
INTRODUCTION
Pulmonary hypertension (PH) commonly occurs in populations that live at high altitudes and in individuals who experience chronic hypoxia due to pulmonary conditions. In people with chronic obstructive pulmonary disease, PH can cause the right side of the heart to fail, resulting in fatality [1–3]. The walls of pulmonary blood vessels can undergo cellular and histological alterations due to sustained hypoxia. The remodeling of pulmonary blood vessels is characterized by increases in pulmonary vessel wall thickness and small artery muscularization. In vivo studies have indicated that hypoxia increases the wall thickness of pulmonary blood vessels by promoting pulmonary artery smooth muscle cell (PASMC) proliferation [4, 5]. Additionally, pulmonary vascular cells have been reported to proliferate in response to hypoxia in vitro [6–8].
Hypoxia activates phosphatidylinositol 3-kinase (PI3K) signal transduction, which leads to the phosphorylation of the serine/threonine kinase Akt. While the induction of PI3K and Akt signaling is important for cells to grow, proliferate, migrate and survive [9], this pathway can also stimulate cellular proliferation in response to hypoxia. Indeed, Akt activation has been observed during the remodeling of blood vessels and the proliferation of PASMCs following vascular injury and hypoxia [10], and has been reported to stabilize hypoxia-inducible factor-1 alpha (HIF-1α) [11]. Notably, HIF-1 and HIF-2 are important transcriptional regulators in response to hypoxia. The transcription factor HIF-1 is a heterodimer: α-subunit (HIF-1α) expression depends on oxygen levels, while β-subunit (HIF-1β/ARNT) expression is constitutive [12]. During normoxia, the degradation of HIF-1α is initiated by the hydroxylation of conserved proline residues [13]. However, during hypoxia, this process is inhibited, such that HIF-1α accumulates, dimerizes with HIF-1β and induces hypoxia-specific gene expression [14]. Recently, the development of pulmonary arterial hypertension was linked to HIF signaling [5, 6, 8, 15]. The hypoxia-induced proliferation of smooth muscle cells is inhibited by HIF-1α knockdown [13], suggesting that HIF-1α contributes to the remodeling of pulmonary blood vessels and the elevation of right ventricular systolic pressure (RVSP) when PASMCs are exposed to chronic hypoxia.
The nucleotide-binding oligomerization domain protein 2 (NOD2) gene has been reported to determine whether individuals will be vulnerable to inflammatory bowel diseases and Crohn’s disease [16, 17]. This gene encodes an intracellular protein that resembles the Toll-like receptors (TLRs) in its leucine-rich repeats. Through its binding to muramyl dipeptide, NOD2 functions intracellularly as a pathogen recognition receptor (PRR) that recognizes the peptidoglycans of gram-positive and -negative bacteria [18]. NOD2 is expressed in myeloid cells such as neutrophils, macrophages and dendritic cells, and in small intestinal Paneth cells [19, 20]. However, NOD2 has been reported to be expressed or to perform different activities in cells such as vascular endothelial cells, gingival and pulp fibroblasts, adipocytes and vascular smooth muscle cells (VSMCs) [21–26].
The function of NOD2 in systemic arterial VSMCs has been published, but its function in VSMCs of the pulmonary circulatory system (including the remodeling of pulmonary blood vessels) has been underexplored. Previously, we demonstrated that arterial VSMCs expressed functional NOD2, but were stimulated to proliferate, migrate and form neointima in a NOD2-deficient animal model [26]. Here, we evaluated the effects of NOD2 on HIF-1α and Akt signaling, pulmonary vascular remodeling and PASMC proliferation in response to hypoxia.
RESULTS
NOD2 deficiency increases RVSP and induces right ventricular hypertrophy following chronic hypoxia
We first investigated the involvement of NOD2 in the pulmonary reaction to hypoxic conditions by analyzing the mRNA expression of Nod2 within mouse lungs. Nod2 expression has been reported in many different cell types, including systemic arterial VSMCs [26]. VSMCs are also very important in the pulmonary circulatory system, and perform a number of tasks that are critical for pulmonary function. However, the contribution of NOD2 to lung disease has yet to be reported. We compared the mRNA levels of Nod2 in the lungs of wild-type mice with those in bone marrow-derived macrophages (BMDMs) stimulated with LPS and IFN-gamma to induce Nod2 expression (a positive control) (Supplementary Figure 1). Notably, the basal level of Nod2 expression was 12-fold higher in mouse lungs than in BMDMs.
Hypoxia is a well-known stimulus of PH, and can occur in individuals with extensive damage of the lung parenchyma due to chronic lung conditions [27]. Hence, we investigated whether hypoxia stimulates the expression of Nod2. We extracted total RNA from NOD2+/+ mouse lungs after exposing them to normoxic or hypoxic conditions for two weeks. As shown in Figure 1A and 1B, the mRNA levels of Nod2 and Nod1 in the lungs were greater in mice subjected to hypoxia than in those exposed to normoxia. However, there were no differences between the two groups in Tlr2, Tlr4, Hif-1α or Hif-1β mRNA levels after two weeks (Figure 1C–1F).
