
INTRODUCTION
Traumatic brain injury (TBI) frequently leads to severe and persistent neurological deficits that can result in considerable morbidity. In addition to neurological deficits, TBI often leads to headache, psychological impairment, and cognitive impairment [1–3], and the combined effects of these disorders produce a substantial burden on society. Temporary or permanent memory loss is seen in many head injury patients [4] and can lead to long-term deficits in learning and cognition [5–7]. However, the nature and extent of damage is variable, and long-term recovery can be difficult to predict. There is presently no known method of preventing memory deficits after TBI or treating them once they occur.
Seizures often occur after TBI, and TBI is a major risk factor for subsequent development of epilepsy [8–10]. There is a strong correlation between posttraumatic epilepsy (PTE) and the loss of brain volume after head injury [11]. Since seizures and TBI are independently associated with memory and learning deficits, it is possible that the two effects may be synergistic such that seizures exacerbate TBI-related physiological dysfunction. There is also evidence that the interval between a TBI and a post-TBI seizure may impact both the severity of TBI comorbidities and epileptogenesis [12, 13], and early seizures (defined as occurring within one week after injury) [14] may be associated with worse memory outcome than seizures that occur remotely. If this is the case, aggressive seizure prophylaxis may be important for improved outcome.
If the combined effect of TBI and seizures on hippocampal processes is greater than either alone, a better understanding of the underlying mechanisms might provide guidance for further clinical therapeutic research efforts. In this study, we report the changes in hippocampal electrophysiological correlates of plasticity of seizures at various intervals after TBI, as well as the capacity of prophylactic antiepileptic medications to ameliorate these effects.
The animals used in this study are a subset of animals used in our previous publication [13]. A diagrammatic representation of manipulations, and treatment times of the various animal groups for this study are shown in Supplementary Data 1.
RESULTS
Short-term plasticity of hippocampal CA1-CA3 Schaffer collateral synapses is affected by KA-induced seizures and by fluid percussion in injury (Figure 1)
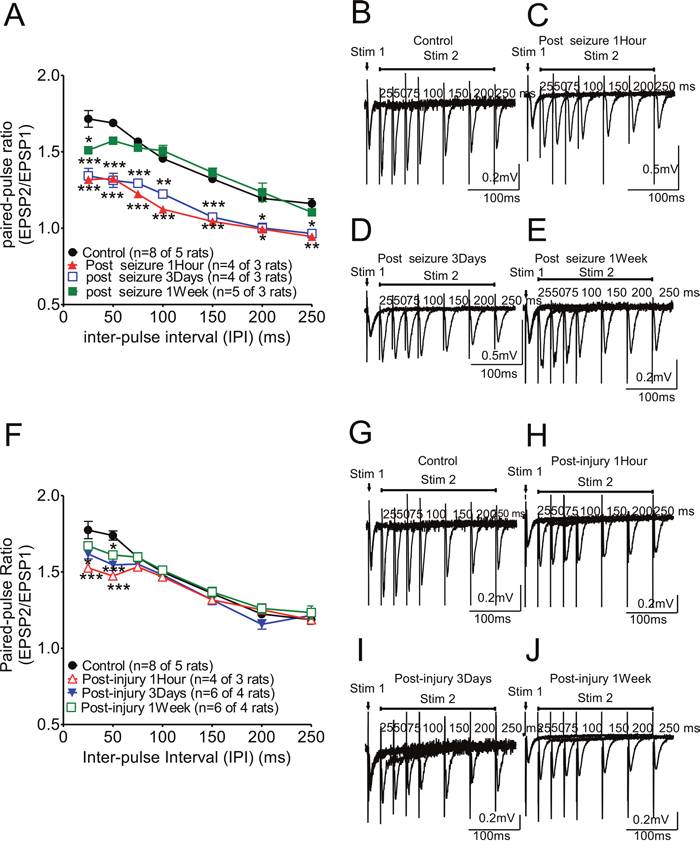
Figure 1: Paired-pulse ratio changes in hippocampal CA1 Schaffer collateral synapses following KA-induced seizures or fluid percussion injury. (A) The ratio was markedly suppressed initially (one hour after KA-induced seizures; red triangle curves) and this effect persisted at three days after KA-induced seizures (open blue box curve) compared with the ratios of control animals (black circle curve). This suppression of this ratio was partially reversed at one week after KA-induced seizures (green box curve). A representative trace from each group is shown in panels (B) to (E). (F) The paired-pulse facilitation after high frequency stimulation (IPI ≤ 75ms) was suppressed at 2Psi in FPI animals while PPR was not affected with longer IPI. This PPR suppression was significant at post-injury 1 hour, and reversed gradually at post-injury 3 days and one week (Figure 1G-1J).
To check the effects of seizures on short-term plasticity only, we measured the paired-pulse ratio (PPR) evoked by different stimulation inter pulse intervals (IPI) (from 10msec to 250msec) in hippocampal CA1 Schaffer collateral synapses after KA-induced seizures with and without FPI. The PPR was affected at very early stages after KA-induced seizures (Figure 1A-1E, one hour after KA-induced seizure; red triangle curve) and was suppressed at three days after KA-induced seizures (blue open box curve) compared with the PPR in the control animals (black circle curve). PPR recovered at one week after KA-induced seizures (green black box curve). Animals given FPI without KA also showed suppression of PPR with gradual recovery by 1 week (Figure 1G-1J). Of note, the combination of TBI and KA seizures showed a significantly greater deficit and longer recovery time (see Figure 2A).
