
INTRODUCTION
Infantile hemangioma (IH) is the most common vascular tumour in childhood, affecting 4% to 5% of infants worldwide [1]. Hemangiomas can show severe progression, which leads to tissue and organ damage that in some cases becomes life-threatening. Clinical treatment varies, including steroids, interferon-alfa, and β-blocker propranolol [2, 3]. However, no definitive therapy is available for IH due to the adverse effects of each drug. The risk factors for IH include preterm birth and placental anomalies [4]. In most cases, IH has a unique clinical course with proliferation and involution phases [5]. Numerous genes involved in IH have been identified. However, the pathogenesis and cause of hemangioma remain largely unknown.
The competitive endogenous RNA (ceRNA) hypothesis proposes that RNA transcripts, both coding and non-coding, compete for post-transcriptional control and coregulate each other using microRNA response elements (MREs) [6, 7]. Mounting evidence has shown that long non-coding RNAs and messenger RNAs can function as ceRNAs in diverse physiological and pathophysiological states such as myogenesis, melanoma development and cancer [8–11]. A recent study profiled the expression of distinct long non-coding RNAs (lncRNAs) in infantile hemangioma using microarray analysis and suggested that lncRNAs regulated several genes with important roles in angiogenesis [12]. Endothelial and circulating C19MC microRNAs are biomarkers of infantile hemangioma [13]. Additionally, integrative meta-analysis identified microRNA-regulated networks in infantile hemangioma [14]. However, the role of the ceRNA network in IH has not been elucidated.
In this study, we used Ribo-Zero RNA-Seq and HiSeq to examine the global expression profiles of protein-coding transcripts and non-coding RNAs, including miRNAs and lncRNAs, in IH and matched normal skin controls. Subsequently, gene ontology and pathway analysis displayed that, compared with the matched normal skin controls, many processes over-represented in IH were related to immune system processes, extracellular region regulation, and carbohydrate derivative binding. Further ceRNA network analysis identified 1256 sponge modulators including 87 miRNA-mediated, 70 lncRNA-mediated and 58 mRNA-mediated interactions. Our study may help expand understanding of the roles of the transcriptome, particularly non-coding transcripts, in the mechanisms underlying IH development and provide new research directions.
RESULTS
Differential expression profiles and bioinformatics analysis of mRNAs in IH compared with matched normal skin controls
To profile differentially expressed mRNAs, lncRNAs and miRNAs in IH, we performed RNA-seq on 3 IH samples and matched normal skin controls. We used an Illumina HiseqXTen platform (Illumina, San Diego, CA) for sequencing with (2 × 150 bp) the paired-end module. Fold changes (IH vs. matched normal skin controls) and p values were calculated from the normalized expression levels. Hierarchical clustering showed distinguishable mRNA expression patterns among the samples (Figure 1A). Up to 144 mRNAs were differentially expressed in the IH samples compared with the matched normal skin controls (fold change ≥2, P ≤ 0.05; for a list of differentially expressed mRNAs, see Table 1). A total of 84 and 60 mRNAs were up-regulated or down-regulated, respectively, by more than two-fold in IH vs. adjacent normal skin tissues (P < 0.05) (Figure 1B). KEGG Pathway analysis indicated that the chemokine, NF-kappa B and TGF-beta signalling pathways, as well as cell adhesion molecules (CAMs), were mostly found in the IH samples compared with matched normal skin (Figure 1C). In addition, gene ontology (GO) analysis revealed that numerous biological processes, molecular functions and cellular components were involved. Many of the processes that are deregulated in IH were related to immune system processes, carbohydrate derivative binding and extracellular region regulation (Figure 1D).
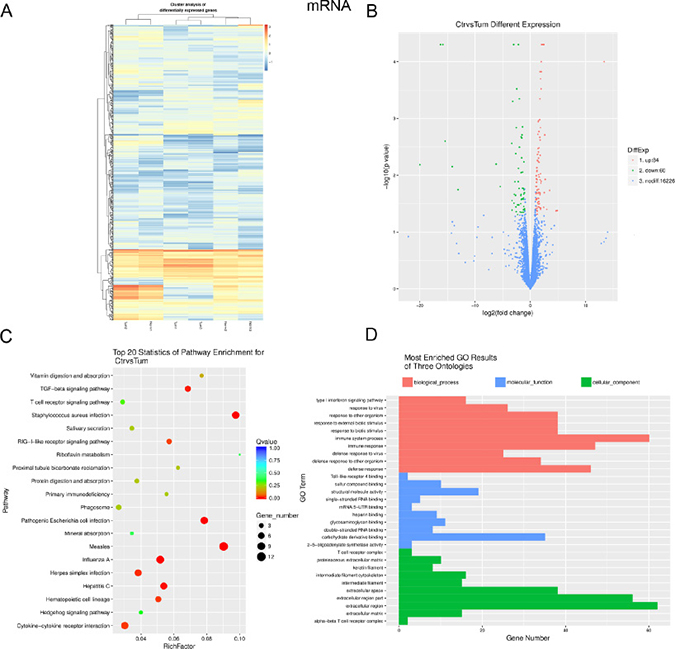
Figure 1: Expression profiles, Gene ontology (GO) terms and pathways for differentially expressed mRNAs between infantile hemangioma and adjacent normal skin tissues. (A) Hierarchical clustering shows a distinguishable mRNA expression profiling among groups. (B) Volcano analysis exhibit differentially expressed mRNAs. Red dots represent up-regulated genes. Green dots illustrate down-regulated genes. (C) The top 20 pathways that are associated with the coding genes are listed. The enrichment Q value or false discovery rate correct the p value for multiple comparisons. P values are calculated using Fisher’s exact test. The term/pathway on the vertical axis is drawn according to the first letter of the pathway in descending order. The horizontal axis represents the enrichment factor, i.e., (the number of dysregulated genes in a pathway/the total number of dysregulated gene)/(the number of genes in a pathway in the database/the total number of genes in the database). Top 20 enriched pathways are selected according to the enrichment factor value. The selection standards were the number of genes in a pathway ≥4. The different colours from green to red represent the Q value (False discovery rate value). The different sizes of the round shapes represent the gene count number in a pathway. (D) Most enriched GO terms of the three ontologies that are associated with the differentially expressed coding genes are listed. The horizontal axis represents the gene number. The term/GO on the vertical axis is drawn according to the first letter of the GO in descending order. Red bar represents the biological process, blue bar displays the molecular function, and green bar illustrates the cellular component. Norm or Ctr, matched normal skin tissue; Tum, infantile hemangioma skin tissue.
