of the chamber were fixed, stained and counted under microscope. Data is expressed as the percent invasion through the Matrigel inserts relative to the control inserts.
Tumor growth
Tumor growth in vivo was performed in NOD-SCID mice as previously described [16, 25, 34, 35]. Briefly, 5 million NSCLC cells were pretreated with or without heat-inactivated E. coli for 24 hours and subcutaneously injected into NOD-SCID mice. Tumor growth was determined by measuring the tumor size with digital calipers. NSCLC cells that failed in tumorigenesis were excluded from the study.
Tumor metastasis
Tumor metastasis in vivo was performed in NOD-SCID mice according to previously described method [25, 70–72]. NOD-SCID mice were intratracheally challenged with LPS (10 μg/mouse, Sigma) and injected with 1 million NSCLC cells through the tail veins after 6 hours. Tumor metastasis was determined by lung weight to reflect the tumor burden at 14th day post NSCLC injection.
Statistical analyses
Data were presented as mean ± SEM. Non-parametric Mann-Whitney U test and Spearman rank correlation coefficient were applied for statistical analysis using GraphPad Prism 6.0 software. P < 0.05 was considered as statistically significant.
Author contributions
M.S, C.M and D.M designed the study. M.S, Y.B, S.Z and X.L were responsible for the experimental work and data analysis. Y.G, L.W and B.L helped in data analysis. M.S, X.L, C.M and D.M wrote the manuscript. All authors have reviewed the manuscript.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This work was supported by grants from National Natural Science Foundation of China (81300186 and 81301035). The funding has no involvement in study design, data collection, data interpretation and manuscript written.
REFERENCES
1. Chen W, Zheng R, Zeng H, Zhang S. Epidemiology of lung cancer in China. Thorac Cancer. 2015; 6:209–15. https://doi.org/10.1111/1759-7714.12169.
2. Inamura K. Lung Cancer: Understanding Its Molecular Pathology and the 2015 WHO Classification. Front Oncol. 2017; 7:193. https://doi.org/10.3389/fonc.2017.00193.
3. Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA. Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc. 2008; 83:584–94. https://doi.org/10.1016/S0025-619660735-0.
4. Paoletti L, Pastis NJ, Denlinger CE, Silvestri GA. A decade of advances in treatment of early-stage lung cancer. Clin Chest Med. 2011; 32:827–38. https://doi.org/10.1016/j.ccm.2011.08.009.
5. Brower V. Adding radiotherapy to chemotherapy in advanced NSCLC. Lancet Oncol. 2017; 18:e645. https://doi.org/10.1016/S1470-204530771-4.
6. Pillai RN, Behera M, Owonikoko TK, Kamphorst AO, Pakkala S, Belani CP, Khuri FR, Ahmed R, Ramalingam SS. Comparison of the toxicity profile of PD-1 versus PD-L1 inhibitors in non-small cell lung cancer: A systematic analysis of the literature. Cancer. 2018; 124:271–277. https://doi.org/10.1002/cncr.31043.
7. Wu YL, Cheng Y, Zhou X, Lee KH, Nakagawa K, Niho S, Tsuji F, Linke R, Rosell R, Corral J, Migliorino MR, Pluzanski A, Sbar EI, et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): a randomised, open-label, phase 3 trial. Lancet Oncol. 2017; 18:1454–66. https://doi.org/10.1016/S1470-204530608-3.
8. Sheng Z, Zhu X, Sun Y, Zhang Y. The efficacy of anti-PD-1/PD-L1 therapy and its comparison with EGFR-TKIs for advanced non-small-cell lung cancer. Oncotarget. 2017; 8:57826–35. https://doi.org/10.18632/oncotarget.18406.
9. Uramoto H, Tanaka F. Recurrence after surgery in patients with NSCLC. Transl Lung Cancer Res. 2014; 3:242–49.
10. Baltayiannis N, Chandrinos M, Anagnostopoulos D, Zarogoulidis P, Tsakiridis K, Mpakas A, Machairiotis N, Katsikogiannis N, Kougioumtzi I, Courcoutsakis N, Zarogoulidis K. Lung cancer surgery: an up to date. J Thorac Dis. 2013; 5:S425–39.