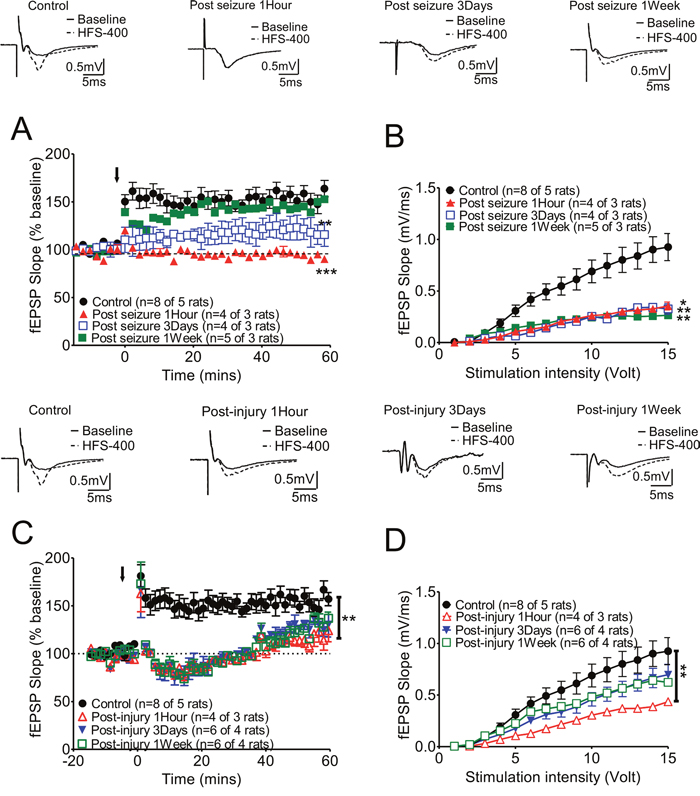
Figure 2: Long-term synaptic plasticity of hippocampal CA3-CA1 Schaffer collateral synapses was affected either by KA-induced seizures or fluid percussion injury alone, and these changes varied at different recording times. (A) Marked suppression of LTP was found at 3 days after KA-induced seizures, while this suppression was partially reversed at 7 days after seizures. A representative trace of each group is shown in the upper panels. (B) The slopes of fEPSPs were generally suppressed after KA-induced seizures. (C) After 2 Psi FPI, the plasticity was impaired in post-1 hour and 3 days animals and mild potentiation was found at the end of the recording time (Post-FPI 1 hour: open red triangles; Post-FPI 3 days: solid blue triangles). LTP could be induced one week after 2 Psi FPI (Open green squares). (D) The slope of fEPSPs were also suppressed by 2Psi FPI initially (1 hour after FPI) and then recovered gradually.
Hippocampal CA1-CA3 Schaffer collateral LTP formation was also impaired by KA-induced seizures initially and then recovered one week later (Figure 3)
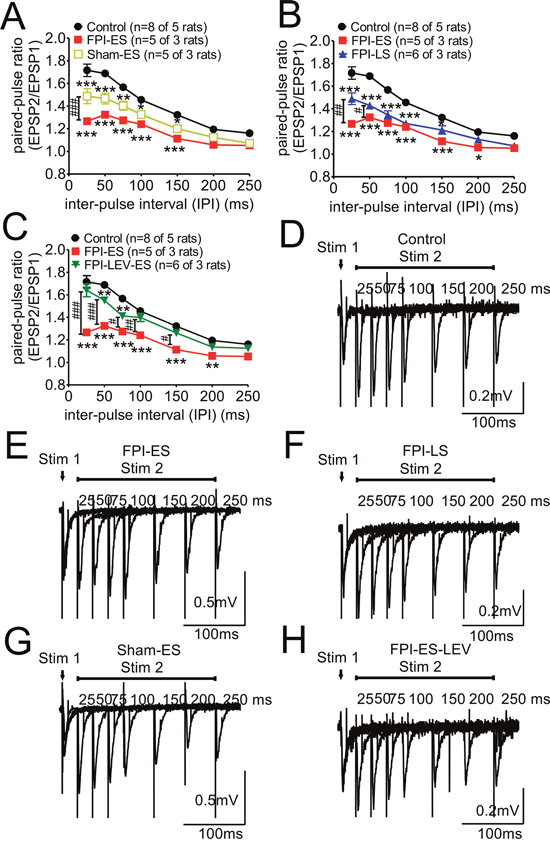
Figure 3: Paired-pulse stimulation responses, impaired in the post-FPI seizure animals, could be partially reversed by prophylactic administration of LEV. Under our experimental conditions (Mg 2+: 1.5 mM and Ca 2+: 2.4 mM and the initial evoked potentials being 30% of the maximal response), the responses were higher in second evoked potentials with a short interval, which is referred as PPF (paired-pulse facilitation). (A) Regardless of whether the animals were injured or not, the extent of PPF in the seizure animals was less than that of the control animals. In addition, profound suppression of PPF was found in the injured animals subjected to seizures (solid red squares). (B) PPF was suppressed in both early and late post-injury seizure animals compared with control animals. The suppression of PPF in the early seizure group was more marked (solid red squares) than that in the later seizure group of injured animals (solid blue triangles) (C) The suppression of PPF in the early seizure group of injured animals (FPI-ES, solid red squares) could be reversed by the prophylactic injection of LEV (FPI-LEV-ES, open green triangles). (D-H) The representative wave forms for paired-pulse stimulation of each group are shown.
To determine the impact of seizures on long-term synaptic plasticity, we analyzed the LTP at hippocampal CA3-CA1 Schaffer collateral synapses at one hour, 3 days, and one week after KA-induced seizures. LTP was affected by KA-induced seizures, and the degree of change varied at different recording times. Initially, LTP was totally suppressed at one hour after seizures (Figure 2A, red triangle). Marked suppression of LTP was found at 3 days after KA-induced seizures (blue open box curve), while this suppression partially recovered at 7 days after seizures (green black box curve). The representative traces from each group are shown in the upper panels of Figure 2. The slopes of the fEPSPs in the KA-induced seizure groups were generally suppressed compared to those of the control group (Figure 2B). FPI also altered LTP with gradual recovery (Figure 2C, 2D). Of note, the combination of TBI and KA seizures showed a much longer recovery time and greater deficit (see Figure 4A, 4D).