Table 1: List of up-regulated and down-regulated mRNAs detected using RNA-seq (FC ≥ 2.83, P < 0.05)
Gene Name |
log2(Tum/Ctr) |
up-or-down |
P_value |
Description |
|---|---|---|---|---|
MPO |
–6.18461 |
down |
0.00635 |
myeloperoxidase |
MAGEB2 |
–5.44166 |
down |
0.0158 |
MAGE family member B2 |
CD8A |
–3.35204 |
down |
0.00275 |
CD8a molecule |
BPI |
–3.31652 |
down |
0.0279 |
bactericidal/permeability-increasing protein |
PGLYRP1 |
–3.3111 |
down |
0.0412 |
peptidoglycan recognition protein 1 |
LOC283788 |
–3.17651 |
down |
0.0005 |
FSHD region gene 1 pseudogene |
IL18R1 |
–3.02589 |
down |
5.00E-05 |
interleukin 18 receptor 1 |
ADCYAP1 |
–2.96929 |
down |
0.031 |
adenylate cyclase activating polypeptide 1 |
CXCL13 |
–2.90547 |
down |
0.02165 |
C-X-C motif chemokine ligand 13 |
MS4A1 |
–2.78553 |
down |
0.013 |
membrane spanning 4-domains A1 |
SERPINB4 |
–2.67117 |
down |
0.02235 |
serpin family B member 4 |
MMP12 |
–2.65413 |
down |
0.02265 |
matrix metallopeptidase 12 |
PIP |
–2.63046 |
down |
0.00195 |
prolactin induced protein |
LEFTY2 |
–2.59114 |
down |
0.0361 |
left-right determination factor 2 |
IL13RA2 |
–2.54593 |
down |
0.04385 |
interleukin 13 receptor subunit alpha 2 |
CSMD3 |
–2.54162 |
down |
0.00145 |
CUB and Sushi multiple domains 3 |
OR51E1 |
–2.45263 |
down |
0.0003 |
olfactory receptor family 51 subfamily E member 1 |
PTH2R |
–2.32814 |
down |
0.03325 |
parathyroid hormone 2 receptor |
ADRB3 |
–2.30562 |
down |
0.0493 |
adrenoceptor beta 3 |
CCL4L2 |
–2.18575 |
down |
0.01295 |
C-C motif chemokine ligand 4 like 2 |
ERAP2 |
–2.14489 |
down |
5.00E-05 |
endoplasmic reticulum aminopeptidase 2 |
LTF |
–2.14431 |
down |
0.00755 |
lactotransferrin |
PADI4 |
–2.08682 |
down |
0.0333 |
peptidyl arginine deiminase 4 |
OR51E2 |
–2.0297 |
down |
0.01705 |
olfactory receptor family 51 subfamily E member 2 |
FKBP5 |
–1.98625 |
down |
0.00045 |
FK506 binding protein 5 |
FOLH1 |
–1.97677 |
down |
0.0032 |
folate hydrolase (prostate-specific membrane antigen) 1 |
CD3G |
–1.862 |
down |
0.04425 |
CD3g molecule |
COL6A5 |
–1.72947 |
down |
0.0316 |
collagen type VI alpha 5 |
TUBBP5 |
–1.70945 |
down |
0.0228 |
tubulin beta pseudogene 5 |
CLEC4M |
–1.6691 |
down |
0.0217 |
C-type lectin domain family 4 member M |
S100A9 |
–1.66717 |
down |
0.0021 |
S100 calcium binding protein A9 |
DIO3 |
–1.65761 |
down |
0.009 |
deiodinase, iodothyronine, type III |
LOC645752 |
–1.64348 |
down |
0.0449 |
golgin A6 family member A pseudogene |
CD3E |
–1.61303 |
down |
0.0266 |
CD3e molecule |
TNNT3 |
–1.55491 |
down |
0.00495 |
troponin T3, fast skeletal type |
S100A8 |
–1.53696 |
down |
0.0022 |
S100 calcium binding protein A8 |
FUT9 |
–1.50479 |
down |
0.00195 |
fucosyltransferase 9 |
KRT31 |
1.50307 |
up |
0.02565 |
keratin 31 |
KRTAP11-1 |
1.51562 |
up |
0.00975 |
keratin associated protein 11-1 |
KRT85 |
1.51808 |
up |
0.01765 |
keratin 85 |
KRT81 |
1.54671 |
up |
0.0292 |
keratin 81 |
KRT34 |
1.54769 |
up |
0.03615 |
keratin 34 |
EPSTI1 |
1.574 |
up |
0.0017 |
epithelial stromal interaction 1 (breast) |
IFI6 |
1.57562 |
up |
0.0033 |
interferon alpha inducible protein 6 |
IFI35 |
1.59026 |
up |
0.0059 |
interferon induced protein 35 |
KRTAP3-2 |
1.61219 |
up |
0.0107 |
keratin associated protein 3-2 |
KC6 |
1.62074 |
up |
0.0318 |
keratoconus gene 6 |
KRT83 |
1.65762 |
up |
0.0203 |
keratin 83 |
OAS3 |
1.69667 |
up |
0.00105 |
2'-5'-oligoadenylate synthetase 3 |
USP18 |
1.72046 |
up |
0.00215 |
ubiquitin specific peptidase 18 |
CYP26B1 |
1.74162 |
up |
0.00045 |
cytochrome P450 family 26 subfamily B member 1 |
LNX1-AS2 |
1.74788 |
up |
0.022 |
LNX1 antisense RNA 2 |
IFI44 |
1.75064 |
up |
0.00015 |
interferon induced protein 44 |
TNFRSF4 |
1.75918 |
up |
0.0375 |
tumor necrosis factor receptor superfamily member 4 |
LOC339975 |
1.76055 |
up |
0.02065 |
uncharacterized LOC339975 |
CLDN11 |
1.7733 |
up |
0.012 |
claudin 11 |
KRT35 |
1.78149 |
up |
0.0024 |
keratin 35 |
KRT33A |
1.80012 |
up |
0.00445 |
keratin 33A |
OAS2 |
1.83729 |
up |
0.00145 |
2'-5'-oligoadenylate synthetase 2 |
KRT86 |
1.85155 |
up |
0.0094 |
keratin 86 |
DCD |
1.85338 |
up |
0.0401 |
dermcidin |
ACAN |
1.85416 |
up |
0.0002 |
aggrecan |
PKD1L2 |
1.90338 |
up |
0.0378 |
polycystin 1 like 2 (gene/pseudogene) |
SCGB1B2P |
1.91013 |
up |
0.01375 |
secretoglobin family 1B member 2, pseudogene |
CMPK2 |
1.92066 |
up |
0.00015 |
cytidine/uridine monophosphate kinase 2 |
ADAMTS18 |
1.95193 |
up |
0.0003 |
ADAM metallopeptidase with thrombospondin type 1 motif 18 |
KRT33B |
1.9594 |
up |
0.0128 |
keratin 33B |
IFIT1 |
1.97285 |
up |
0.0001 |
interferon induced protein with tetratricopeptide repeats 1 |
MX1 |
1.99777 |
up |
0.00015 |
MX dynamin like GTPase 1 |
RSAD2 |
2.07171 |
up |
5.00E-05 |
radical S-adenosyl methionine domain containing 2 |
IFI44L |
2.31465 |
up |
5.00E-05 |
interferon induced protein 44 like |
LINC00487 |
2.42803 |
up |
0.0367 |
long intergenic non-protein coding RNA 487 |
ISG15 |
2.47094 |
up |
5.00E-05 |
ISG15 ubiquitin-like modifier |
SULT1A2 |
2.52975 |
up |
0.02025 |
sulfotransferase family 1A member 2 |
DMC1 |
2.61725 |
down |
0.00415 |
DNA meiotic recombinase 1 |
FAM132B |
2.69101 |
up |
0.0118 |
- |
NRIR |
2.97361 |
up |
0.0399 |
negative regulator of interferon response (non-protein coding) |
MTHFD2P1 |
3.49824 |
up |
0.01845 |
methylenetetrahydrofolate dehydrogenase (NADP+ dependent) 2, methenyltetrahydrofolate cyclohydrolase pseudogene 1 |
OR8B2 |
4.67189 |
up |
0.0423 |
olfactory receptor family 8 subfamily B member 2 |
LOC101929128 |
4.88337 |
up |
0.0419 |
uncharacterized LOC101929128 |
Ctr, matched normal skin tissue; Tum, infantile hemangioma skin tissue.
Bioinformatics analysis of lncRNAs in IH compared with matched normal skin controls
Using the Gencode, RefSeq and UCSC Knowngene databases for non-coding transcripts, we identified 256 differentially expressed lncRNAs with greater than two-fold changes in IH and p values < 0.05 (Figure 2A). Of these, 177 were overexpressed and 79 were underexpressed in IH relative to the matched normal skin controls (fold change ≥ 2, P ≤ 0.05; for a list of differentially expressed lncRNAs, see Table 2). LncRNAs (long non-coding RNAs), are defined as greater than 200 nucleotides in length, transcribed by RNA polymerase II (RNA PII), and lacking an open reading frame [15]. LncRNAs have been found to regulate protein-coding (pc) gene expression at both the transcriptional and post-transcriptional levels [16]. To identify the potential mRNA targets of lncRNAs, we use RNAplex to predict the binding of lncRNAs with the antisense target mRNAs. mRNAs 10 kb upstream or downstream of lncRNAs were considered to be the conceivable lncRNA targets and defined as cis target mRNAs. Gene ontology (GO) analysis revealed that cis target mRNAs of differentially expressed lncRNAs were mostly involved in regulatory mechanisms related to transcription, nucleic acid binding transcription factor activity and intracellular components (Figure 2B). KEGG Pathway analysis indicated that the MAPK signalling pathway, regulation of autophagy and metabolic pathways were implicated for the cis target mRNAs of differentially expressed lncRNAs (Figure 2C).