11. Kim ES, Kim YT, Kang CH, Park IK, Bae W, Choi SM, Lee J, Park YS, Lee CH, Lee SM, Yim JJ, Kim YW, Han SK, et al. Prevalence of and risk factors for postoperative pulmonary complications after lung cancer surgery in patients with early-stage COPD. Int J Chron Obstruct Pulmon Dis. 2016; 11:1317–26. https://doi.org/10.2147/COPD.S105206.
12. Lugg ST, Agostini PJ, Tikka T, Kerr A, Adams K, Bishay E, Kalkat MS, Steyn RS, Rajesh PB, Thickett DR, Naidu B. Long-term impact of developing a postoperative pulmonary complication after lung surgery. Thorax. 2016; 71:171–76. https://doi.org/10.1136/thoraxjnl-2015-207697.
13. Subotic D, Van Schil P, Grigoriu B. Optimising treatment for post-operative lung cancer recurrence. Eur Respir J. 2016; 47:374–78. https://doi.org/10.1183/13993003.01490-2015.
14. Berghmans T, Sculier JP, Klastersky J. A prospective study of infections in lung cancer patients admitted to the hospital. Chest. 2003; 124:114–20. https://doi.org/10.1378/chest.124.1.114.
15. Mei J, Liu L, Tang M, Xu N, Pu Q, Liu C, Ma L, Shi H, Che G. Airway bacterial colonization in patients with non-small cell lung cancer and the alterations during the perioperative period. J Thorac Dis. 2014; 6:1200–08.
16. Ye M, Gu X, Han Y, Jin M, Ren T. Gram-negative bacteria facilitate tumor outgrowth and metastasis by promoting lipid synthesis in lung cancer patients. J Thorac Dis. 2016; 8:1943–55. https://doi.org/10.21037/jtd.2016.06.47.
17. Chow SC, Gowing SD, Cools-Lartigue JJ, Chen CB, Berube J, Yoon HW, Chan CH, Rousseau MC, Bourdeau F, Giannias B, Roussel L, Qureshi ST, Rousseau S, et al. Gram negative bacteria increase non-small cell lung cancer metastasis via Toll-like receptor 4 activation and mitogen-activated protein kinase phosphorylation. Int J Cancer. 2015; 136:1341–50. https://doi.org/10.1002/ijc.29111.
18. Hodzic Z, Schill EM, Bolock AM, Good M. IL-33 and the intestine: The good, the bad, and the inflammatory. Cytokine. 2017; 100:1–10. https://doi.org/10.1016/j.cyto.2017.06.017.
19. Drake LY, Kita H. IL-33: biological properties, functions, and roles in airway disease. Immunol Rev. 2017; 278:173–84. https://doi.org/10.1111/imr.12552.
20. Griesenauer B, Paczesny S. The ST2/IL-33 Axis in Immune Cells during Inflammatory Diseases. Front Immunol. 2017; 8:475. https://doi.org/10.3389/fimmu.2017.00475.
21. Wang Y, Wang L, Hua S. Interleukin-33 in children with asthma: A systematic review and meta-analysis. Allergol Immunopathol (Madr). 2017; 45:387–92. https://doi.org/10.1016/j.aller.2016.12.007.
22. Hardman C, Ogg G. Interleukin-33, friend and foe in type-2 immune responses. Curr Opin Immunol. 2016; 42:16–24. https://doi.org/10.1016/j.coi.2016.05.004.
23. Liew FY, Girard JP, Turnquist HR. Interleukin-33 in health and disease. Nat Rev Immunol. 2016; 16:676–89. https://doi.org/10.1038/nri.2016.95.
24. Hu LA, Fu Y, Zhang DN, Zhang J. Serum IL-33 as a diagnostic and prognostic marker in non- small cell lung cancer. Asian Pac J Cancer Prev. 2013; 14:2563–66. https://doi.org/10.7314/APJCP.2013.14.4.2563.