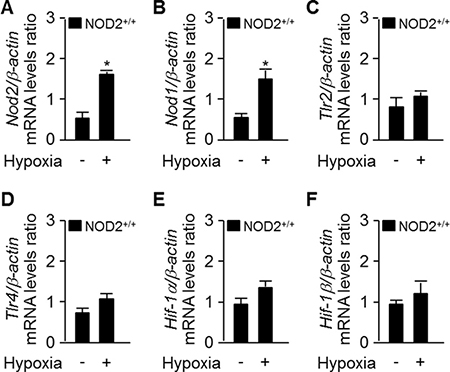
Figure 1: Nod2 expression is enhanced in the mouse lung after chronic hypoxia. -Total RNA was extracted from NOD2+/+ lungs after two weeks of exposure to normoxic (21% O2) or normobaric hypoxic (10% O2) conditions. Quantitative real-time RT-PCR was performed to compare the mRNA levels of Nod2 (A), Nod1 (B), Tlr2 (C), Tlr4 (D), Hif-1α (E) and Hif-1β (F) between the groups. *P < 0.05, upregulation of mRNA levels in the hypoxia vs. normoxia group. For all real-time PCR analyses, mouse β-actin was used as a control for normalization. The expression of each mRNA was normalized to that of β-actin. Values are presented as means ± SDs, n = 3.
Next, we subjected NOD2−/− and NOD2+/+ mice to normoxic or normobaric hypoxic conditions for two weeks. We then assessed whether the pulmonary arteries were becoming hypertensive by determining the RVSP and Fulton’s index (the ratio of the right ventricular mass to the sum of the left ventricular and septal masses), a measure of hypertrophy in the right ventricle due to elevated right ventricular pressure and afterload. Following hypoxic treatment, the RVSP was significantly higher in NOD2−/− mice (30.13 ± 0.9 mmHg) than in NOD2+/+ mice (23.6 ± 1.3 mmHg; P < 0.05) and normoxic mice (Figure 2A). Additionally, Fulton’s index values were higher in NOD2−/− mice than in NOD2+/+ mice (0.37 ± 0.002 vs. 0.29 ± 0.003, P < 0.05) after normobaric hypoxic treatment (Figure 2B). However, the total body weights and whole heart weights did not differ between NOD2−/− (21.59 ± 0.4 g and 111.53 ± 3.4 mg, respectively) and NOD2+/+ (20.27 ± 0.75 g and 122.12 ± 5.6 mg, respectively) mice after exposure to normobaric hypoxic conditions (Figure 2C and 2D). Our results indicate that increased NOD2 levels improve the reaction of pulmonary blood vessels to hypoxic conditions.
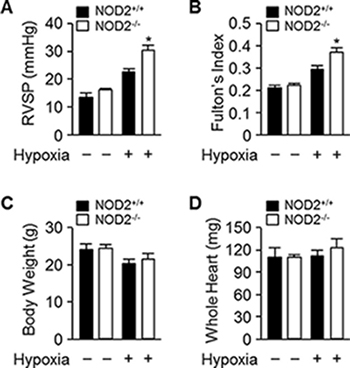
Figure 2: Exaggerated elevation of RVSP and Fulton’s index values in NOD2−/− mice after chronic hypoxia. (A) RVSP in NOD2+/+ (black bars) and NOD2−/− (white bars) mice after two-week exposure to hypoxic (n = 18 per group) or normoxic (n = 10 per group) conditions. (B) Fulton’s index values (ratio of right ventricular weight to left ventricular plus septal weight) of NOD2+/+ (black bars) and NOD2−/− (white bars) mice after two-week exposure to hypoxic (n = 18 per group) or normoxic (n = 15 per group) conditions. C and D, The body weights (C) and whole heart weights (D) of NOD2+/+ (black bars) and NOD2−/− (white bars) mice after exposure to hypoxic (n = 18 per group) or normoxic (n = 15 per group) conditions for two weeks. Data are expressed as means ± SDs; *P < 0.05 for hypoxic NOD2−/− mice vs. hypoxic NOD2+/+ mice.
Absence of NOD2 promotes the remodeling of pulmonary blood vessels and the hypertrophy of PASMCs following chronic hypoxia.
We then performed histology to investigate the effects of NOD2 expression on the remodeling of pulmonary blood vessels during PH caused by hypoxia. Staining with hematoxylin and eosin demonstrated that the blood vessels were remodeled to a greater extent in NOD2−/− mice than in NOD2+/+ mice after hypoxic treatment (Figure 3A). Moreover, under hypoxic conditions, the thickness of the pulmonary arteriole walls increased further in NOD2−/− mice (64.19 ± 6.8%) than in NOD2+/+ mice (38.85 ± 3.6%; P < 0.05) or normoxic controls (Figure 3B).
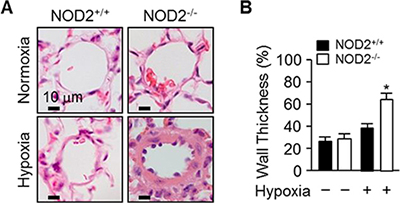
Figure 3: NOD2 deficiency enhances pulmonary vascular remodeling after chronic hypoxia. (A) Representative 5-μm hematoxylin and eosin-stained sections from NOD2+/+ (left) and NOD2−/− (right) mice, generated after two weeks of normoxic (top) or hypoxic (bottom) treatment. (B) Quantification of pulmonary arteriole wall thickness within the lungs of NOD2+/+ (black bars) and NOD2−/− (white bars) mice after exposure to normoxic (n = 7 per group) or hypoxic (n = 10 per group) conditions for two weeks (values expressed as percentages). Ten vessels were analyzed per mouse. Data are expressed as means ± SDs; *P < 0.05 for hypoxic NOD2−/− mice vs. hypoxic NOD2+/+ mice.