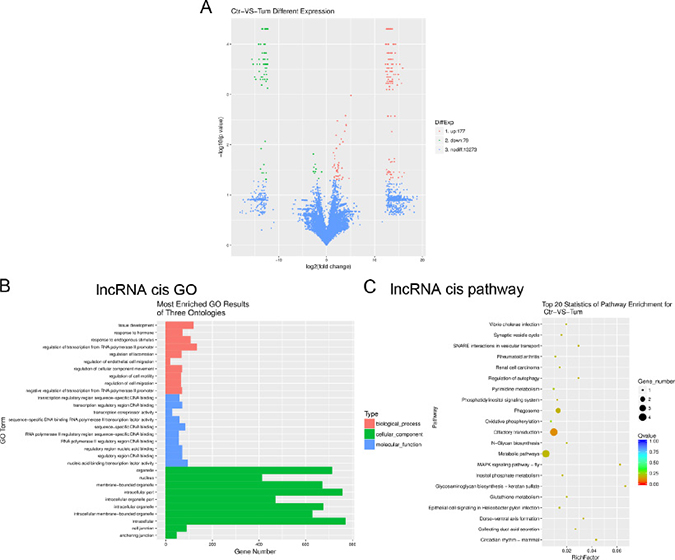
Figure 2: Gene ontology (GO) terms and pathways for target mRNAs of differentially expressed lncRNAs between infantile hemangioma and adjacent normal skin tissues. (A) Hierarchical clustering shows a distinguishable lncRNA expression profiling among groups. (B) Most enriched GO terms of the three ontologies that are associated with the cis target mRNAs of differentially expressed lncRNAs are listed. (C) The top 20 pathways that are associated with the cis target mRNAs of differentially expressed lncRNAs are listed. (B) The horizontal axis represents the gene number. The term/GO on the vertical axis is drawn according to the first letter of the GO in descending order. Red bar represents the biological process, blue bar displays the molecular function, and green bar illustrates the cellular component. (C) The enrichment Q value or false discovery rate correct the p value for multiple comparisons. P values are calculated using Fisher’s exact test. The term/pathway on the vertical axis is drawn according to the first letter of the pathway in descending order. The horizontal axis represents the enrichment factor, i.e., (the number of dysregulated genes in a pathway/the total number of dysregulated gene)/(the number of genes in a pathway in the database/the total number of genes in the database). Top 20 enriched pathways are selected according to the enrichment factor value. The selection standards were the number of genes in a pathway ≥4. The different colours from green to red represent the Q value (False discovery rate value). The different sizes of the round shapes represent the gene count number in a pathway. Ctr, matched normal skin tissue; Tum, infantile hemangioma skin tissue.
Table 2: List of up-regulated and down-regulated lncRNAs detected using RNA-seq (FC ≥ 2.83, P < 0.05)
LncRNAID |
GenePos |
log2 (Tum/Ctr) |
up-or-down |
p_value |
LncRNA GeneID |
|---|---|---|---|---|---|
TCONS_00116532 |
chr5:101515283-101519050 |
–2.74762 |
down |
0.0153 |
XLOC_074583 |
TCONS_00049881 |
chr16:76805140-76807266 |
–2.74208 |
down |
0.03405 |
XLOC_031473 |
TCONS_00140157 |
chr8:111620546-111622506 |
–2.65655 |
down |
0.02805 |
XLOC_090179 |
TCONS_00108080 |
chr4:97512593-97516549 |
–2.51518 |
down |
0.03695 |
XLOC_068366 |
TCONS_00049878 |
chr16:76790731-76793194 |
–2.36369 |
down |
0.0245 |
XLOC_031470 |
TCONS_00116531 |
chr5:101510927-101514253 |
–2.31673 |
down |
0.03415 |
XLOC_074582 |
TCONS_00125444 |
chr6:77144877-77146646 |
–2.19865 |
down |
0.0304 |
XLOC_080698 |
TCONS_00047386 |
chr15:95209116-95212765 |
–1.62607 |
down |
0.04695 |
XLOC_030120 |
TCONS_00099093 |
chr3:117310248-117315662 |
1.60464 |
up |
0.026 |
XLOC_061648 |
TCONS_00094390 |
chr3:115548706-115554914 |
1.63654 |
up |
0.045 |
XLOC_058344 |
TCONS_00112159 |
chr5:104342703-104346581 |
1.6409 |
up |
0.0436 |
XLOC_071442 |
TCONS_00099090 |
chr3:117295535-117304861 |
1.65949 |
up |
0.0244 |
XLOC_061645 |
TCONS_00125869 |
chr6:92532111-92539501 |
1.72153 |
up |
0.01895 |
XLOC_081015 |
TCONS_00092337 |
chr3:21220256-21226664 |
1.73612 |
up |
0.01655 |
XLOC_056961 |
TCONS_00036940 |
chr13:105981280-105999840 |
1.8649 |
up |
0.0291 |
XLOC_023137 |
TCONS_00022826 |
chr11:26799455-26802752 |
1.995 |
up |
0.04175 |
XLOC_013761 |
TCONS_00092345 |
chr3:21256300-21262413 |
2.01955 |
up |
0.0065 |
XLOC_056969 |
TCONS_00125870 |
chr6:92539642-92542765 |
2.05275 |
up |
0.0118 |
XLOC_081016 |
TCONS_00144439 |
chr9:13841053-13843543 |
2.11565 |
up |
0.04215 |
XLOC_093201 |
TCONS_00132673 |
chr7:94029530-94036098 |
2.1175 |
up |
0.03365 |
XLOC_085154 |
TCONS_00088818 |
chr22:11878998-11880540 |
2.12569 |
up |
0.04895 |
XLOC_054900 |
TCONS_00099084 |
chr3:117263272-117265249 |
2.13489 |
up |
0.0322 |
XLOC_061639 |
TCONS_00089156 |
chr22:23536916-23548776 |
2.23141 |
up |
0.00335 |
XLOC_055095 |
TCONS_00112938 |
chr5:136193267-136196387 |
2.2618 |
up |
0.03135 |
XLOC_072057 |
TCONS_00093724 |
chr3:76746891-76748374 |
2.30541 |
up |
0.02505 |
XLOC_057834 |
TCONS_00021543 |
chr11:124180846-124186471 |
2.31143 |
up |
0.04245 |
XLOC_012894 |
TCONS_00099092 |
chr3:117307664-117309582 |
2.31645 |
up |
0.0351 |
XLOC_061647 |
TCONS_00077659 |
chr2:12602918-12606149 |
2.33745 |
up |
0.02735 |
XLOC_047338 |
TCONS_00106552 |
chr4:19193285-19198298 |
2.3654 |
up |
0.02555 |
XLOC_067212 |
TCONS_00109906 |
chr4:187247968-187249825 |
2.40602 |
up |
0.02915 |
XLOC_069754 |
TCONS_00106583 |
chr4:19283454-19288399 |
2.44245 |
up |
0.0258 |
XLOC_067243 |
TCONS_00142894 |
chr8:98381305-98382530 |
2.46222 |
up |
0.04195 |
XLOC_092151 |
TCONS_00092347 |
chr3:21263525-21265295 |
2.50214 |
up |
0.0228 |
XLOC_056971 |
TCONS_00126048 |
chr6:104495681-104497908 |
2.57552 |
up |
0.04765 |
XLOC_081156 |
TCONS_00021618 |
chr11:124429567-124433267 |
2.6993 |
up |
0.00985 |
XLOC_012968 |
TCONS_00142579 |
chr8:89240840-89241898 |
2.72566 |
up |
0.0405 |
XLOC_091914 |
TCONS_00116213 |
chr5:86348589-86351550 |
3.02011 |
up |
0.0077 |
XLOC_074350 |
TCONS_00013577 |
chr10:64133769-64134877 |
3.14074 |
up |
0.04865 |
XLOC_008097 |
TCONS_00021614 |
chr11:124419031-124423063 |
3.17524 |
up |
0.02925 |
XLOC_012964 |
TCONS_00138013 |
chr8:9196728-9203476 |
3.31685 |
up |
0.04465 |
XLOC_088644 |
TCONS_00117017 |
chr5:130223107-130225700 |
3.34741 |
up |
0.0088 |
XLOC_074967 |
TCONS_00032295 |
chr12:91627173-91631603 |
3.74738 |
up |
0.0054 |
XLOC_019826 |
TCONS_00116260 |
chr5:86539574-86547371 |
3.90234 |
up |
0.00265 |
XLOC_074397 |
TCONS_00116261 |
chr5:86548066-86552126 |
4.02636 |
up |
0.004 |
XLOC_074398 |
TCONS_00032240 |
chr12:91452883-91455961 |
4.05433 |
up |
0.00415 |
XLOC_019771 |
TCONS_00087133 |
chr21:38178744-38181900 |
4.62793 |
up |
0.04685 |
XLOC_053685 |
TCONS_00116241 |
chr5:86436761-86447930 |
5.05555 |
up |
0.00105 |
XLOC_074378 |
Ctr, matched normal skin tissue; Tum, infantile hemangioma skin tissue.