25. Wang C, Chen Z, Bu X, Han Y, Shan S, Ren T, Song W. IL-33 signaling fuels outgrowth and metastasis of human lung cancer. Biochem Biophys Res Commun. 2016; 479:461–68. https://doi.org/10.1016/j.bbrc.2016.09.081.
26. Zaidi N, Lupien L, Kuemmerle NB, Kinlaw WB, Swinnen JV, Smans K. Lipogenesis and lipolysis: the pathways exploited by the cancer cells to acquire fatty acids. Prog Lipid Res. 2013; 52:585–89. https://doi.org/10.1016/j.plipres.2013.08.005.
27. Dragu DL, Necula LG, Bleotu C, Diaconu CC, Chivu-Economescu M. Therapies targeting cancer stem cells: current trends and future challenges. World J Stem Cells. 2015; 7:1185–201.
28. Plaks V, Kong N, Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell. 2015; 16:225–38. https://doi.org/10.1016/j.stem.2015.02.015.
29. Lanoix JP, Pluquet E, Lescure FX, Bentayeb H, Lecuyer E, Boutemy M, Dumont P, Jounieaux V, Schmit JL, Dayen C, Douadi Y. Bacterial infection profiles in lung cancer patients with febrile neutropenia. BMC Infect Dis. 2011; 11:183. https://doi.org/10.1186/1471-2334-11-183.
30. Joosten LA, Abdollahi-Roodsaz S, Dinarello CA, O’Neill L, Netea MG. Toll-like receptors and chronic inflammation in rheumatic diseases: new developments. Nat Rev Rheumatol. 2016; 12:344–57. https://doi.org/10.1038/nrrheum.2016.61.
31. Gay NJ, Symmons MF, Gangloff M, Bryant CE. Assembly and localization of Toll-like receptor signalling complexes. Nat Rev Immunol. 2014; 14:546–58. https://doi.org/10.1038/nri3713.
32. Guha M. Anticancer TLR agonists on the ropes. Nat Rev Drug Discov. 2012; 11:503–05. https://doi.org/10.1038/nrd3775.
33. Rakoff-Nahoum S, Medzhitov R. Toll-like receptors and cancer. Nat Rev Cancer. 2009; 9:57–63. https://doi.org/10.1038/nrc2541.
34. Hattar K, Savai R, Subtil FS, Wilhelm J, Schmall A, Lang DS, Goldmann T, Eul B, Dahlem G, Fink L, Schermuly RT, Banat GA, Sibelius U, et al. Endotoxin induces proliferation of NSCLC in vitro and in vivo: role of COX-2 and EGFR activation. Cancer Immunol Immunother. 2013; 62:309–20. https://doi.org/10.1007/s00262-012-1341-2.
35. Zhang X, Wang C, Shan S, Liu X, Jiang Z, Ren T. TLR4/ROS/miRNA-21 pathway underlies lipopolysaccharide instructed primary tumor outgrowth in lung cancer patients. Oncotarget. 2016; 7:42172–82. https://doi.org/10.18632/oncotarget.9902.
36. Zhan Z, Xie X, Cao H, Zhou X, Zhang XD, Fan H, Liu Z. Autophagy facilitates TLR4- and TLR3-triggered migration and invasion of lung cancer cells through the promotion of TRAF6 ubiquitination. Autophagy. 2014; 10:257–68. https://doi.org/10.4161/auto.27162.
37. Xu L, Wen Z, Zhou Y, Liu Z, Li Q, Fei G, Luo J, Ren T. MicroRNA-7-regulated TLR9 signaling-enhanced growth and metastatic potential of human lung cancer cells by altering the phosphoinositide-3-kinase, regulatory subunit 3/Akt pathway. Mol Biol Cell. 2013; 24:42–55. https://doi.org/10.1091/mbc.E12-07-0519.
38. Cherfils-Vicini J, Platonova S, Gillard M, Laurans L, Validire P, Caliandro R, Magdeleinat P, Mami-Chouaib F, Dieu-Nosjean MC, Fridman WH, Damotte D, Sautès-Fridman C, Cremer I. Triggering of TLR7 and TLR8 expressed by human lung cancer cells induces cell survival and chemoresistance. J Clin Invest. 2010; 120:1285–97. https://doi.org/10.1172/JCI36551.