To determine whether the enhanced pulmonary vascular remodeling after normobaric hypoxia involved PASMC hypertrophy, we analyzed the prevalence of alpha-smooth muscle actin (α-SMA)-positive cells in the lungs of NOD2−/− and NOD2+/+ mice. In both NOD2−/− and NOD2+/+ mice, little collagen was deposited in the distal blood vessels that had undergone remodeling (Trichrome staining; data not shown). On the other hand, α-SMA immunostaining indicated that the blood vessels of NOD2−/− mice were profoundly remodeled in response to hypoxic conditions; the distal pulmonary arterioles were neomuscularized, exhibiting neointimal enlargement and α-SMA-positivity (Figure 4A). Conversely, the vessels of NOD2+/+ mice were remodeled to a significantly lesser degree and harbored fewer α-SMA-positive cells. Morphometric analysis revealed significant PASMC hypertrophy after hypoxia in NOD2−/− mice, corresponding to a significantly greater area per cell (1540 ± 98 pixels) than their NOD2+/+ counterparts (950 ± 158 pixels) (Figure 4B).
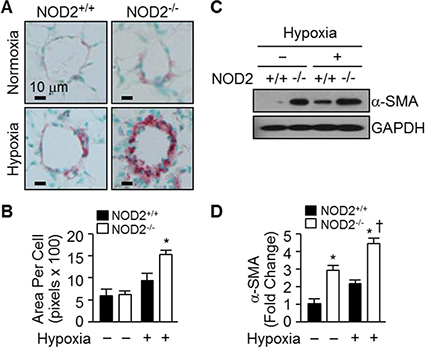
Figure 4: NOD2−/− mice exhibit enhanced hypertrophy of PASMCs after hypoxia. (A) α-SMA immunostaining of the lungs of NOD2+/+ (left) and NOD2−/− (right) mice after exposure to normoxic (top) or hypoxic (bottom) conditions for two weeks. (B) Quantification of PASMC size in NOD2+/+ (black bars) and NOD2−/− (white bars) mice. Ten vessels per mouse were analyzed after exposure to normoxic (n = 5 per group) or hypoxic (n = 8 per group) conditions for two weeks. Data are expressed as means ± SDs; *P < 0.05 for hypoxic NOD2−/− mice vs. hypoxic NOD2+/+ mice. (C) Western blot analysis of α-SMA levels in the lungs of NOD2+/+ and NOD2−/− mice after two weeks of exposure to hypoxic or normoxic conditions. GAPDH was used as control for normalization. The experiment was conducted three times, and the images depict representative results. (D) Normalized α-SMA levels represented graphically. *P < 0.05 for hypoxic mice vs. normoxic controls; † P < 0.05 for hypoxic NOD2−/− mice vs. hypoxic NOD2+/+ mice.
Subsequently, we performed Western blotting to quantify the amount of α-SMA protein in the lungs of NOD2−/− and NOD2+/+ mice after exposing them to hypoxic and normoxic conditions (Figure 4C). Interestingly, the baseline levels of α-SMA protein were slightly higher in NOD2−/− mice than in NOD2+/+ mice. After hypoxia, however, α-SMA protein expression increased an additional 4.5-fold in NOD2−/− lungs, significantly greater than the 2.2-fold upregulation observed in NOD2+/+ lungs (Figure 4D). Thus, NOD2 may confer protective effects with respect to pulmonary vascular remodeling and PASMC hypertrophy after hypoxia.
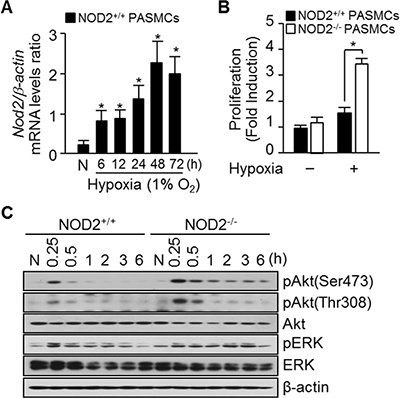
Figure 5: Absence of NOD2 during hypoxia enhances PASMC proliferation. (A) Total RNA was extracted from NOD2+/+ PASMCs exposed to normoxic (N) or hypoxic conditions for the indicated lengths of time. Nod2 mRNA levels were analyzed by quantitative real-time RT-PCR, and mouse β-actin was used as a control for normalization. Values are presented as means ± SDs, n = 3. (B) NOD2+/+ and NOD2−/− PASMCs were plated and incubated for 72 hours under normoxic (21% O2) or hypoxic (1% O2) conditions. Proliferation was then assessed with a BrdU Cell Proliferation Assay Kit. *P < 0.05, increased cell proliferation of NOD2−/− vs. NOD2+/+ PASMCs in response to hypoxia. Values are presented as means ± SDs, n = 12. (C) Total protein was extracted from NOD2+/+ and NOD2−/− PASMCs exposed to normoxic (N) or hypoxic conditions for the indicated lengths of time. The levels of Akt, phosphorylated Akt (Ser473 and Thr308), total ERK and pERK were then assessed by Western blot; β-actin was used as a loading control. Experiments were performed independently at least three times.
Enhanced PASMC proliferation contributes to the hypoxic vascular remodeling of the NOD2−/− strain
We next investigated the function of NOD2 in vitro by subjecting PASMCs to hypoxic conditions and analyzing Nod2 mRNA expression. While Nod2 mRNA levels began to increase in PASMCs 6 hours after the cells were exposed to hypoxia, this increase was much more striking after 48 hours (Figure 5A).