Differential expression and bioinformatics analysis of miRNAs in IH compared with matched normal skin controls
We also determines the miRNA expression profiles between IH and matched normal skin controls using HiSeq. One hundred forty-two miRNA candidates were found to be differentially expressed (fold change ≥ 2, P ≤ 0.05; for a list of differentially expressed miRNAs, see Table 3). Of these, 42 miRNAs were up-regulated in IH, whereas 100 miRNAs were down-regulated (Figure 3A). To examine the potential biological functions of the miRNAs of interest in IH, we use miRanda, targetscan and PITA software to identify the target genes of known miRNAs with differential expression profiles and extracted intersections or unions of target genes as the final prediction result. Gene ontology (GO) analysis revealed that the target mRNAs of differentially expressed miRNAs were mostly involved in cellular processes, cell components and binding (Figure 3B).
Table 3: List of up-regulated and down-regulated miRNAs detected using small RNA-seq (FC ≥ 2.83, P < 0.05)
miR_name |
fold-change(log2 Tum/Ctr) |
up-or-down |
p_value |
sig-lable |
|---|---|---|---|---|
hsa-miR-9-3p |
–7.10538475 |
down |
0.000122742 |
** |
hsa-miR-1303 |
–6.86430997 |
down |
0.000490585 |
** |
hsa-miR-223-3p |
–3.53749571 |
down |
1.65E-266 |
** |
hsa-miR-509-3-5p |
–3.49076386 |
down |
4.54E-10 |
** |
hsa-miR-509-5p |
–2.50137905 |
down |
0.00149862 |
** |
hsa-miR-450a-2-3p |
–2.22132264 |
down |
0.007574552 |
** |
hsa-miR-337-5p |
–2.13638952 |
down |
0.000918863 |
** |
hsa-miR-135a-5p |
–2.11442989 |
down |
0.012782924 |
* |
hsa-miR-513c-5p |
–2.11442989 |
down |
0.012782924 |
* |
hsa-miR-2355-3p |
–2.08634155 |
down |
0.004368201 |
** |
hsa-miR-202-5p |
–1.99889374 |
down |
0.007235532 |
** |
hsa-miR-200c-5p |
–1.99886536 |
down |
0.021358055 |
* |
hsa-miR-664b-3p |
–1.99886536 |
down |
0.021358055 |
* |
hsa-miR-3648 |
–1.93753391 |
down |
0.001441353 |
** |
hsa-miR-429 |
–1.93708866 |
down |
1.47E-256 |
** |
hsa-miR-26a-1-3p |
–1.92492452 |
down |
0.004102228 |
** |
hsa-miR-3611 |
–1.92492452 |
down |
0.004102228 |
** |
hsa-miR-187-3p |
–1.89198508 |
down |
0.000828053 |
** |
hsa-miR-664a-3p |
–1.87801528 |
down |
1.82E-41 |
** |
hsa-miR-383-5p |
–1.87335512 |
down |
0.03528718 |
* |
hsa-miR-335-3p |
–1.84083467 |
down |
7.07E-87 |
** |
hsa-miR-203a-3p |
–1.72913403 |
down |
0 |
** |
hsa-miR-3912-3p |
–1.71155782 |
down |
2.01E-06 |
** |
hsa-miR-183-3p |
–1.69939239 |
down |
0.031012226 |
* |
hsa-miR-135b-5p |
–1.69929888 |
down |
3.86E-09 |
** |
hsa-miR-141-5p |
–1.69460943 |
down |
6.85E-21 |
** |
hsa-miR-377-5p |
–1.65878433 |
down |
7.03E-08 |
** |
hsa-miR-16-5p |
–1.65065115 |
down |
0 |
** |
hsa-miR-150-5p |
–1.62494834 |
down |
2.07E-49 |
** |
hsa-miR-627-5p |
–1.6233869 |
down |
0.000309094 |
** |
hsa-miR-6510-3p |
–1.58388025 |
down |
0.000271487 |
** |
hsa-miR-548p |
–1.58387326 |
down |
0.026725738 |
* |
hsa-miR-203b-3p |
–1.58383438 |
down |
1.74E-09 |
** |
hsa-miR-195-5p |
–1.5823761 |
down |
0 |
** |
hsa-miR-141-3p |
–1.5772266 |
down |
0 |
** |
hsa-miR-200b-3p |
–1.57584304 |
down |
0 |
** |
hsa-miR-944 |
–1.5589637 |
down |
4.65E-10 |
** |
hsa-miR-200b-5p |
–1.53135387 |
down |
2.72E-13 |
** |
hsa-miR-31-5p |
–1.5239109 |
down |
6.16E-159 |
** |
hsa-miR-183-5p |
–1.52252398 |
down |
2.78E-140 |
** |
hsa-miR-92a-1-5p |
–1.52249641 |
down |
0.007046614 |
** |
hsa-miR-493-5p |
–1.51904529 |
down |
6.20E-36 |
** |
hsa-miR-320d |
1.58604035 |
up |
0.004519782 |
** |
hsa-miR-524-3p |
1.69892552 |
up |
4.81E-139 |
** |
hsa-miR-7704 |
1.8311765 |
up |
0.000268926 |
** |
hsa-miR-450a-1-3p |
2.00090764 |
up |
0.021183756 |
* |
hsa-miR-185-5p |
2.03429793 |
up |
1.64E-39 |
** |
hsa-miR-503-5p |
2.10316253 |
up |
1.37E-29 |
** |
hsa-miR-122-5p |
2.13865463 |
up |
0.00090565 |
** |
hsa-miR-1-3p |
2.40555691 |
up |
2.02E-13 |
** |
hsa-miR-7-5p |
2.40555691 |
up |
2.02E-13 |
** |
hsa-miR-520e |
2.41594514 |
up |
0.002562365 |
** |
hsa-miR-1269b |
3.39374391 |
up |
3.56E-05 |
** |
hsa-miR-1268a |
3.8087166 |
up |
0.000515933 |
** |
hsa-miR-1268b |
3.8087166 |
up |
0.000515933 |
** |
Ctr, matched normal skin tissue; Tum, infantile hemangioma skin tissue.
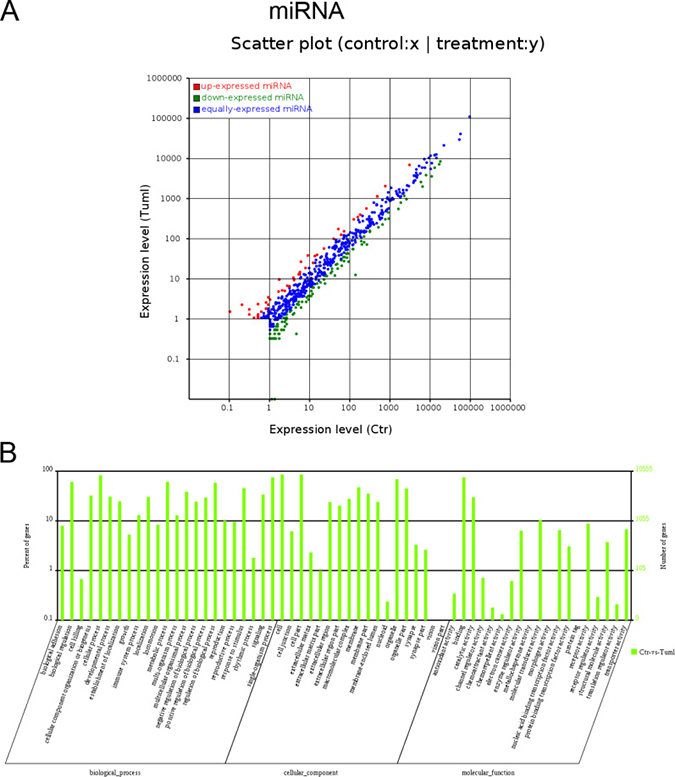
Figure 3: Expression profiles of differentially expressed miRNAs and Gene ontology (GO) terms for target mRNAs of differentially expressed miRNAs between infantile hemangioma and adjacent normal skin tissues. (A) Scatter plot shows the differentially expressed miRNAs. Red dots represent up-regulated miRNAs. Green dots illustrate down-regulated miRNAs. (B) Most enriched GO terms of the three ontologies that are associated with the target mRNAs of differentially expressed miRNAs are listed. The term/GO on the horizontal axis is drawn according to the first letter of the GO in ascending order from left to right. The vertical axis represents the percent of genes and gene number. Ctr, matched normal skin tissue; Tum, infantile hemangioma skin tissue.