39. De S, Zhou H, DeSantis D, Croniger CM, Li X, Stark GR. Erlotinib protects against LPS-induced endotoxicity because TLR4 needs EGFR to signal. Proc Natl Acad Sci USA. 2015; 112:9680–85. https://doi.org/10.1073/pnas.1511794112.
40. Schwartz C, O’Grady K, Lavelle EC, Fallon PG. Interleukin 33: an innate alarm for adaptive responses beyond Th2 immunity-emerging roles in obesity, intestinal inflammation, and cancer. Eur J Immunol. 2016; 46:1091–100. https://doi.org/10.1002/eji.201545780.
41. Lu B, Yang M, Wang Q. Interleukin-33 in tumorigenesis, tumor immune evasion, and cancer immunotherapy. J Mol Med (Berl). 2016; 94:535–43. https://doi.org/10.1007/s00109-016-1397-0.
42. Mertz KD, Mager LF, Wasmer MH, Thiesler T, Koelzer VH, Ruzzante G, Joller S, Murdoch JR, Brümmendorf T, Genitsch V, Lugli A, Cathomas G, Moch H, et al. The IL-33/ST2 pathway contributes to intestinal tumorigenesis in humans and mice. OncoImmunology. 2015; 5:e1062966. https://doi.org/10.1080/2162402X.2015.1062966.
43. Liu X, Zhu L, Lu X, Bian H, Wu X, Yang W, Qin Q. IL-33/ST2 pathway contributes to metastasis of human colorectal cancer. Biochem Biophys Res Commun. 2014; 453:486–92. https://doi.org/10.1016/j.bbrc.2014.09.106.
44. Zhang Y, Davis C, Shah S, Hughes D, Ryan JC, Altomare D, Peña MM. IL-33 promotes growth and liver metastasis of colorectal cancer in mice by remodeling the tumor microenvironment and inducing angiogenesis. Mol Carcinog. 2017; 56:272–87. https://doi.org/10.1002/mc.22491.
45. Kim JY, Lim SC, Kim G, Yun HJ, Ahn SG, Choi HS. Interleukin-33/ST2 axis promotes epithelial cell transformation and breast tumorigenesis via upregulation of COT activity. Oncogene. 2015; 34:4928–38. https://doi.org/10.1038/onc.2014.418.
46. Jovanovic IP, Pejnovic NN, Radosavljevic GD, Pantic JM, Milovanovic MZ, Arsenijevic NN, Lukic ML. Interleukin-33/ST2 axis promotes breast cancer growth and metastases by facilitating intratumoral accumulation of immunosuppressive and innate lymphoid cells. Int J Cancer. 2014; 134:1669–82. https://doi.org/10.1002/ijc.28481.
47. Tong X, Barbour M, Hou K, Gao C, Cao S, Zheng J, Zhao Y, Mu R, Jiang HR. Interleukin-33 predicts poor prognosis and promotes ovarian cancer cell growth and metastasis through regulating ERK, JNK signaling pathways. Mol Oncol. 2016; 10:113–25. https://doi.org/10.1016/j.molonc.2015.06.004.
48. Yu XX, Hu Z, Shen X, Dong LY, Zhou WZ, Hu WH. IL-33 Promotes Gastric Cancer Cell Invasion and Migration Via ST2-ERK1/2 Pathway. Dig Dis Sci. 2015; 60:1265–72. https://doi.org/10.1007/s10620-014-3463-1.
49. Hu W, Wu C, Li X, Zheng Z, Xie Q, Deng X, Jiang J, Wu C. Serum IL-33 level is a predictor of progression-free survival after chemotherapy. Oncotarget. 2017; 8:35116–23. https://doi.org/10.18632/oncotarget.16627.