To elucidate the potential mechanisms whereby NOD2 influences pulmonary vascular remodeling, we investigated the proliferation, migration and contractility of NOD2−/− PASMCs exposed to hypoxia in vitro. The proliferation of NOD2−/− PASMCs was 2.23 ± 0.42-fold greater than that of NOD2+/+ PASMCs under hypoxic conditions (Figure 5B). We subsequently used gelatin-coated 24-Transwell chambers to assess the effect of NOD2 on cellular migration. Notably, the migration of NOD2−/− and NOD2+/+ PASMCs did not differ significantly (132.97 ± 4.2 cells/mm2 and 92.64 ± 5.7 cells/mm2, respectively) in response to hypoxia (Supplementary Figure 2A and 2B). Next, we investigated cell contractility using a three-dimensional collagen matrix assay. Hypoxic NOD2−/− and NOD2+/+ PASMCs exhibited similar levels of gel contraction 4 hours after matrix release (64 ± 3.0% and 56 ± 3.3% of original gel size, respectively) (Supplementary Figure 2C and 2D).
To determine the molecular pathways that induced the growth of NOD2-deficient PASMCs, we investigated the effects of NOD2 deficiency on PI3K and Akt signaling under hypoxic conditions. For these experiments, total protein was harvested from NOD2+/+ and NOD2−/− PASMCs at 0.25, 0.5, 1, 2, 3 and 6 hours after hypoxic treatment. Assessment of the mitogen-activated protein kinase pathway revealed that phosphorylated extracellular signal-regulated kinase (pERK) levels in NOD2−/− PASMCs were not altered compared with NOD2+/+ PASMCs after hypoxia. Also, p-p38 and stress-activated protein kinase/c-Jun NH2-terminal kinase (SAPK/JNK) were not detected in response to hypoxia (data not shown). In contrast, Akt phosphorylation (i.e., activation) was enhanced in NOD2−/− PASMCs exposed to hypoxic conditions compare with NOD2+/+ PASMCs (Figure 5C).
HIF-1α is another important mediator of arterial remodeling and PH in response to chronic hypoxia [15]. To investigate the effects of NOD2 deficiency on HIF-1α expression, we harvested total RNA and protein from NOD2+/+ and NOD2−/− PASMCs exposed to hypoxia for 6, 12, 24, 48 and 72 hours. HIF-1α protein was maintained until 24 hours post-exposure and then it was decreased in NOD2+/+ PASMCs. On the other hand, an increase in the expression of this protein was observed in NOD2−/− PASMCs by 12 hours after hypoxia exposure, and a more striking increase was evident after 48 hours (Figure 6A). Furthermore, in NOD2−/− PASMCs, we observed reduced protein expression of hydroxyl-HIF-1α (Pro564) and prolyl hydroxylase domain protein 2 (PHD2), which facilitates the attachment of HIF-1α to the von Hippel-Lindau (VHL) E3 ubiquitin ligase and its subsequent ubiquitin-dependent degradation [13, 28]. Overall, however, there were no changes in HIF-1β and HIF-2α protein expression. Interestingly, increased VHL protein expression was detected in NOD2−/− PASMCs. Lastly, we observed a time-dependent increase in Hif-1α, Hif-1β and Hif-2α mRNA levels in both NOD2+/+ and NOD2−/− PASMCs upon exposure to hypoxic conditions; however, the expression of these factors did not differ between NOD2+/+ and NOD2−/− PASMCs after hypoxia exposure (Figure 6B–6D).
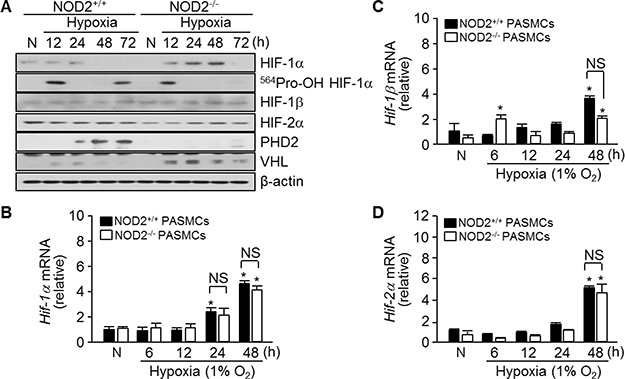
Figure 6: Absence of NOD2 enhances the stability of HIF-1α protein in PASMCs exposed to hypoxic conditions. (A) Total protein was extracted from NOD2+/+ and NOD2−/− PASMCs exposed to normoxic (N) or hypoxic conditions for the indicated lengths of time. The protein levels of HIF-1α, hydroxylated HIF-1α (Pro564), HIF-1β, HIF-2α, PHD2 and VHL were then assessed by Western blot; β-actin was used as a loading control. Experiments were performed at least three independent times. (B–D), Total RNA was extracted from NOD2+/+ and NOD2−/− PASMCs exposed to normoxic (N) or hypoxic conditions for the indicated lengths of time. The mRNA levels of Hif-1α (B), Hif-1β (C) and Hif-2α (D) were then analyzed by quantitative real-time RT-PCR; mouse β-actin was used as a control for normalization. *P < 0.05, upregulation after hypoxia vs. after normoxia (N). NS, not significant. Values are presented as means ± SDs, n = 3.
Together, our data indicate that greater PASMC proliferation, not migration or contractility, is responsible for the remodeling of pulmonary blood vessels in hypoxia-exposed NOD2−/− mice. Moreover, prolonged Akt phosphorylation and HIF-1α expression are important contributors to the enhanced proliferation of NOD2−/− PASMCs during hypoxia.