Real-time quantitative PCR validation
To validate the RNA-seq data, we randomly selected 9 differentially expressed RNAs (bold-face type in Tables 1–3). Real-time quantitative PCR (qRT-PCR) and Bulge-LoopTM qRT-PCR analyses were performed on an additional 9 independent IH skin samples (Table 4). The results revealed that similar up-regulation or down-regulation patterns were observed in both the RNA-seq and qRT-PCR samples for the 9 RNAs (Figure 4, bold in Tables 1–3). Therefore, our RNA-seq data were reliable and stable. Among the 9 RNAs, IFI44L, ISG15, TCONS_00088818, TCONS_000112159, TCONS_000125870, miR-503-5p and miR-524-3p were all expressed to a greater extent in the IH tissues than in the matched normal skin controls.
Table 4: Demographic and clinical characteristics of infantile hemangioma (IH) patients (capillary hemangioma)
No. |
Age |
Gender |
Position |
Pathology |
Sample use |
|---|---|---|---|---|---|
1 |
3 months 8 days |
Male |
Left axilla |
IH |
RNA-seq |
2 |
7 months 5 days |
Female |
Head |
IH |
RNA-seq |
3 |
12 months |
Female |
Left Abdomen |
IH |
RNA-seq |
4 |
9 months |
Female |
Right thoracic wall |
IH |
qRT-PCR |
5 |
5 months |
Male |
Thoracic wall |
IH |
qRT-PCR |
6 |
6 months 9 days |
Female |
Occiput |
IH |
qRT-PCR |
7 |
8 months 13 days |
Female |
Right posterior neck |
IH |
qRT-PCR |
8 |
10 months |
Female |
Left abdomen |
IH |
qRT-PCR |
9 |
4 months |
Female |
Right thoracic wall |
IH |
qRT-PCR |
10 |
11 months |
Male |
Thoracic wall |
IH |
qRT-PCR |
11 |
8 months 21 days |
Female |
Occiput |
IH |
qRT-PCR |
12 |
5 months 12 days |
Female |
Head |
IH |
qRT-PCR |
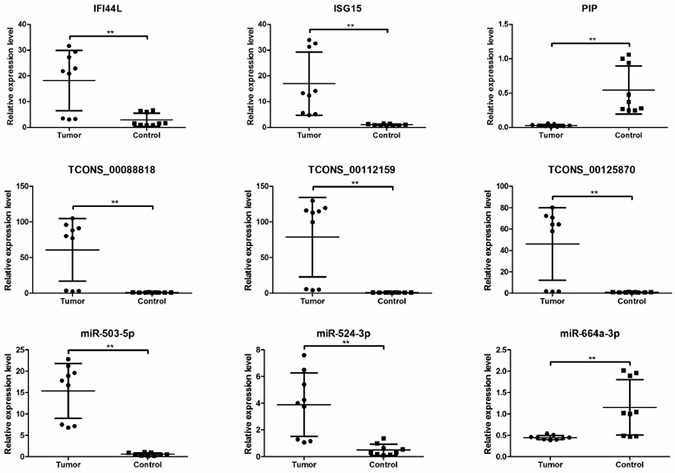
Figure 4: The differential expression of mRNAs, lncRNAs and miRNAs between additional IH skin (n = 9) and matched normal skin tissues (n = 9). mRNAs and lncRNAs expression were validated by quantitative real-time PCR using 2(–△△Ct) method. miRNAs expression was validated by Bulge-LoopTM qRT-PCR. *P < 0.05, **P < 0.01.
The ceRNA network construction
Recently, lncRNAs and mRNAs have been demonstrated to be function as ceRNAs in diverse physiological and pathophysiological states. It is known that mRNAs or lncRNAs can bind miRNAs through microRNA response elements (MREs). Therefore, we use rna22 and targetscan to screen the lncRNAs and mRNAs with MREs. To ascertain the associations of differentially expressed lncRNAs and miRNAs with mRNAs, based on the 142 differentially expressed miRNAs, 144 differentially expressed mRNAs, and 256 differentially expressed lncRNAs, an lncRNA-miRNA-mRNA correlation network was constructed (Figure 5). The network displayed the associations among 87 miRNAs, 70 lncRNAs and 58 mRNAs mediated interactions. For example, miR-26a-1-3p could bind to lncRNAs TCONS_00074621 and mRNA CD24, miR-24-3p bind with TCONS_00000006 and LEFTY2, moreover, miR-514a-3p bind to TCONS_00040753 and FLT1. As shown in Figure 5, one miRNA was associated with one to tens of mRNAs and vice versa. One lncRNA was related to one to tens of miRNAs. In total, 1256 sponge modulators participated in 87 miRNA-mediated, 70 lncRNA-mediated and 58 mRNAs’ transcripts-mediated interactions. These findings indicated that the expression profiles of miRNAs, mRNAs and lncRNAs were significantly correlated.
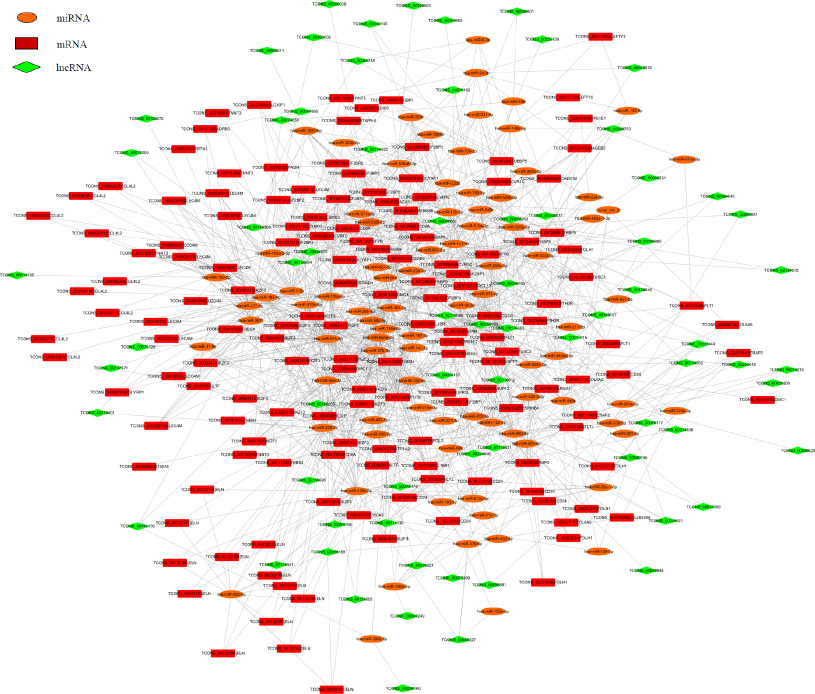
Figure 5: The ceRNA network of the differentially expressed miRNA-mediated lncRNAs and mRNAs interactions. Red color represents the mRNA name, green color displays the lncRNA name, and orange color illustrates the miRNA name. Taken TCONS_00126150 | CD24 and TCONS_00126153 | CD24 as an example, the TCONS_ number before | means the ID number of each transcript, one gene has many transcripts.