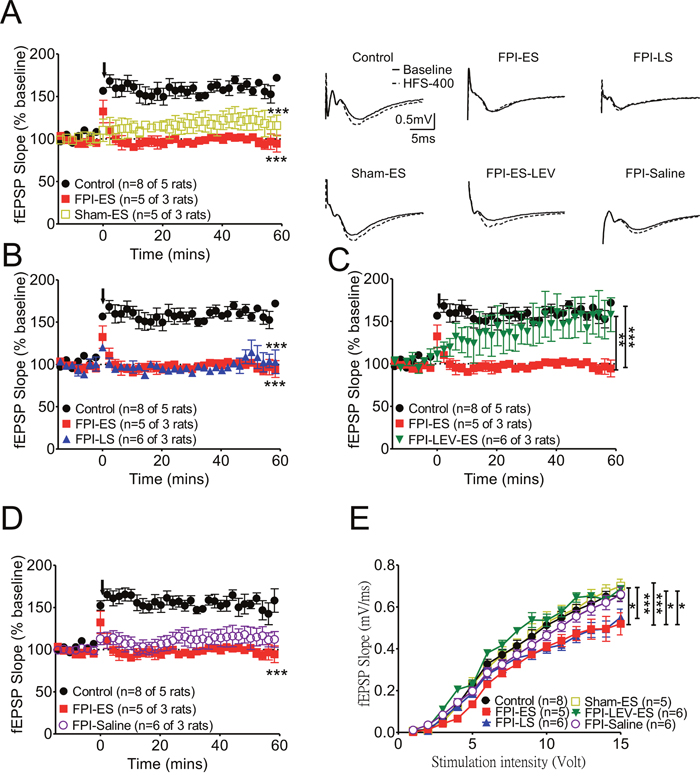
Figure 4: After brain injury, the suppression of synaptic plasticity as manifested by LTP in the hippocampus in seizure animals was prominent. (A) Synaptic plasticity at CA3-CA1 synapses was compared in control, FPI-ES, and sham–SE groups. LTP, readily induced in control animals, was virtually absent in seizure animals with brain injury (FPI-ES group, red squares). (B) After brain injury, neither the early seizure (FPI-ES) nor late seizure (FPI-LS, blue triangles) groups manifested LTP in fEPSPs from the CA3-CA1 pathway. (C) If the brain injury animals received prophylactic levetiracetam infusion for one week after injury, synaptic plasticity was partially restored (FPI-LEV-ES group, inverted green triangles). (D) LTP formation in the FPI-only group (FPI-saline group, open purple circles) was impaired, being significantly suppressed by early seizure induction after FPI (FPI-ES group, red squares). (E) The evoked potentials used in the LTP experiments were 45~50% of the maximum response to stimulation after we performed input-output (I/O) curves at each stimulation site in each hippocampal brain slice. *:P<0.05, ***:P<0.001 Control vs other groups, ##:P<0.01, ###:P<0.001 Sham+ES vs other groups, $:P<0.05 FPI+Saline vs FPI+ES.
Paired-pulse stimulation responses were impaired in post-FPI + KA seizure animals but could be partially reversed by prophylactic injection of LEV (Figure 3)
We next assessed the effect of FPI combined with seizures on short-term plasticity. By applying paired-pulse stimulation at different stimulation intervals (from 10 to 250 msec) to each brain slice, the paired-pulse ratio (PPR) was calculated. Under our experimental conditions (Mg 2+: 1.5 mM and Ca 2+: 2.4 mM and the initial evoked potentials being 30% of the maximal response), the responses were higher for second evoked potentials with a short interval, which is referred as PPF (paired-pulse facilitation). Regardless of whether the animal was injured or not, the extent of PPF in the seizure animals was less than that in the control animals (Figure 3A), whereas a more profound suppression of PPF was found in the injured animals subjected to seizures (Figure 3A, red solid squares). Although PPF was suppressed in both post-injury early and late onset seizure animals in comparison to the control animals, the suppression was greater in the early seizure group (Figure 3B, red solid squares) than in the late seizure group of injured animals (Figure 3B, blue solid triangles). When prophylactic LEV was administered after injury, the suppression of PPF in the early seizure group of injured animals (Figure 3C, FPI-ES; red solid squares) could be reversed (Figure 3C, FPI-LEV-ES, green open triangles). The representative wave forms for paired-pulse stimulation for each group are shown in Figure 3D-3H.
Post-TBI early seizures profoundly impaired LTP formation compared with late post-TBI seizures (Figure 4)
Next, to determine the effect of brain injury and seizure on long-term synaptic plasticity, the long-term potentiation of CA1 Schaffer collateral synapses was assessed in the FPI groups with early or late onset of seizures induced by KA. Our data indicate that the suppression of synaptic plasticity, manifested by LTP in the hippocampus in seizure animals, was prominent (Figure 4A). LTP, which was readily induced in control animals, was virtually absent in seizure animals after brain injury (Figure 4A FPI-ES group, red square). After brain injury, LTP could not be induced in either the early seizure (Figure 4B, FPI-ES) or late seizure (Figure 4B, FPI-LS, blue triangle) groups. However, when prophylactic levetiracetam was administered by pump infusion for one week after injury, synaptic plasticity was partially restored (Figure 4C, FPI-LEV-ES group, reverse green triangles). LTP formation in the FPI-only group (Figure 4D, FPI-saline group, open purple circle) was impaired, but these impairments were more profound with early seizure induction after FPI (Figure 4D, FPI-ES group, red squares). The evoked potentials used in the LTP experiments were 45~50% of the maximum response to stimulation after we generated input-output (I/O) curves for each stimulation site in the hippocampal brain slices. The I/O curve of the FPI-only group (FPI-saline group, open purple circles) was shifted to the right (i.e. moderately suppressed by the injury), and the I/O curves of the injured animals with early onset seizure (Figure 4E, FPI-ES, red squares) or late onset seizure (Figure 4E, FPI-LS, blue triangles) were suppressed more significantly and shifted further to the right compared with the seizure-only (Figure 4E, sham-ES, open yellow squares) and control groups (Figure 4E, black circles).
DISCUSSION
TBI has been implicated in memory loss and other deficits [5, 15, 16], and seizures are also known to contribute to learning impairments [17, 18]. Memory and learning may be impaired by both TBIs and seizures; therefore, in patients with posttraumatic seizures, deficits to memory and learning processes may be profound. The current study sought to test this hypothesis by using FPI and KA-induced seizure animal models to determine the mechanisms underlying such changes in order to better inform future clinical applications.
In this study, we measured presynaptic as well as postsynaptic transmission in the hippocampus through the use of short-term and long-term synaptic plasticity experiments conducted with rats subjected to FPI followed by early or late KA-induced seizures.