50. Maywald RL, Doerner SK, Pastorelli L, De Salvo C, Benton SM, Dawson EP, Lanza DG, Berger NA, Markowitz SD, Lenz HJ, Nadeau JH, Pizarro TT, Heaney JD. IL-33 activates tumor stroma to promote intestinal polyposis. Proc Natl Acad Sci USA. 2015; 112:E2487–96. https://doi.org/10.1073/pnas.1422445112.
51. Xiao P, Wan X, Cui B, Liu Y, Qiu C, Rong J, Zheng M, Song Y, Chen L, He J, Tan Q, Wang X, Shao X, et al. Interleukin 33 in tumor microenvironment is crucial for the accumulation and function of myeloid-derived suppressor cells. OncoImmunology. 2015; 5:e1063772. https://doi.org/10.1080/2162402X.2015.1063772.
52. Hu H, Sun J, Wang C, Bu X, Liu X, Mao Y, Wang H. IL-33 facilitates endocrine resistance of breast cancer by inducing cancer stem cell properties. Biochem Biophys Res Commun. 2017; 485:643–50. https://doi.org/10.1016/j.bbrc.2017.02.080.
53. Wang K, Shan S, Yang Z, Gu X, Wang Y, Wang C, Ren T. IL-33 blockade suppresses tumor growth of human lung cancer through direct and indirect pathways in a preclinical model. Oncotarget. 2017; 8:68571–82. https://doi.org/10.18632/oncotarget.19786.
54. Polumuri SK, Jayakar GG, Shirey KA, Roberts ZJ, Perkins DJ, Pitha PM, Vogel SN. Transcriptional regulation of murine IL-33 by TLR and non-TLR agonists. J Immunol. 2012; 189:50–60. https://doi.org/10.4049/jimmunol.1003554.
55. Tjota MY, Williams JW, Lu T, Clay BS, Byrd T, Hrusch CL, Decker DC, de Araujo CA, Bryce PJ, Sperling AI. IL-33-dependent induction of allergic lung inflammation by FcγRIII signaling. J Clin Invest. 2013; 123:2287–97. https://doi.org/10.1172/JCI63802.
56. Li J, Zhang L, Chen X, Chen D, Hua X, Bian F, Deng R, Lu F, Li Z, Pflugfelder SC, Li DQ. Pollen/TLR4 Innate Immunity Signaling Initiates IL-33/ST2/Th2 Pathways in Allergic Inflammation. Sci Rep. 2016; 6:36150. https://doi.org/10.1038/srep36150.
57. Chang J, Xia Y, Wasserloos K, Deng M, Blose KJ, Vorp DA, Turnquist HR, Billiar TR, Pitt BA, Zhang MZ, Zhang LM. Cyclic stretch induced IL-33 production through HMGB1/TLR-4 signaling pathway in murine respiratory epithelial cells. PLoS One. 2017; 12:e0184770. https://doi.org/10.1371/journal.pone.0184770.
58. Espinassous Q, Garcia-de-Paco E, Garcia-Verdugo I, Synguelakis M, von Aulock S, Sallenave JM, McKenzie AN, Kanellopoulos J. IL-33 enhances lipopolysaccharide-induced inflammatory cytokine production from mouse macrophages by regulating lipopolysaccharide receptor complex. J Immunol. 2009; 183:1446–55. https://doi.org/10.4049/jimmunol.0803067.
59. Tokunaga T, Naruke Y, Shigematsu S, Kohno T, Yasui K, Ma Y, Chua KJ, Katayama I, Nakamura T, Hishikawa Y, Koji T, Yatabe Y, Nagayasu T, et al. Aberrant expression of interferon regulatory factor 3 in human lung cancer. Biochem Biophys Res Commun. 2010; 397:202–07. https://doi.org/10.1016/j.bbrc.2010.05.085.
60. Chen W, Li Z, Bai L, Lin Y. NF-kappaB in lung cancer, a carcinogenesis mediator and a prevention and therapy target. Front Biosci (Landmark Ed). 2011; 16:1172–85. https://doi.org/10.2741/3782.
61. Youn JH, Oh YJ, Kim ES, Choi JE, Shin JS. High mobility group box 1 protein binding to lipopolysaccharide facilitates transfer of lipopolysaccharide to CD14 and enhances lipopolysaccharide-mediated TNF-alpha production in human monocytes. J Immunol. 2008; 180:5067–74. https://doi.org/10.4049/jimmunol.180.7.5067.