DISCUSSION
Currently, with the advent of next-generation sequencing technologies, RNA-Seq is gradually replacing microarrays for the detection of transcript expression profiling. IH is one of the most common tumors diagnosed in young children. The pathogenesis of hemangioma is largely unknown due to its sophisticated etiology. Although a lot of papers report the RNA networks in IH [12, 14, 17–19], those work all used microarrays methods. In this study, we used Ribo-Zero RNA-Seq and HiSeq means to examine the global expression profiles of protein-coding transcripts and non-coding RNAs, including miRNAs and lncRNAs, in IH and matched normal skin controls. Totally, 144 mRNAs, 256 lncRNAs and 142 miRNAs were found to be differentially expressed (fold change ≥ 2, P ≤ 0.05) in IH compared to matched normal skin. Further integrated ceRNA network analysis revealed that 353 sponge modulators participate in 39 miRNA, 29 lncRNA and 147 mRNA mediated interactions.
Competitive endogenous (ce) RNAs cross-regulate each other through sequestration of shared microRNAs and form complex regulatory networks based on their microRNA signatures [20]. Genome-wide transcriptional profiling of vessels from proliferating and involuting hemangiomas has been used to identify differentially expressed genes [19]. A lncRNA microarray study reported that a large number of genes are differentially expressed in IH [12]. Integrative meta-analysis identifies miRNA-regulated networks in IH [14]. Here, based on RNA-seq technology, using IH tissues and matched normal skin, we presented a new ceRNA network that determined the functions of particular miRNAs, lncRNAs and mRNAs in IH development (Figure 5). Alterations in one ceRNA may have striking effects on the integrated ceRNA and transcriptional networks. Taken miR-664a-3p as an example, it was down-regulated in IH tissues from the small RNA-seq data (Table 3). Bulge-LoopTM qRT-PCR demonstrated that expression of miR-664a-3p was decreased in another nine IH tissues (Figure 4). The ceRNA network analysis revealed that three lncRNAs TCONS_00020616, TCONS_00058199 and TCONS_00108595 could bind to miR-664a-3p. Moreover, nine mRNAs including ADCYAP1, CSMD3, IL18R1, IKZF3, CD8A, FGL2, FUT9, IGF2BP1, and TMEM38B were predicted to bind with miR-664a-3p (Figure 5). This ceRNA network indicated that those nine mRNAs’ expression could be regulated by miR-664a-3p, and three lncRNAs TCONS_00020616, TCONS_00058199, TCONS_00108595 could compete to bind with miR-664a-3p and then affect those nine mRNAs’ expression.
The pathogenesis of IH has been linked to pathways affecting angiogenesis and vasculogenesis [21]. Those microarray analyses concluded that angiogenin may be a useful serum marker for hemangiomas [22], IH endothelial cells (HEMECs) reflects a pro-proliferative cell type with altered adhesive characteristics [17], proliferating hemangiomas display increased expression of genes involved in endothelial-pericyte interactions, as well as those involved in neural and vascular patterning [19], lncRNAs likely regulate several genes in angiogenesis [12], miRNA-mRNA expression networks display that deregulated genes play roles in cell growth and differentiation, cell signaling, angiogenesis and vasculogenesis [14]. In the present study, using RNA-seq technology, we found that deregulated mRNAs related to immune system processes, carbohydrate derivative binding, extracellular region regulation, chemokine, NF-kappa B and TGF-beta signalling pathways, as well as cell adhesion molecules (CAMs) (Figure 1). Moreover, cis target mRNAs of differentially expressed lncRNAs were mostly involved in regulatory mechanisms related to transcription, nucleic acid binding transcription factor activity, intracellular components, MAPK signalling pathway, regulation of autophagy and metabolic pathways (Figure 2). Additionally, target mRNAs of differentially expressed miRNAs were mostly involved in cellular processes, cell components and binding (Figure 3). These results are partly consistent with those of previous studies in that CAMs are involved. Besides, RNA-seq data show some new findings in that regulation of autophagy and metabolic pathways, TGF-beta, NF-kappa B signalling and chemokine signalling were involved in the pathogenesis of IH.
Endothelial TGF-β signalling has been implicated in the regulation of angiogenesis [23]. Expression of NF-kappa B target genes was demonstrated in proliferating IH. Targeting NF-kappa B in infantile hemangioma-derived stem cells reduced VEGF-A expression [24]. The chemokine CXCL-14 has been reported to be involved in the occurrence and development of infantile hemangioma [25]. Our RNA-seq data found that TGF-beta, NF-kappa B signalling and chemokine signalling were involved in the pathogenesis of IH. The results are consistent with those of previous studies, which suggested that the RNA-seq data are reliable.
By carefully comparing our data with other’s, IGF2, FOXF1 and EGFL7 were reported to be up-regulated in IH, FOXC1 and EGFR were down-regulated in IH [12]. In this study, we found that IGF2 mRNA-binding proteins (IGF2BPs) including IGF2BP1, IGF2BP2 and IGF2BP3 were all downregualted in IH. Although recent publication reported that results obtained by RNA-seq and microarrays were highly reproducible [26], some discrepancy may be existed in the differentially expressed RNAs. Therefore, further demonstrating the function of particular RNA in IH development is urgently needed. In addition, larger samples are needed to perform receiver operating characteristic (ROC) curve analysis to prove that some of the IH RNAs are promising biomarkers.
Taken together, understanding the functional interactions among lncRNAs, miRNAs and mRNAs could lead to new explanations for IH disease pathogenesis [7]. Further elucidating the underlying mechanisms of the functions of miRNAs, lncRNAs and mRNAs in IH would be helpful in revealing the biological aetiology and potentially provide useful information for IH evaluation and treatment.
MATERIALS AND METHODS
Ethics statement
This study was approved by the Medical Ethics Committee of the Obstetrics and Gynaecology Hospital affiliated with Nanjing Medical University (No. [2015] 91). Children with IH underwent surgery at out hospital. The IH samples and matched normal skin controls were collected from patients who underwent surgery and whose parents provided written consent.
Tissue samples
Proliferating capillary infantile hemangioma (IH) and matched normal skin tissues were obtained from 12 different patients who were admitted to the Obstetrics and Gynaecology Hospital affiliated with Nanjing Medical University for IH removal. Patient information is listed in Table 4. A diagnosis of proliferative infantile hemangioma was confirmed by routine pathological examination. The collected skin samples were immediately frozen in liquid nitrogen for total RNA preparation.
Total RNA isolation
Total RNA was extracted from biopsy samples using the Qiagen RNeasy mini kit (Qiagen, Valencia, CA). After ribosomal RNA depletion, RNA-seq libraries were prepared using ScriptSeq complete kits from Epicenter (Madison, WI). RNA purity was assessed using the Nano Photometer® spectrophotometer (IMPLEN, CA, USA), and RNA concentration was measured using the Qubit® RNA Assay Kit in Qubit® 2.0 Flurometer (Life Technologies, CA, USA). RNA integrity was assessed using the RNA Nano 6000 Assay Kit from the Bioanalyser 2100 system (Agilent Technologies, CA, USA).
Library preparation, quality examination and sequencing for mRNAs and lncRNAs
The sequencing libraries were prepared following manufacturer recommendations from the VAHTSTM Total RNA-seq (H/M/R) Library Prep Kit for Illumina®. The details of library construction were patented by the company (Vazyme, China). After cluster generation, the libraries were sequenced on an Illumina Hiseq X10 platform, and 150-bp paired-end reads were generated.
Raw reads in fastq format were first processed using in-house perl scripts. Clean reads were obtained by removing reads with adapters, reads in which unknown bases were more than 5% and low quality reads (if the percentage of low quality bases was greater than 50% in a given read, we defined the low quality base to be the base whose sequencing quality was no more than 10). At the same time, Q20, Q30, and GC contents were calculated for the clean reads. All downstream analyses were based on the clean reads.
The reference genome and gene model annotation files were downloaded directly from the genome website. The reference genome index was built using Bowtie (v2.1.0) [27], and the paired-end clean reads were aligned to the reference genome using TopHat (v2.1.1) [28].