First, in the short-term plasticity experiment, PPRs were markedly suppressed at the acute stage (at 1 hour and 3 days after KA-induced seizure), and this suppression partially recovered (Figure 1A). In the long-term plasticity experiment, the seizures showed acute effects on LTP. The suppression of LTP could be observed at 3 days after a seizure, but this suppression partially recovered at one week after the seizure (Figure 2). However, the input-output curves (Figure 2B, I/O curve), representing the slope of fEPSPs with different stimulation intensities from 1 to 15 volts, in seizure animals were all shifted significantly to right even at one week after seizures. This shift to the right may relate to impaired glutamaturgic transmission. Biochemical findings have demonstrated that injury leads to significantly smaller N-methyl-D-aspartate (NMDA) potentials and glutamate-induced excitatory currents, increased dendritic spine size, and decreased expression of alpha-calcium calmodulin kinase II, which is consistent with electrophysiological findings demonstrating that NMDA and AMPA currents are decreased at 7 days post-injury [19]. In most studies, the current levels used to tetanize the hippocampal slices for LTP are 40-50% of those needed for maximum EPSP, and fewer fibers are stimulated in TBI animals due to the maximum elicited EPSP being lower in TBI hippocampal slices [20–22]. Therefore, shifting the I/O curve in our data to the right may relate to impaired glutamatergic transmission and LTP in TBI animals may be difficult to induce and maintain. Fluid percussion injury at 2 Psi, which is a very low level of injury, also elicited profound but reversible suppression of PPR and LTP. This suggests that even low levels of concussive injury may have significant effects on hippocampal mechanisms which have been related to learning and memory.
LTP CA1 Schaffer collateral mechanisms
The major components of CA1 LTP formation include glutamatergic transmission, NMDA glutamate receptors, and Ca2+ channels, which are normally blocked by magnesium (Mg2+) but which open when glutamate activates the NMDA receptors, with one important qualifier [23]. Traditionally, LTP from CA1 Schaffer collateral stimulation induced by NMDA receptors was thought to be activated by the membrane of the post synaptic neuron being partially depolarized coincident with presynaptic input, with neurotransmitter and voltage dependent Ca2+ channels then being activated [24]. Recent studies, however, have indicated that much more complex mechanisms are involved in LTP, including changes in gene expression and protein synthesis; of particular importance is the induction of CREB, PKA, and MAPK signaling pathways and synthesis of postsynaptic AMPA receptors [25–28]. In addition, there are both pre and postsynaptic structural changes at the level of dendritic spines [29, 30].
The factors that affect short-term plasticity formation (PPF)
Paired-pulse stimulation in CA1 Schaffer collateral synapses was used to determine presynaptic neurotransmission, and our data (Figures 1 and 3) indicate that short-term synaptic plasticity was profoundly suppressed by TBI followed by KA-induced seizures. Short-term synaptic plasticity is primarily a presynaptic function that is influenced by several factors [31]. First, the role of presynaptic calcium signaling in many use-dependent forms of plasticity are important [32, 33]. Action potentials arrive at the presynaptic terminal and induce calcium influx and calcium binding to multiple low-affinity sites on synaptotagmin that trigger vesicle fusion. Therefore, short-term plasticity appears to depend primarily on the fluctuations and homeostasis in calcium levels at the synaptic site.
Synaptic plasticity, TBI, and seizures
There are many possible causes of synaptic plasticity deficits exacerbated by seizures after TBI.
1. Altered calcium homeostasis: Animal studies have revealed that Ca2+ homeostasis in TBI-impacted hippocampal neurons is altered, a change which may result from necrotic or apoptotic cell death and abnormalities in Ca2+ influx and efflux. In addition, long-term changes in Ca2+ buffering or Ca2+ sequestration/release mechanisms may underlie these changes in Ca2+ homeostasis after TBI [34, 35]. Moreover, seizures have also been found to alter hippocampal neuron calcium homeostasis [36, 37]. Relatedly, FPI has been found to induce impairment of neurotransmission, including the transmission of dopamine and acetylcholine [38, 39], as well as glutamate. These findings may explain why FPI exacerbates the synaptic plasticity impairment caused by a KA-induced seizure.
2. Neuron loss in the hippocampus: Hippocampal involvement in PTE has been reported to include reduced bilateral hippocampal volume in humans after TBI [40–42]. In addition, extensive cortical and subcortical neuronal loss has also been reported after TBI [43–45]. The hippocampal CA3 and hilus have been found to be damaged initially after cortical impact [46, 47]; which results, in turn, in neuronal loss resembling “hippocampal sclerosis”, a hallmark of temporal-lobe epilepsy [45, 48].
3. Altered synapse strength: Time-dependent changes in synaptic proteins occur well after levels of oxidants peak following brain injury, which suggests that the depletion of antioxidant systems following trauma affects synaptic functioning and plasticity [49].
4. Suppression of excitatory transmission: The long-term depression of excitatory neurotransmission due to alterations in synaptic GluR2-containing, calcium-impermeable AMPARs after head injury may contribute to cognitive deficits resulting from TBI [50].
5. In addition, an early post-TBI seizure promotes epileptogenesis and also profoundly suppresses synaptic plasticity: The early onset of post-TBI seizures affected the severity and duration of those seizures, which may contribute to epileptogenesis [13]. However, the infusion of prophylactic levetiracetam could provide significant neuroprotection, a finding which was also previously documented [13, 51]. Such infusions may protect the cellular structure of CA1 Schaffer collateral synapses and preserve not only short-term but also long-term synaptic plasticity [52, 53].
The antiepileptic effects of LEV may involve several mechanisms [51, 52]: (1) interaction with SV2A which is one isoform of synaptic vesicle protein 2 (SV2) (2) removal of the Zn++-induced suppression of GABA-mediated presynaptic inhibition. (3) the combination of inhibitory effects on depolarization-induced and Ca2+ release-associated neurotransmitter releases. (4) modulation of the presynaptic P/Q-type voltage-dependent calcium (Ca2+) channel to reduce glutamate release in the dentate gyrus of the hippocampus that regulates seizure activities. (5) via intracellular inhibition of presynaptic Ca2+ channels to inhibit neurotransmitter release. Furthermore, daily LEV treatment may have beneficial effects on the histological, molecular, and behavioral elements of neurological recovery after TBI, in part, via modulation of neuroinflammatory and excitatory pathways.
MATERIALS AND METHODS
Ethics statement
6-week-old male Sprague-Dawley rats (N=40) were used in the present study. The animals were housed under a 12 hr light/dark cycle and provided with food and water ad libitum. All animal procedures were conducted in accordance with NIH guidelines and were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of the National Defense Medical Center (Taiwan Protocol Number IACUC-13-145). The number of animals used was the minimum number required, based on power calculations (α=0.05 and 1-ß=0.80). All surgery was done under aseptic conditions and postoperative analgesia was provided by administration of NSAIDs. Perioperative opiates were not used as they have been shown to be neuroprotective and might have confounded interpretation of the results.