DISCUSSION
TLRs, which comprise a class of transmembrane PRR, were suggested to connect inflammation with PH, thus contributing to the emergence of PH, while being dispensable for lesion formation [29–31]. TLR signaling, especially involving TLR2 and 4, has been described in human pulmonary vascular cells and patients with idiopathic pulmonary arterial hypertension [29–32]. Moreover, the TLR Pro631His variant correlates robustly with PH incidence in patients with systemic sclerosis [30]. Conversely, other studies have suggested that NOD2 and TLR3 have protective functions in vascular diseases, including atherosclerosis [26, 33]; indeed, we previously demonstrated such a function of NOD2 in arterial VSMCs. Mouse arterial VSMC proliferation and migration were associated negatively with the induction of NOD2 expression, and NOD2-deficient arterial VSMCs proliferated, migrated and formed neointima following vascular damage [26]. Using a perivascular collar model, Cole et al. demonstrated that activated TLR3 suppressed the development of the neointima following carotid damage. In contrast, atherosclerotic lesions in the aortic root formed earlier in TLR3/apolipoprotein E double-knockout mice (ApoE−/−/TLR3−/−) than in control mice (ApoE+/+/TLR3+/+). Considered along with the models of vascular damage, these results indicate that PRRs like NOD2 and TLR3 may not merely be innate immune receptors, but also have other functions. While PRRs are necessary for innate immunity, they may perform markedly different activities in response to noninfectious stimuli. As an example, PRRs such as TLR3 and NOD2 protect the walls of the blood vessels, while others such as TLR2 and TLR4 can damage them. We hypothesized that NOD2 may also have a protective function during hypoxia, preventing an exaggerated increase in pulmonary vascular remodeling and RVSP.
Consistent with this hypothesis, we observed enhanced Nod2 expression in the lungs following prolonged exposure of mice to hypoxic conditions (Figure 1A). Interestingly, the RVSP and Fulton’s index values were significantly greater in NOD2-deficient mice than in wild-type mice after chronic hypoxic treatment (Figure 2A and 2B). We subsequently assessed pulmonary arterial remodeling after two weeks of normobaric hypoxic (10% O2) treatment to determine whether the observed changes in the RVSP and Fulton’s index values were associated with PH. Following two weeks of chronic hypoxia, the medial layer of small pulmonary arteries exhibited greater thickening in NOD2−/− mice than in NOD2+/+ mice (Figure 3A). In addition, the PASMCs of NOD2-deficient mice became significantly hypertrophic, contributing to the striking remodeling of the blood vessels (Figure 4A and 4B). Interestingly, while baseline expression of α-SMA was greater in NOD2−/− mice than in NOD2+/+ mice, the RVSP and Fulton’s index values did not differ significantly between ten- to twelve-week-old NOD2+/+ and NOD2−/− mice exposed to normoxic conditions. Thus, in the absence of vascular remodeling (Figure 3), the source of α-SMA does not appear to be the VSMCs of small pulmonary vessels. In older mice, however, enhanced α-SMA expression might alter the RVSP or Fulton’s index of NOD2−/− mice under normoxic conditions.
In animals, hypoxia stimulates the proliferation of PASMCs, thus increasing the wall thickness of the pulmonary blood vessels [4, 5]. The pathologic process whereby pulmonary blood vessels are remodeled intensifies as PASMCs proliferate increasingly. Here, we used mouse primary PASMCs to study the involvement of NOD2 in PASMC proliferation in response to hypoxia. In general, hypoxia promoted Nod2 expression in PASMCs; however, NOD2−/− PASMCs proliferated at a higher level than NOD2+/+ PASMCs under hypoxic conditions (Figure 5A and 5B). While PASMC migration and contractility are also involved in the pathogenesis of pulmonary vascular remodeling [1, 34], the migratory and contractility capacities of NOD2−/− and NOD2+/+ PASMCs did not differ significantly after hypoxic treatment (Supplementary Figure 2). These results suggest that NOD2 protects against pulmonary vascular remodeling under hypoxic conditions by preventing PASMC proliferation.
Generally, hypoxia initiates PI3K signaling, which leads to Akt phosphorylation [9, 10]. These pathways, along with the HIF signaling pathway, are thought to contribute to the proliferation of PASMCs under hypoxic conditions. Muramyl dipeptide, a ligand recognized by NOD2, independently induces Akt phosphorylation in different cell types [35, 36], but also inhibits insulin-stimulated Akt phosphorylation in myotubes [37]. Moreover, we previously observed sustained phosphorylation of Akt (Ser473 and Thr308) in the presence of platelet-derived growth factor-BB in NOD2−/− VSMCs [26]. In the present study, we examined the influence of NOD2 on PI3K and Akt signaling to determine whether these pathways were responsible for NOD2−/− PASMC proliferation under hypoxic conditions. Akt phosphorylation increased in NOD2−/− PASMCs after hypoxia exposure, and a similar response was observed in the ERK signaling pathway (Figure 5C). Moreover, HIF-1α protein expression, which correlates with sustained Akt phosphorylation, was significantly higher in NOD2−/− PASMCs than in NOD2+/+ PASMCs under hypoxic conditions (Figure 6A), although there were no differences in the mRNA levels of Hif-family genes between NOD2−/− and NOD2+/+ PASMCs after hypoxic treatment (Figure 6B-6D). HIF-1α is a key protein in the lung response to reduced oxygen availability, and is thought to be involved in PH development [5, 8, 15]. Indeed, HIF-1 is necessary for smooth muscle hypertrophy, and HIF-1α significantly contributes to the remodeling of pulmonary blood vessels under chronic hypoxic conditions [8]. The hydroxylation of HIF-1α (at proline 564) and the expression of PHD2 protein were lower in NOD2−/− PASMCs than in NOD2+/+ PASMCs after hypoxia exposure (Figure 6A). Interestingly, the levels of VHL, which participates in the ubiquitin-dependent degradation of HIF-1α, were greater in NOD2−/− PASMCs than in NOD2+/+ PASMCs after hypoxia exposure (Figure 6A).