Transcriptome assembly, lncRNA prediction and target gene prediction
The mapped reads from each sample were assembled using Cufflinks (v2.2.1) [29] with a reference-based approach. Cufflinks uses a probabilistic model to simultaneously assemble and quantify the expression levels of a minimal set of isoforms, which provides a maximum likelihood explanation of the expression data in a given locus. Then, Cuffmerge was used to merge these sample assemblies into a master transcriptome, which was compared to known transcripts via Cuffcompare. The lncRNAs were predicted by several strict steps based on RNA structural characteristics and non-coding properties. The steps were as follows: 1) transcripts, not in any class code of “ j, i, o, u, x ”, were filtered out; 2) transcripts shorter than 200 bp were filtered out; 3) transcripts aligned to sequences in the NONCODE database [30] by blastn were identified as known lncRNAs; 4) the retained transcripts (known lncRNAs were not included) were used to predict protein coding potential by Coding Potential Calculator (CPC) [31] and TransDecoder (http://transdecoder.github.io/), transcripts with coding potential were removed, and those without coding potential were identified as novel lncRNAs. The known lncRNAs and novel lncRNAs were together used for subsequent analyses.
LncRNAs can negatively or positively affect expression of the downstream gene via an upstream noncoding promoter. Genes within 10 kb upstream or downstream of lncRNAs were abstracted by bedtools (http://bedtools.readthedocs.io/en/latest/) as lncRNA target genes. However, antisense lncRNAs can regulate overlapping sense transcripts. Transcripts that overlapped with LncRNAs on the opposite strand were also identified as lncRNA target genes, and the interactions between lncRNAs and transcripts were revealed by RNAplex [32].
Quantification of gene expression levels and differential expression analysis
Cuffdiff (v2.2.1) [33] was used to calculate FPKMs for both lncRNAs and coding genes in each group. Gene FPKMs were computed by summing the FPKMs of the transcripts in each gene group. FPKM stands for “fragments per kilobase of exon per million fragments mapped” and is calculated based on the length of the fragments and the reads count mapped to each fragment.
Cuffdiff (v2.2.1) provides statistical routines for determining differential expression in digital transcripts or gene expression datasets using a model based on a negative binomial distribution. Transcripts or genes with corrected p values less than 0.05 and absolute values of log2 (fold change) <1 were classified as significantly differentially expressed.
Small RNA sequencing and bioinformatics analysis
Total RNA was separated by 15% agarose gels to extract the small RNA (18–30 nt). After ethanol precipitation and centrifugal enrichment of small RNA samples, the library was prepared according to the methods and processes described in the Small RNA Sample Preparation Kit (Illumina, RS-200-0048). Insert size was assessed using the Agilent Bioanalyser 2100 system (Agilent Technologies, CA, USA), and after the insert size was consistent with expectations, qualified insert size was accurately quantitated using a Taqman fluorescence probe from the AB Step One Plus Real-Time PCR system (Library valid concentration > 2 nM). The qualified libraries were sequenced using an Illumina Hiseq 2500 platform, and 50-bp single-end reads were generated.
First, the tags were mapped to the reference genome by SOAP [34] to analyse their distributions within the genome and were aligned to the miRBase database [35] using blast. The tags were identified as known miRNAs when they satisfied the following criteria: 1) there were no mismatches when aligned to the miRNA precursors in the miRBase database; 2) based on the first criteria, the tags were aligned to the mature miRNAs in the miRBase database with at least 16-nt overlap while allowing offsets. The miRNA target genes were predicted using two software programs (targetscan and miRanda) as we previously described [36], and the intersection of target genes (the intersections were the same target genes of the same miRNAs) were the final target genes.
The miRNA expression levels were measured by “Transcripts Per Kilobase Million” (TPM).
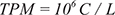
where C is the read count of a miRNA and L is the total count of clean reads in sample.
Differentially expressed miRNAs were evaluated using the following statistical tests:
1) Statistical algorithm developed by Audic and Claverie (1997) [37]
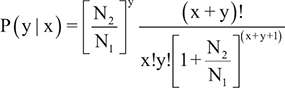
where N1 is the total clean reads from sample 1, N2 is the total clean reads from sample 2, x is the number of reads from sample 1 mapped to miRNA A and y is the number of reads from sample 2 mapped to miRNA A.
Gene Ontology (GO) and KEGG enrichment analysis
GO enrichment analysis of differentially expressed genes or target genes of differentially expressed lncRNAs was implemented using a perl module (GO::TermFinder) [38]. GO terms with corrected p values less than 0.05 were considered to be significantly enriched among the differentially expressed genes or the target genes of differentially expressed lncRNAs. R functions (phyper and qvalue) were used to test for statistical enrichment of the differentially expressed genes or target genes of the differentially expressed lncRNAs among the KEGG pathways. KEGG pathways with corrected p values less than 0.05 were considered to be significantly enriched among the differentially expressed genes or the target genes of the differentially expressed lncRNAs.
Validation of RNA-seq data
To confirm the RNA-seq data, the expression profiles of randomly selected mRNAs and lncRNAs were tested in another 9 IH patients using quantitative real-time polymerase chain reactions (qRT-PCR) with the SYBR green method on an Applied Biosystems ViiA™ 7 Dx (Life Technologies, USA). Patient information is listed in Table 4. The sequences of the specific PCR primer sets used for qRT-PCR are listed in Table 5. The RNA expression levels were normalized to the internal control gene, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), using the 2(–△△Ct) method as we previously described [39]. Three selected miRNAs were further examined by Bulge-LoopTM qRT-PCR according to the manufacturer’s protocol (RIBOBIO, Guangzhou, China) with the SYBR green method on an Applied Biosystems ViiATM 7 Dx (Life Technologies, USA). The miRNA expression levels were normalized to u6 (RIBOBIO, Guangzhou, China), using the 2(–△△Ct) method.
Table 5: Details of primer pairs used in analysis of mRNAs and lncRNAs expression by qRT-PCR
Gene name |
Forward primer (5′-3′) |
Reverse primer (5′-3′) |
|---|---|---|
IFI44L |
ACAGAGCCAAATGATTCCCTATG |
TCGATAAACGACACACCAGTTG |
ISG15 |
CGCAGATCACCCAGAAGATCG |
TTCGTCGCATTTGTCCACCA |
PIP |
GTCAGTACGTCCAAATGACGAA |
CTGTTGGTGTAAAAGTCCCAGT |
TCONS_00088818 |
GCCTTGTGGTGTCTCCTCAG |
TAGACCAGGCGTCATAGCAGAA |
TCONS_00112159 |
GAAACAGCCACGGAGGGAAC |
GATTTCTGCAATGCCGTGCC |
TCONS_00125870 |
CCTAGAACCAGGGGCCACAA |
TTTGCTGGGCACTCTGTAGC |
CeRNA network analysis
The miRanda and TargetScan assessments were used to identify ceRNAs (competing endogenous RNAs, including protein-coding messenger RNAs, long non-coding RNAs and circular RNAs), containing microRNA response elements (MREs). Then, ceRNAs with common miRNAs were selected to predict the global interactions between miRNAs and ceRNAs. Additionally, the ceRNAs with common miRNAs that were up- or down-regulated by miRNAs were abstracted based on differential expression to predict the co-regulated interactions of miRNAs and ceRNAs. The co-regulated ceRNA network was generated by Cytoscape (V. 3.4.0) [40].
Statistical analysis
Data were analysed using the SPSS 20.0 software package (SPSS, Chicago, IL, USA) with an independent-samples T test performed between the two groups. All values are represented as the mean ± standard deviation (SD) from at least three independent experiments. Statistical significance was defined as P < 0.05.
Author contributions
Jun Li projected the experiment. Jingyun Li performed the sample preparation and wrote the manuscript. Qian Li, Ling Chen, Yanli Gao and Bei Zhou performed the bioinformatics analysis. Jun Li edited the manuscript.
ACKNOWLEDGMENTS AND FUNDING
This study was supported by the Nanjing Science and Technology project (201503047); the Jiangsu Provincial Medical Youth Talent; and the Jiangsu Maternal and Child Health Research Project (F201608).
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interests.
REFERENCES
1. Munden A, Butschek R, Tom WL, Marshall JS, Poeltler DM, Krohne SE, Alio AB, Ritter M, Friedlander DF, Catanzarite V, Mendoza A, Smith L, Friedlander M, et al. Prospective study of infantile haemangiomas: incidence, clinical characteristics and association with placental anomalies. Br J Dermatol. 2014; 170:907–913.