Experimental protocols and groups of animals (Supplementary Data 1)
Two separate experiments were performed to examine the effect of single and repeat seizures, respectively. In the first experiment, rats underwent fluid percussion injury at 2 Psi and were subsequently induced to have a single seizure at 1 hour (FPI+1hr, N=3), 3 days (FPI+3d, N=4), or 7 days after TBI (FPI+7d, N=4), and were compared with control animals not exposed to TBI (control, N=5). For the second experiment, rats underwent fluid percussion injury followed by induction of three seizures at weekly intervals starting at 1 week after TBI (FPI+ES, N=3), 2 weeks after TBI (FPI+LS, N=3), and 1 week after TBI with prophylactic levetiracetam infusion (25.2 mg/kg/day) via ALZET mini-pump for 7 days (FPI+LEV+ES, N=3); these were compared with uninjured rats (control, N=5), rats exposed to TBI but not induced to have seizures (FPI-saline, N=3), and rats not exposed to TBI but induced to have three seizures at one week intervals (Sham-ES, N=3).
Seizure induction
Kainic Acid (Sigma-Aldrich, Co., St Louis, MO, US, 7 mg/kg) was injected via the tail to induce seizures in each of the seizure groups. All the animals underwent electrode implantation into the M1 cortex to further identify the onset of seizures and the epileptic spikes. To control for postictal apnea, all the animals received manual artificial respiration via a tube placed over the nose until spontaneous respiration was seen. The mortality rate in each group with three times of KA-induced seizure protocol were: FPI-ES: 3/6 (50%); FPI-LS: 0/3; Sham-ES: 0/3; FPI-LEV-ES: 0/3; and total mortality rate was 3/15 (20%).
Fluid percussion traumatic brain injury
The fluid percussion device (model HPD-1700, Dragonfly R&D, USA) used to produce TBIs in the rats has previously been described by Matsushita et al.[54] The specific methods used to produce midline FPIs are detailed in our previous papers [55, 56] but are briefly reiterated here to facilitate reproduction of our results. Animals were anesthetized using Zoletil 50mg/kg IP; a 4.8 mm diameter burr hole was then drilled at the midline between the coronal and lambdoid sutures, and a Leur-Loc hub was affixed to the perimeter of the burr hole using cyanoacrylate. Dental acrylic and two small nickel-plated screws were then used to anchor the hub to the skull. The rats were anesthetized, the surgical site exposed, and the animal was connected to the injury device. Injury was induced by striking the piston with a weighted metal pendulum released from a pre-determined height or angle. The resulting rapid injection of a small volume of saline into the closed cranial cavity produces a pulse of increased intracranial pressure that is associated with a deformation of the brain. The force of the injury administered was 2 Psi of pressure and resulted in the suppression of the righting reflex for a period of between 6 and 9 min. Sham animals were connected to the injury device but no injury was delivered, and suppression of the righting reflex lasted less than 60 sec. Pressure pulses were measured extra-cranially with a pressure transducer, recorded on a digital real-time oscilloscope (TDS210, Sony Tektronix Corp., Japan), and analyzed by Wave Star software (Sony Tektronix Corp., Japan). The fluid percussion device delivered transient pressure fluid pulses with constant wave form and duration (17–21 ms) to cause brain injury [57].
Prophylactic levetiracetam were administered by infusion pump implanted after TBI for one week (Infusion of LEV (25.2mg/kg/day) via ALZET mini-pumps for 7 days after TBI. The LEV+FPI+ES group received levetiracetam infusion pump implantation right after FPI for one week and the KA induced seizures were performed after we removed the pump. The control group for these animals were TBI + KA without LEV. This is also shown in Supplementary Data 1. Many of the other details on the control groups, LEV administration, and timing were detailed in our previous paper [13]. The animals used here on hippocampal plasticity, which is new, were a subset of those animals.
Hippocampal brain slice preparation
Hippocampal slices from the rat brains were prepared according to the procedure described in previous reports [58, 59]. Each animal was decapitated and its brain quickly removed and rapidly immersed in cold (4°C), oxygenated, high-sucrose, low-Ca2+-containing artificial cerebrospinal fluid (aCSF) with the following composition: 87 mM NaCl, 2.5 mM KCl, 7 mM MgCl2, 0.5 mM CaCl2, 1.25 mM NaH2PO4, 25 mM glucose, 75 mM sucrose, and 25 mM NaHCO3. Using a vibrating tissue slicer (VT1000S; Leica Instruments), the brain was sliced into 300–350-μm thick coronal slices. Incubation of the hemisectioned brain slices containing the hippocampus in normal aCSF consisting of 126 mM NaCl, 3.0 mM KCl, 1.5 mM MgCl2, 2.4 mM CaCl2, 1.2 mM NaH2PO4, 11.0 mM glucose, and 25 mM NaHCO3 saturated with 95% O2 and 5% CO2 at room temperature was then carried out for ≥90 min before electrophysiological recordings of the slices were made. After being transferred to a recording chamber, individual brain slices were continuously superfused with normal aCSF (2 mL/min) and maintained at a temperature of 30°–32°C using a temperature-controlled solution heater (TC-324B; Warner Instruments). A syringe pump (Razel) was then utilized to deliver drugs to the slices via superfusion.
In vitro hippocampal electrophysiology
Following preparation procedures described in previous studies [58, 60], field excitatory postsynaptic potentials (fEPSP) of each individual brain slice were recorded, with all of the recordings being conducted in the CA1 region of the hippocampus. Specifically, extracellular fEPSPs were obtained using 3 M NaCl-filled electrodes and an AC amplifier (A-M Systems Model 1800), with the signals being high- (10 Hz) and low-pass (10 kHz) filtered. Using an A/D board (National Instruments PCI 6024E, or Digidata 1320A; Axon Instruments), the fEPSP data were transmitted at 4 kHz to a personal computer running Windows-based software (WCP, courtesy of John Dempster, University of Strathclyde, Glasgow, UK; http://www.strath.ac.uk/sipbs/; or pCLAMP 9.0, Axon Instruments). Using single, 0.1-msec pulses, delivered at a frequency of 0.033 Hz through a bipolar electrode constructed with formvar-insulated nichrome wire, responses were elicited from each brain slice via electrical stimulation of the stratum radiatum. The intensity of these stimuli was adjusted as necessary to yield fEPSPs with peak amplitudes of 0.5–1 mV (30%–40% of the maximal response to avoid ceiling effects). Prior to electrical stimulation, at least 10 min of stable baseline recordings were obtained. Using the data acquisition software, the peak amplitude and slope of the initial (1–2 msec) rising phase of each fEPSP were calculated off-line, with any changes to the synaptic response being normalized to the aforementioned baseline period. High-frequency stimulation (HFS), consisting of three 1-sec trains of 100 Hz delivered at 10-sec intervals at twice the stimulation intensity utilized to evoke LTP was then administered. Following these HFS trains, the stimulation intensity was then returned to the level previously utilized to produce baseline fEPSPs.