In conclusion, our findings indicate that NOD2 is important for the response of PASMCs to hypoxia. While we found that Akt activation leads to HIF-1α stabilization and PASMC proliferation in NOD2−/− mice, future investigations will be required to characterize the molecular pathways governing the relationship between NOD2 and the Akt signaling pathway. The findings presented here have expanded our comprehension of the involvement of PRRs in hypoxia-induced PH, and demonstrate that NOD2 may be a promising target for therapies to prevent the remodeling of blood vessels in pulmonary vascular conditions.
MATERIALS AND METHODS
Animals
NOD2−/− mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA) on a pure C57BL/6 genetic background, and these mice were bred within our animal facility at Harvard Medical School. The Standing Committee on Animal Care at Harvard Medical School approved all the animal experimentation protocols of this study under the guidelines of our approved Institutional Animal Care and Use Committee protocol.
Hypoxic exposure and hemodynamic measurements
Eight- to ten-week-old NOD2−/− and NOD2+/+ littermates were exposed to normobaric hypoxia (10% O2, OxyCycler chamber, Biospherix Ltd, Redfield, NY, USA) or normoxia (21% O2) for two weeks. After exposure, mice were anesthetized with sodium pentobarbital (60 mg/kg), and hemodynamic measurements were performed. The hearts were excised, and the ventricles were dissected and weighed. Right ventricular hypertrophy was calculated as the right ventricular weight normalized to the total body weight.
Histological analysis and morphometry
Lungs were inflated, harvested, fixed in methyl Carnoy’s solution and embedded in paraffin. Sections were stained with hematoxylin and eosin and immunostained for α-SMA (1:50, Sigma-Aldrich Co. LLC, St Louis, MO, USA). Remodeling was quantified as described previously [38]. The percent wall thickness was calculated as follows: wall thickness (%) = (areaext − areaint) ÷ areaext × 100, where areaext represents the external diameter and areaint represents the internal diameter of each vessel. PASMC hypertrophy was calculated as the vessel wall area divided by the number of nuclei per vessel, and reported as the area per cell.
Cell culture
Primary PASMCs were isolated from adult (eight- to ten-week-old) NOD2−/− and NOD2+/+ mice as described [39], with modification. Hypoxia experiments were performed in a New Brunswick™ Galaxy® 48 R (1% O2, Eppendorf AG, Hamburg, Germany). The NOD2−/− and NOD2+/+ PASMCs were grown in Dulbecco’s Modified Eagle’s Medium (Life Technologies, Grand Island, NY, USA) supplemented with 20% fetal bovine serum, penicillin (100 U/mL) and streptomycin (100 μg/mL). In all experiments, cells between passages three and nine were used. All cells were incubated at 37°C in a humidified atmosphere of 5% CO2 and 95% air.
Western blot analysis
Protein extracts from lungs exposed to hypoxia or normoxia were analyzed by Western blot analysis. Cell extracts from 100-mm dishes were harvested with radioimmunoprecipitation assay buffer (Tris/Cl, pH 7.6; 100 mmol/L ethylenediaminetetraacetic acid; 5 mmol/L NaCl; 50 mmol/L β-glycerophosphate; 50 mmol/L NaF; 50 mmol/L Na3VO4; 0.1 mmol/L NP-40; and 0.5% sodium deoxycholate) with 1× Complete™ Protease Inhibitor Cocktail (Roche Applied Science, Mannheim, Germany). Tissue extracts were homogenized with radioimmunoprecipitation assay buffer. The protein concentrations of the cell lysates were determined with a Pierce BCA protein assay kit (Thermo Fisher Scientific, Inc., Waltham, MA) and were resolved on 12% sodium dodecyl sulfate-polyacrylamide gels. Proteins were transferred onto pure polyvinylidene difluoride membranes by means of a wet transfer system (GE Healthcare Bio-Sciences, Pittsburgh, PA, USA). Membranes were blocked for 2 hours at room temperature with a 5% nonfat milk solution in Tris-buffered saline Tween (TBST) buffer (20 mM Tris-HCl, pH 7.4; 500 mM NaCl; 0.1% Tween20). The blots were then incubated with various antibodies, including monoclonal α-SMA (Sigma-Aldrich Co. LLC), polyclonal anti-HIF-1α, anti-HIF-2α, anti-HIF-1β (Bethyl Laboratories, Montgomery, TX, USA), anti-phospho-Akt (Ser473, Thr308), total Akt (Cell Signaling Technology, Inc., Danvers, MA, USA), anti-PHD2, anti-VHL (Santa Cruz Biotechnology, Inc., Dallas, TX, USA) and anti-hydroxy-HIF-1α (Cell Signaling Technology), in TBST overnight at room temperature. Equal loading was confirmed with an anti-GAPDH (glyceraldehyde 3-phosphate dehydrogenase) antibody (Santa Cruz Biotechnology, Inc.) and anti-β-actin (Sigma-Aldrich Co. LLC). The blots were then washed three times in TBST and incubated with an anti-rabbit or anti-mouse secondary antibody in TBST for 1 hour at room temperature. Finally, immunoblots were detected with the SuperSignal® West Pico Chemiluminescent Substrate (Thermo Fisher Scientific, Inc.) and visualized after exposure to X-ray film.
Proliferation assay
The cell proliferation rate at 72 hours was measured by BrdU incorporation with the Cell Proliferation ELISA, BrdU (colorimetric) kit (Roche Diagnostics, Basel, Switzerland) according to the manufacturer’s instructions.