2. Kim KH, Choi TH, Choi Y, Park YW, Hong KY, Kim DY, Choe YS, Lee H, Cheon JE, Park JB, Park KD, Kang HJ, Shin HY, et al. Comparison of Efficacy and Safety Between Propranolol and Steroid for Infantile Hemangioma: A Randomized Clinical Trial. JAMA Dermatol. 2017; 153:529–536.
3. Frieden IJ. Infantile hemangioma research: looking backward and forward. J Invest Dermatol. 2011; 131:2345–2348.
4. Smith C, Friedlander SF, Guma M, Kavanaugh A, Chambers CD. Infantile Hemangiomas: An Updated Review on Risk Factors, Pathogenesis, and Treatment. Birth Defects Res. 2017; 109:809–815.
5. Leaute-Labreze C, Prey S, Ezzedine K. Infantile haemangioma: part I. Pathophysiology, epidemiology, clinical features, life cycle and associated structural abnormalities. J Eur Acad Dermatol Venereol. 2011; 25:1245–1253.
6. Li LJ, Zhao W, Tao SS, Leng RX, Fan YG, Pan HF, Ye DQ. Competitive endogenous RNA network: potential implication for systemic lupus erythematosus. Expert Opin Ther Targets. 2017; 21:639–648.
7. Thomson DW, Dinger ME. Endogenous microRNA sponges: evidence and controversy. Nat Rev Genet. 2016; 17:272–283.
8. Sun Y, Cheng H, Wang G, Yu G, Zhang D, Wang Y, Fan W, Yang W. Deregulation of miR-183 promotes melanoma development via lncRNA MALAT1 regulation and ITGB1 signal activation. Oncotarget. 2017; 8:3509–3518. https://doi.org/10.18632/oncotarget.13862.
9. Cao C, Zhang T, Zhang D, Xie L, Zou X, Lei L, Wu D, Liu L. The long non-coding RNA, SNHG6–003, functions as a competing endogenous RNA to promote the progression of hepatocellular carcinoma. Oncogene. 2017; 36:1112–1122.
10. Han X, Yang F, Cao H, Liang Z. Malat1 regulates serum response factor through miR-133 as a competing endogenous RNA in myogenesis. FASEB J. 2015; 29:3054–3064.
11. Liu XH, Sun M, Nie FQ, Ge YB, Zhang EB, Yin DD, Kong R, Xia R, Lu KH, Li JH, De W, Wang KM, Wang ZX, et al. Lnc RNA HOTAIR functions as a competing endogenous RNA to regulate HER2 expression by sponging miR-331–3p in gastric cancer. Mol Cancer. 2014; 13:92.
12. Liu X, Lv R, Zhang L, Xu G, Bi J, Gao F, Zhang J, Xue F, Wang F, Wu Y, Fu C, Wang Q, Huo R. Long noncoding RNA expression profile of infantile hemangioma identified by microarray analysis. Tumour Biol. 2016.
13. Strub GM, Kirsh AL, Whipple ME, Kuo WP, Keller RB, Kapur RP, Majesky MW, Perkins JA. Endothelial and circulating C19MC microRNAs are biomarkers of infantile hemangioma. JCI Insight. 2016; 1:e88856.
14. Bertoni N, Pereira LM, Severino FE, Moura R, Yoshida WB, Reis PP. Integrative meta-analysis identifies microRNA-regulated networks in infantile hemangioma. BMC Med GeneT. 2016; 17:4.
15. Batista PJ, Chang HY. Long noncoding RNAs: cellular address codes in development and disease. Cell. 2013; 152:1298–1307.
16. Kornienko AE, Guenzl PM, Barlow DP, Pauler FM. Gene regulation by the act of long non-coding RNA transcription. Bmc Biol. 2013; 11:59.
17. Stiles JM, Rowntree RK, Amaya C, Diaz D, Kokta V, Mitchell DC, Bryan BA. Gene expression analysis reveals marked differences in the transcriptome of infantile hemangioma endothelial cells compared to normal dermal microvascular endothelial cells. Vasc Cell. 2013; 5:6.
18. Ritter MR, Moreno SK, Dorrell MI, Rubens J, Ney J, Friedlander DF, Bergman J, Cunningham BB, Eichenfield L, Reinisch J, Cohen S, Veccione T, Holmes R, et al. Identifying potential regulators of infantile hemangioma progression through large-scale expression analysis: a possible role for the immune system and indoleamine 2,3 dioxygenase (IDO) during involution. Lymphat Res Biol. 2003; 1:291–299.
19. Calicchio ML, Collins T, Kozakewich HP. Identification of signaling systems in proliferating and involuting phase infantile hemangiomas by genome-wide transcriptional profiling. Am J Pathol. 2009; 174:1638–1649.
20. Ala U, Karreth FA, Bosia C, Pagnani A, Taulli R, Leopold V, Tay Y, Provero P, Zecchina R, Pandolfi PP. Integrated transcriptional and competitive endogenous RNA networks are cross-regulated in permissive molecular environments. Proc Natl Acad Sci U S A. 2013; 110:7154–7159.
21. Greenberger S, Bischoff J. Pathogenesis of infantile haemangioma. Br J Dermatol. 2013; 169:12–19.
22. Jiang C, Lin X, Hu X, Chen H, Jin Y, Ma G, Chen D, Chen X, Gu W. Angiogenin: a potential serum marker of infantile hemangioma revealed by cDNA microarray analysis. Plast Reconstr Surg. 2014; 134:231e–239e.
23. Wang X, Abraham S, McKenzie J, Jeffs N, Swire M, Tripathi VB, Luhmann U, Lange C, Zhai Z, Arthur HM, Bainbridge J, Moss SE, Greenwood J, et al. LRG1 promotes angiogenesis by modulating endothelial TGF-beta signalling. NATURE. 2013; 499:306–311.
24. Greenberger S, Adini I, Boscolo E, Mulliken JB, Bischoff J. Targeting NF-kappaB in infantile hemangioma-derived stem cells reduces VEGF-A expression. Angiogenesis. 2010; 13:327–335.
25. Xu GQ, Lu RR, Huo R, Guo X. [The expression of CEACAM-1 and CXCL-14 in infantile hemangioma]. Zhonghua Zheng Xing Wai Ke Za Zhi. 2010; 26:195–198.
26. Chen L, Sun F, Yang X, Jin Y, Shi M, Wang L, Shi Y, Zhan C, Wang Q. Correlation between RNA-Seq and microarrays results using TCGA data. Gene. 2017.
27. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012; 9:357–359.
28. Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013; 14:R36.
29. Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012; 7:562–578.
30. Zhao Y, Li H, Fang S, Kang Y, Wu W, Hao Y, Li Z, Bu D, Sun N, Zhang MQ, Chen R. NONCODE 2016: an informative and valuable data source of long non-coding RNAs. Nucleic Acids Res. 2016; 44:D203–D208.
31. Kong L, Zhang Y, Ye ZQ, Liu XQ, Zhao SQ, Wei L, Gao G. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. NUCLEIC ACIDS RES. 2007; 35:W345–W349.
32. Tafer H, Hofacker IL. RNAplex: a fast tool for RNA-RNA interaction search. Bioinformatics. 2008; 24:2657–2663.
33. Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013; 31:46–53.
34. Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, Wang J. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009; 25:1966–1967.
35. Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014; 42:D68–D73.
36. Li J, Zou J, Li Q, Chen L, Gao Y, Yan H, Zhou B, Li J. Assessment of differentially expressed plasma microRNAs in nonsyndromic cleft palate and nonsyndromic cleft lip with cleft palate. Oncotarget. 2016; 7:86266–86279. https://doi.org/10.18632/oncotarget.13379.
37. Audic S, Claverie JM. The significance of digital gene expression profiles. GENOME RES. 1997; 7:986–995.
38. Boyle EI, Weng S, Gollub J, Jin H, Botstein D, Cherry JM, Sherlock G. GO::TermFinder—open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics. 2004; 20:3710–3715.
39. Li J, Long W, Li Q, Zhou Q, Wang Y, Wang H, Zhou B, Li J. Distinct expression profiles of lncRNAs between regressive and mature scars. Cell Physiol Biochem. 2015; 35:663–675.
40. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003; 13:2498–2504.