Statistical analysis (Supplementary Data 2)
Data in the text and figures are expressed as means + SEM. Statistical analyses of data for the dopamine release input/output curves, fEPSP slope curves, and paired-pulse ratio were performed using a two-way analysis of variance (ANOVA) followed by a Bonferroni post hoc test for multiple comparisons. All statistical tests were two-tailed and were performed using GraphPad Prism 5.02 (GraphPad Scientific, San Diego, CA, USA). A p-value < 0.05 was considered significant for all analyses. *p<.05, **p<.01, ***p<.001 in all figures.
CONCLUSIONS
In this study, we investigated short-term and long-term synaptic plasticity suppression after TBI and KA-induced seizures. Our data indicate that general synaptic (presynaptic and postsynaptic) suppression occurs in TBI animals experiencing a subsequent seizure. Such synaptic suppression affects neurotransmission in terms of both PPF or LTP, with the presynaptic and postsynaptic transmission suppression after a seizure alone being transient, whereas such transmission suppression persists for a longer time when a seizure is combined with a TBI. Moreover, prophylactic levetiracetam infusion partially reverses such long term suppression of synaptic plasticity.
Abbreviations
2 Psi: 2 Pounds per square inch; aCSF: artificial cerebrospinal fluid; AMPARs: AMPA receptors; ANOVA: Analysis of variance; CA1: cornu ammonis 1; Ca: Calcium; CA3: cornu ammonis 3; EEG: Electroencephalography; fEPSP: field excitatory post synaptic potentials; FPI: fluid percussion injury; HFS: High-frequency stimulation; I/O curves: input-output curves; IACUC: Institutional Animal Care and Use Committee; IPI: interpulse intervals; KA: kainic acid; LEV: levetiracetam; LTP: long-term potentiation; M1 cortex: primary motor cortex; Mg: magnesium; NSAIDs: nonsteroidal anti-inflammatory drugs; PPF: paired pulse facilitation; PPR: paired-pulse ratio; PTE: posttraumatic epilepsy; TBI: traumatic brain injury.
Author contributions
Yuan-Hao Chen was responsible for study design, experiments performance, data analysis, and article writing, Tung-Tai Kuo was responsible for experiment performance, data collection. Eagle Yi-Kung Huang was responsible for study design, data analysis. Barry J. Hoffer was responsible for study design, data analysis and manuscript editing. Yu-Ching Chou was responsible for data analysis and statistics, Yung-Hsiao Chiang and Hsin-I Ma were responsible for financially supporting the work, Jonathan P. Miller was responsible for study design, data analysis and manuscript editing.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to declare.
FUNDING
This work was supported by the National Science Council of Taiwan under grant MOST105-2314-B-016-001-MY3, and by Medical Research Project grants TSGH-C106-066 and TSGH-C106-065 from the Tri-Service General Hospital of Taiwan and MAB-106-035 from the National Defense Medical Center. This project was also supported in part by philanthropic contributions from the George R. and Constance P. Lincoln family.
REFERENCES
1. Formisano R, Bivona U, Catani S, D'Ippolito M, Buzzi MG. Post-traumatic headache: facts and doubts. J Headache Pain. 2009; 10: 145-52. https://doi.org/10.1007/s10194-009-0108-4.
2. Jorge RE, Robinson RG, Starkstein SE, Arndt SV. Influence of major depression on 1-year outcome in patients with traumatic brain injury. J Neurosurg. 1994; 81: 726-33. https://doi.org/10.3171/jns.1994.81.5.0726.
3. May M, Milders M, Downey B, Whyte M, Higgins V, Wojcik Z, Amin S, O'Rourke S. Social behavior and impairments in social cognition following traumatic brain injury. J Int Neuropsychol Soc. 2017; 23: 400-11. https://doi.org/10.1017/S1355617717000182.
4. Zhao ZA, Ning YL, Li P, Yang N, Peng Y, Xiong RP, Zhao Y, Liu D, Zeng XJ, Chen JF, Zhou YG. Widespread hyperphosphorylated tau in the working memory circuit early after cortical impact injury of brain (Original study). Behav Brain Res. 2017; 323: 146-53. https://doi.org/10.1016/j.bbr.2017.02.002.
5. Kline AE, Massucci JL, Marion DW, Dixon CE. Attenuation of working memory and spatial acquisition deficits after a delayed and chronic bromocriptine treatment regimen in rats subjected to traumatic brain injury by controlled cortical impact. J Neurotrauma. 2002; 19: 415-25. https://doi.org/10.1089/08977150252932370.
6. Walker KR, Tesco G. Molecular mechanisms of cognitive dysfunction following traumatic brain injury. Front Aging Neurosci. 2013; 5: 29. https://doi.org/10.3389/fnagi.2013.00029.
7. Scheff SW, Price DA, Hicks RR, Baldwin SA, Robinson S, Brackney C. Synaptogenesis in the hippocampal CA1 field following traumatic brain injury. J Neurotrauma. 2005; 22: 719-32. https://doi.org/10.1089/neu.2005.22.719.
8. Feeney DM, Walker AE. The prediction of posttraumatic epilepsy. A mathematical approach. Arch Neurol. 1979; 36: 8-12.
9. Annegers JF, Coan SP. The risks of epilepsy after traumatic brain injury. Seizure. 2000; 9: 453-7. https://doi.org/10.1053/seiz.2000.0458.
10. Salazar AM, Grafman J. Post-traumatic epilepsy: clinical clues to pathogenesis and paths to prevention. Handb Clin Neurol. 2015; 128: 525-38. https://doi.org/10.1016/B978-0-444-63521-1.00033-9.
11. Salazar AM, Jabbari B, Vance SC, Grafman J, Amin D, Dillon JD. Epilepsy after penetrating head injury. I. Clinical correlates: a report of the Vietnam Head Injury Study. Neurology. 1985; 35: 1406-14.