62. Kim MS, Kim E, Heo JS, Bae DJ, Lee JU, Lee TH, Lee HJ, Chang HS, Park JS, Jang AS, Koh ES, Hwang HG, Lim G, et al. Circulating IL-33 level is associated with the progression of lung cancer. Lung Cancer. 2015; 90:346–51. https://doi.org/10.1016/j.lungcan.2015.08.011.
63. Akimoto M, Hayashi JI, Nakae S, Saito H, Takenaga K. Interleukin-33 enhances programmed oncosis of ST2L-positive low-metastatic cells in the tumour microenvironment of lung cancer. Cell Death Dis. 2016; 7:e2057. https://doi.org/10.1038/cddis.2015.418.
64. Gao X, Wang X, Yang Q, Zhao X, Wen W, Li G, Lu J, Qin W, Qi Y, Xie F, Jiang J, Wu C, Zhang X, et al. Tumoral expression of IL-33 inhibits tumor growth and modifies the tumor microenvironment through CD8+ T, NK cells. J Immunol. 2015; 194:438–45. https://doi.org/10.4049/jimmunol.1401344.
65. Lucarini V, Ziccheddu G, Macchia I, La Sorsa V, Peschiaroli F, Buccione C, Sistigu A, Sanchez M, Andreone S, D’Urso MT, Spada M, Macchia D, Afferni C, et al. IL-33 restricts tumor growth and inhibits pulmonary metastasis in melanoma-bearing mice through eosinophils. OncoImmunology. 2017; 6:e1317420. https://doi.org/10.1080/2162402X.2017.1317420.
66. Dominguez D, Ye C, Geng Z, Chen S, Fan J, Qin L, Long A, Wang L, Zhang Z, Zhang Y, Fang D, Kuzel TM, Zhang B. Exogenous IL-33 Restores Dendritic Cell Activation and Maturation in Established Cancer. J Immunol. 2017; 198:1365–75. https://doi.org/10.4049/jimmunol.1501399.
67. Wasmer MH, Krebs P. The Role of IL-33-Dependent Inflammation in the Tumor Microenvironment. Front Immunol. 2017; 7:682. https://doi.org/10.3389/fimmu.2016.00682.
68. Ha CT, Li XH, Fu D, Moroni M, Fisher C, Arnott R, Srinivasan V, Xiao M. Circulating interleukin-18 as a biomarker of total-body radiation exposure in mice, minipigs, and nonhuman primates (NHP). PLoS One. 2014; 9:e109249. https://doi.org/10.1371/journal.pone.0109249.
69. Wen Z, Shen Y, Berry G, Shahram F, Li Y, Watanabe R, Liao YJ, Goronzy JJ, Weyand CM. The microvascular niche instructs T cells in large vessel vasculitis via the VEGF-Jagged1-Notch pathway. Sci Transl Med. 2017; 9:9. https://doi.org/10.1126/scitranslmed.aal3322.
70. Yan L, Cai Q, Xu Y. The ubiquitin-CXCR4 axis plays an important role in acute lung infection-enhanced lung tumor metastasis. Clin Cancer Res. 2013; 19:4706–16. https://doi.org/10.1158/1078-0432.CCR-13-0011.
71. Liu X, Pei C, Yan S, Liu G, Liu G, Chen W, Cui Y, Liu Y. NADPH oxidase 1-dependent ROS is crucial for TLR4 signaling to promote tumor metastasis of non-small cell lung cancer. Tumour Biol. 2015; 36:1493–502. https://doi.org/10.1007/s13277-014-2639-9.
72. Li Q, Han Y, Wang C, Shan S, Wang Y, Zhang J, Ren T. MicroRNA-125b promotes tumor metastasis through targeting tumor protein 53-induced nuclear protein 1 in patients with non-small-cell lung cancer. Cancer Cell Int. 2015; 15:84. https://doi.org/10.1186/s12935-015-0233-x.