Quantitative real-time reverse-transcription polymerase chain reaction (RT-PCR)
Total RNA was isolated with TRIzol reagent (Thermo Fisher Scientific, Inc.), and reverse transcription was performed with a SuperScript™ III First-Strand Synthesis System (Thermo Fisher Scientific, Inc.). Quantitative real-time RT-PCR was conducted with iQ SYBR Green Supermix (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The primer sequences were as follows: mouse Nod1 forward primer 5ʹ- GAAGGCACCCCATTGGGTT -3ʹ and reverse primer 5ʹ-
AATCTCTGCATCTTCGGCTGA -3ʹ; Nod2 forward primer 5ʹ-
CAGGTCTCCGAGAGGGTACTG -3ʹ and reverse primer 5ʹ-
GCTACGGATGAGCCAAATGAAG -3ʹ; Tlr2 forward primer 5ʹ-
GCAAACGCTGTTCTGCTCAG -3ʹ and reverse primer 5ʹ-
AGGCGTCTCCCTCTATTGTATT -3ʹ; Tlr4 forward primer 5ʹ-
ATGGCATGGCTTACACCACC -3ʹ and reverse primer 5ʹ-
GAGGCCAATTTTGTCTCCACA -3ʹ; Hif-1α forward primer 5ʹ-
TCATCAGTTGCCACTTCCCCAC -3ʹ and reverse primer 5ʹ-
CCGTCATCTGTTAGCACCATCAC -3ʹ; Hif-1β forward primer 5ʹ-
TAGACCATCGTTGTGTGGCT -3ʹ and reverse primer 5ʹ-
CACCTGCTGAAAGCTGTCTC -3ʹ; Hif-2α forward primer 5ʹ-
TAAAGCGGCAGCTGGAGTAT -3ʹ and reverse primer 5ʹ-
ACTGGGAGGCATAGCACTGT -3ʹ.
Amplification of cDNA started with 10 minutes at 95°C, followed by 40 cycles of 15 seconds at 95°C and 1 minute at 60°C.
Statistical analysis
Data are shown as the mean ± standard deviation (SD). For comparisons between two groups, we used Student’s two-tailed unpaired t test. For comparisons between more than two groups, and for multiple comparisons, we used analysis of variance. Statistically significant differences were accepted at P < 0.05.
Abbreviations
α-SMA: alpha-smooth muscle actin; ApoE: apolipoprotein E; BMDMs: bone marrow-derived macrophages; ERK: extracellular signal-regulated kinase; HIF: hypoxia-inducible factor; NOD2: nucleotide-binding oligomerization domain protein 2; PASMCs: pulmonary artery smooth muscle cells; PH: pulmonary hypertension; PHD2: prolyl hydroxylase domain protein 2; PI3K: phosphatidylinositol 3-kinase; PRR: pathogen recognition receptor; RVSP: right ventricular systolic pressure; SAPK/JNK: stress-activated protein kinase/c-Jun NH2-terminal kinase; TLRs: toll-like receptors; VHL: von Hippel-Lindau; VSMCs: vascular smooth muscle cells.
Author contributions
Min-Young Kwon and Narae Hwang performed experiments; Young-Jun Park contributed to the experimental design; Mark A. Perrella and Su Wol Chung contributed to the experimental design and wrote the manuscript.
ACKNOWLEDGMENTS
None.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Education (NRF-2014R1A6A1030318 and 2013008302).
REFERENCES
1. MacNee W. Pathophysiology of corpulmonale in chronic obstructive pulmonary disease. Part one. Am J Respir Crit Care Med. 1994; 150:833–852.
2. Rabinovitch M, Gamble W, Nadas AS, Miettinen OS, Reid L. Rat pulmonary circulation after chronic hypoxia: hemodynamic and structural features. Am J Physiol Heart Circ Physiol. 1979; 236:H818–H827.
3. Rabinovitch M, Konstam MA, Gamble WJ, Papanicolaou N, Aronovitz MJ, Treves S, Reid L. Changes in pulmonary blood flow affect vascular response to chronic hypoxia in rats. Circ Res. 1983; 52:432–441.
4. Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, McWilliams R, Beaty T, Sham JS, Wiener CM, Sylvester JT, Semenza GL. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1 alpha. J Clin Invest. 1999; 103:691–696.
5. Yu L, Quinn DA, Garg HG, Hales CA. Cyclin-dependent kinase inhibitor p27Kip1, but not p21WAF1/Cip1, is required for inhibition of hypoxia-induced pulmonary hypertension and remodeling by heparin in mice. Circ Res. 2005; 97:937–945.
6. Cooper AL, Beasley D. Hypoxia stimulates proliferation and interleukin-1alpha production in human vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 1999; 277:H1326–H1337.
7. Frid MG, Aldashev AA, Dempsey EC, Stenmark KR. Smooth muscle cells isolated from discrete compartments of the mature vascular media exhibit unique phenotypes and distinct growth capabilities. Circ Res. 1997; 81:940–952.
8. Schultz K, Fanburg BL, Beasley D. Hypoxia and hypoxia-inducible facter-1α promote growth facter-induced proliferation of human vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2006; 290:H2528–H2534.
9. Garat CV, Crossno JT Jr, Sullivan TM, Reusch JE, Klemm DJ. Inhibition of phosphatidylinositol 3-kinase/Akt signaling attenuates hypoxia-induced pulmonary artery remodeling and suppresses CREB depletion in arterial smooth muscle cells. J Cardiovasc Pharmacol. 2013; 62:539–548.
10. Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res. 2006; 99:675–691.
11. Zundel W, Schindler C, Haas-Kogan D, Koong A, Kaper F, Chen E, Gottschalk AR, Ryan HE, Johnson RS, Jefferson AB, Stokoe D, Giaccia AJ. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev. 2000; 14:391–396.