12. Eslami M, Ghanbari E, Sayyah M, Etemadi F, Choopani S, Soleimani M, Amiri Z, Hadjighassem M. Traumatic brain injury accelerates kindling epileptogenesis in rats. Neurol Res. 2016; 38: 269-74. https://doi.org/10.1179/1743132815Y.039108R10086.
13. Chen YH, Huang EY, Kuo TT, Hoffer BJ, Wu PJ, Ma HI, Tsai JJ, Chou YC, Chiang YH. Levetiracetam prophylaxis ameliorates seizure epileptogenesis after fluid percussion injury. Brain Res. 2016; 1642: 581-9. https://doi.org/10.1016/j.brainres.2016.04.013.
14. Ong LC, Dhillon MK, Selladurai BM, Maimunah A, Lye MS. Early post-traumatic seizures in children: clinical and radiological aspects of injury. J Paediatr Child Health. 1996; 32: 173-6.
15. Himanen L, Portin R, Tenovuo O, Taiminen T, Koponen S, Hiekkanen H, Helenius H. Attention and depressive symptoms in chronic phase after traumatic brain injury. Brain Inj. 2009; 23: 220-7. https://doi.org/10.1080/02699050902748323.
16. Smith DH, Okiyama K, Thomas MJ, Claussen B, McIntosh TK. Evaluation of memory dysfunction following experimental brain injury using the Morris water maze. J Neurotrauma. 1991; 8: 259-69. https://doi.org/10.1089/neu.1991.8.259.
17. Brace HM, Jefferys JG, Mellanby J. Long-term changes in hippocampal physiology and learning ability of rats after intrahippocampal tetanus toxin. J Physiol. 1985; 368: 343-57.
18. Coras R, Pauli E, Li J, Schwarz M, Rossler K, Buchfelder M, Hamer H, Stefan H, Blumcke I. Differential influence of hippocampal subfields to memory formation: insights from patients with temporal lobe epilepsy. Brain. 2014; 137: 1945-57. https://doi.org/10.1093/brain/awu100.
19. Schwarzbach E, Bonislawski DP, Xiong G, Cohen AS. Mechanisms underlying the inability to induce area CA1 LTP in the mouse after traumatic brain injury. Hippocampus. 2006; 16: 541-50. https://doi.org/10.1002/hipo.20183.
20. Witgen BM, Lifshitz J, Smith ML, Schwarzbach E, Liang SL, Grady MS, Cohen AS. Regional hippocampal alteration associated with cognitive deficit following experimental brain injury: a systems, network and cellular evaluation. Neuroscience. 2005; 133: 1-15. https://doi.org/10.1016/j.neuroscience.2005.01.052.
21. Norris CM, Scheff SW. Recovery of afferent function and synaptic strength in hippocampal CA1 following traumatic brain injury. J Neurotrauma. 2009; 26: 2269-78. https://doi.org/10.1089/neu.2009.1029.
22. Zhang B, Chen X, Lin Y, Tan T, Yang Z, Dayao C, Liu L, Jiang R, Zhang J. Impairment of synaptic plasticity in hippocampus is exacerbated by methylprednisolone in a rat model of traumatic brain injury. Brain Res. 2011; 1382: 165-72. https://doi.org/10.1016/j.brainres.2011.01.065.
23. Siegelbaum SA, Kandel ER. Learning-related synaptic plasticity: LTP and LTD. Curr Opin Neurobiol. 1991; 1: 113-20.
24. O'Dell TJ, Kandel ER, Grant SG. Long-term potentiation in the hippocampus is blocked by tyrosine kinase inhibitors. Nature. 1991; 353: 558-60. https://doi.org/10.1038/353558a0.
25. Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003; 40: 361-79.
26. Malenka RC, Nicoll RA. Long-term potentiation--a decade of progress? Science. 1999; 285: 1870-4.
27. Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002; 25: 103-26. https://doi.org/10.1146/annurev.neuro.25.112701.142758.
28. Song I, Huganir RL. Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci. 2002; 25: 578-88.
29. Malik R, Chattarji S. Enhanced intrinsic excitability and EPSP-spike coupling accompany enriched environment-induced facilitation of LTP in hippocampal CA1 pyramidal neurons. J Neurophysiol. 2012; 107: 1366-78. https://doi.org/10.1152/jn.01009.2011.
30. Wang W, Trieu BH, Palmer LC, Jia Y, Pham DT, Jung KM, Karsten CA, Merrill CB, Mackie K, Gall CM, Piomelli D, Lynch G. A Primary cortical input to hippocampus expresses a pathway-specific and endocannabinoid-dependent form of long-term potentiation. eNeuro. 2016. https://doi.org/10.1523/ENEURO.0160-16.2016.
31. Regehr WG. Short-term presynaptic plasticity. Cold Spring Harb Perspect Biol. 2012; 4: a005702. https://doi.org/10.1101/cshperspect.a005702.
32. Burnashev N, Rozov A. Presynaptic Ca2+ dynamics, Ca2+ buffers and synaptic efficacy. Cell Calcium. 2005; 37: 489-95. https://doi.org/10.1016/j.ceca.2005.01.003.
33. Neher E, Sakaba T. Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron. 2008; 59: 861-72. https://doi.org/10.1016/j.neuron.2008.08.019.
34. Niu LJ, Xu RX, Zhang P, Du MX, Jiang XD. Suppression of Frizzled-2-mediated Wnt/Ca(2)(+) signaling significantly attenuates intracellular calcium accumulation in vitro and in a rat model of traumatic brain injury. Neuroscience. 2012; 213: 19-28. https://doi.org/10.1016/j.neuroscience.2012.03.057.
35. Sun DA, Deshpande LS, Sombati S, Baranova A, Wilson MS, Hamm RJ, DeLorenzo RJ. Traumatic brain injury causes a long-lasting calcium (Ca2+)-plateau of elevated intracellular Ca levels and altered Ca2+ homeostatic mechanisms in hippocampal neurons surviving brain injury. Eur J Neurosci. 2008; 27: 1659-72. https://doi.org/10.1111/j.1460-9568.2008.06156.x.
36. Raza M, Pal S, Rafiq A, DeLorenzo RJ. Long-term alteration of calcium homeostatic mechanisms in the pilocarpine model of temporal lobe epilepsy. Brain Res. 2001; 903: 1-12.
37. Bravo-Martinez J, Delgado-Coello B, Garcia DE, Mas-Oliva J. Analysis of plasma membrane Ca2+-ATPase gene expression during epileptogenesis employing single hippocampal CA1 neurons. Exp Biol Med (Maywood). 2011; 236: 409-17. https://doi.org/10.1258/ebm.2011.010342.
38. Verbois SL, Scheff SW, Pauly JR. Chronic nicotine treatment attenuates alpha 7 nicotinic receptor deficits following traumatic brain injury. Neuropharmacology. 2003; 44: 224-33.
39. Kelso ML, Wehner JM, Collins AC, Scheff SW, Pauly JR. The pathophysiology of traumatic brain injury in alpha7 nicotinic cholinergic receptor knockout mice. Brain Res. 2006; 1083: 204-10. https://doi.org/10.1016/j.brainres.2006.01.127.
40. Bigler ED, Abildskov TJ, Petrie J, Farrer TJ, Dennis M, Simic N, Taylor HG, Rubin KH, Vannatta K, Gerhardt CA, Stancin T, Owen Yeates K. Heterogeneity of brain lesions in pediatric traumatic brain injury. Neuropsychology. 2013; 27: 438-51. https://doi.org/10.1037/a0032837.
41. Bigler ED, Anderson CV, Blatter DD. Temporal lobe morphology in normal aging and traumatic brain injury. AJNR Am J Neuroradiol. 2002; 23: 255-66.
42. Tate DF, Bigler ED. Fornix and hippocampal atrophy in traumatic brain injury. Learn Mem. 2000; 7: 442-6.
43. Weisend MP, Feeney DM. The relationship between traumatic brain injury-induced changes in brain temperature and behavioral and anatomical outcome. J Neurosurg. 1994; 80: 120-32. https://doi.org/10.3171/jns.1994.80.1.0120.
44. Cortez SC, McIntosh TK, Noble LJ. Experimental fluid percussion brain injury: vascular disruption and neuronal and glial alterations. Brain Res. 1989; 482: 271-82.
45. Lowenstein DH, Thomas MJ, Smith DH, McIntosh TK. Selective vulnerability of dentate hilar neurons following traumatic brain injury: a potential mechanistic link between head trauma and disorders of the hippocampus. J Neurosci. 1992; 12: 4846-53.
46. Hicks RR, Smith DH, Lowenstein DH, Saint Marie R, McIntosh TK. Mild experimental brain injury in the rat induces cognitive deficits associated with regional neuronal loss in the hippocampus. J Neurotrauma. 1993; 10: 405-14. https://doi.org/10.1089/neu.1993.10.405.
47. Toth K, Freund TF, Miles R. Disinhibition of rat hippocampal pyramidal cells by GABAergic afferents from the septum. J Physiol. 1997; 500: 463-74.
48. Golarai G, Greenwood AC, Feeney DM, Connor JA. Physiological and structural evidence for hippocampal involvement in persistent seizure susceptibility after traumatic brain injury. J Neurosci. 2001; 21: 8523-37.
49. Ansari MA, Roberts KN, Scheff SW. Oxidative stress and modification of synaptic proteins in hippocampus after traumatic brain injury. Free Radic Biol Med. 2008; 45: 443-52. https://doi.org/10.1016/j.freeradbiomed.2008.04.038.
50. Goforth PB, Ren J, Schwartz BS, Satin LS. Excitatory synaptic transmission and network activity are depressed following mechanical injury in cortical neurons. J Neurophysiol. 2011; 105: 2350-63. https://doi.org/10.1152/jn.00467.2010.
51. Caudle KL, Lu XC, Mountney A, Shear DA, Tortella FC. Neuroprotection and anti-seizure effects of levetiracetam in a rat model of penetrating ballistic-like brain injury. Restor Neurol Neurosci. 2016; 34: 257-70. https://doi.org/10.3233/RNN-150580.
52. Ge YX, Tian XZ, Lin YY, Liu XY. Chronic treatment with levetiracetam reverses deficits in hippocampal LTP in vivo in experimental temporal lobe epilepsy rats. Neurosci Lett. 2016; 628: 194-200. https://doi.org/10.1016/j.neulet.2016.06.043.
53. West PJ, Saunders GW, Remigio GJ, Wilcox KS, White HS. Antiseizure drugs differentially modulate theta-burst induced long-term potentiation in C57BL/6 mice. Epilepsia. 2014; 55: 214-23. https://doi.org/10.1111/epi.12524.
54. Matsushita Y, Shima K, Nawashiro H, Wada K, Tsuzuki N, Miyazawa T. Real time monitoring of glutamate following fluid percussion brain injury with hypoxia in the rat. Acta Neurochir Suppl. 2000; 76: 207-12.
55. Chen YH, Huang EY, Kuo TT, Ma HI, Hoffer BJ, Tsui PF, Tsai JJ, Chou YC, Chiang YH. Dopamine release impairment in striatum after different levels of cerebral cortical fluid percussion injury. Cell Transplant. 2014. https://doi.org/10.3727/096368914X683584.
56. Eakin K, Li Y, Chiang YH, Hoffer BJ, Rosenheim H, Greig NH, Miller JP. Exendin-4 ameliorates traumatic brain injury-induced cognitive impairment in rats. PLoS One. 2013; 8: e82016. https://doi.org/10.1371/journal.pone.0082016.
57. Shojo H, Kibayashi K. Changes in localization of synaptophysin following fluid percussion injury in the rat brain. Brain Res. 2006; 1078: 198-211. https://doi.org/10.1016/j.brainres.2006.01.063.
58. Hoffman AF, Oz M, Yang R, Lichtman AH, Lupica CR. Opposing actions of chronic Delta9-tetrahydrocannabinol and cannabinoid antagonists on hippocampal long-term potentiation. Learn Mem. 2007; 14: 63-74. https://doi.org/10.1101/lm.439007.
59. Chen YH, Kuo TT, Chu MT, Ma HI, Chiang YH, Huang EY. Postnatal systemic inflammation exacerbates impairment of hippocampal synaptic plasticity in an animal seizure model. Neuroimmunomodulation. 2013; 20: 223-32. https://doi.org/10.1159/000348440.
60. Chen YH, Harvey BK, Hoffman AF, Wang Y, Chiang YH, Lupica CR. MPTP-induced deficits in striatal synaptic plasticity are prevented by glial cell line-derived neurotrophic factor expressed via an adeno-associated viral vector. FASEB J. 2008; 22: 261-75. https://doi.org/10.1096/fj.07-8797com.