12. Semenza GL, Nejfelt MK, Chi SM, Antonarakis SE. Hypoxia-inducible nuclear factors bind to an enhancer element located 3′ to the human erythropoietin gene. Proc Natl Acad Sci USA. 1991; 88:5680–5684.
13. Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG Jr. HIF alpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001; 292:464–468.
14. Huang LE, Gu J, Schau M, Bunn HF. Regulation of hypoxia-inducible factor 1α is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci USA. 1998; 95:7987–7992.
15. Shimoda LA, Semenza GL. HIF and the lung: role of hypoxia-inducible factors in pulmonary development and disease. Am J Respir Crit Care Med. 2011; 183:152–156.
16. Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP. Association of NOD2 LRR variants with susceptibility to Crohn’s disease. Nature. 2001; 411:599–603.
17. Ogura Y, Bonen DK, Inohara N, Nicolae L, Chen FF. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001; 411:603–606.
18. Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A. NOD2 is a general sensor of peptidoglycan through MDP detection. J Biol Chem. 2003; 278:8869–8872.
19. Rosenstiel P, Kuhbacher T, Seegert D, Schreiber S. TNF-α and IFN-γ regulate the expression of the NOD2 in human intestinal epithelial cells. Gastroenterology. 2003; 124:1001–1009.
20. Lala S, Ogura Y, Osborne C, Hor SY, Bromfield A, Davies S, Ogunbiyi O, Nunez G, Keshav S. Crohn’s disease and the NOD2 gene: a role for paneth cells. Gastroenterology. 2003; 125:47–57.
21. Stroh T, Batra A, Glauben R, Bereswill S, Siegmund B. NOD1 and 2: regulation of expression and function in preadipocytes. J Immunol. 2008; 181:3620 –3627.
22. Hirao K, Yumoto H, Nakanishi T, Matsuo T. Roles of TLR2, TLR4, and NOD2 in pulp fibroblasts. J Dent Res. 2009; 88:762–767.
23. Hosokawa I, Hosokawa Y, Ozaki K, Yumoto H, Nakae H, Matsuo T. Proinflammatory effects of MDP on human gingival fibroblasts. J Periodontal Res. 2010; 45:193–199.
24. Davey MP, Martin TM, Planck SR, Lee J, Zamora D, Rosenbaum JT. Human endothelial cells express NOD2/CARD15 and increase IL-6 secretion in response to muramyl dipeptide. Microvasc Res. 2006; 71:103–107.
25. Oh HM, Lee HJ, Seo GS, Choi EY, Kweon SH, Chun CH, Han WC, Lee KM, Lee MS, Choi SC, Jun CD. Induction and localization of NOD2 protein in human endothelial cells. Cell Immunol. 2005; 237:37– 44.
26. Kwon MY, Liu X, Lee SJ, Kang YH, Choi AM, Lee KU, Perrella MA, Chung SW. Nucleotide-binding oligomerization domain protein 2 deficiency enhances neointimal formation in response to vascular injury. Arterioscler Thromb Vasc Biol. 2011; 31:2441–2447.
27. Fishman AP. Hypoxia on the pulmonary circulation: how and where it acts. Circ Res. 1976; 38:221–231.
28. Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001; 292:468–472.
29. George PM, Badiger R, Shao D, Edwards MR, Wort SJ, Paul-Clark MJ, Mitchell JA. Viral Toll Like Receptor activation of pulmonary vascular smooth muscle cells results in endothelin-1 generation; relevance to pathogenesis of pulmonary arterial hypertension. Biochem Biophys Res Commun. 2012; 426:486–491.
30. Broen JC, Bossini-Castillo L, van Bon L, Vonk MC, Knaapen H, Beretta L, Rueda B, Hesselstrand R, Herrick A, Worthington J, Hunzelman N, Denton CP, Fonseca C. A rare polymorphism in the gene for Toll-like receptor 2 is associated with systemic sclerosis phenotype and increases the production of inflammatory mediators. Arthritis Rheum. 2012; 64:264–271.
31. Rupasree Y, Naushad SM, Rajasekhar L, Uma A, Kutala VK. Association of TLR4 (D299G, T399I), TLR9 -1486T > C, TIRAP S180L and TNF-α promoter (-1031, -863, -857) polymorphisms with risk for systemic lupus erythematosus among South Indians. Lupus. 2015; 24:50–57.
32. Hilbert T, Dornbusch K, Baumgarten G, Hoeft A, Frede S, Klaschik S. Pulmonary vascular inflammation: effect of TLR signalling on angiopoietin/TIE regulation. Clin Exp Pharmacol Physiol. 2017; 44:123–131.
33. Cole JE, Navin TJ, Cross AJ, Goddard ME, Alexopoulou L, Mitra AT, Davies AH, Flavell RA, Feldmann M, Monaco C. Unexpected protective role for TLR3 in the arterial wall. Proc Natl Acad Sci U S A. 2011; 108:2372–2377.
34. Shimoda LA, Laurie SS. Vascular remodeling in pulmonary hypertension. J Mol Med (Berl). 2013; 91:297–309.
35. Zhao L, Lee JY, Hwang DH. The phosphatidylinositol 3kinase/Akt pathway negatively regulates Nod2mediated NFkappaB pathway. Biochem Pharmacol. 2008; 75:1515–1525.
36. Tokuhira N, Kitagishi Y, Suzuki M, Minami A, Nakanishi A, Ono Y, Kobayashi K, Matsuda S, Ogura Y. PI3K/AKT/PTEN pathway as a target for Crohn’s disease therapy. Int J Mol Med. 2015; 35:10–6.
37. Tamrakar AK, Schertzer JD, Chiu TT, Foley KP, Bilan PJ, Philpott DJ, Klip A. NOD2 activation induces muscle cell-autonomous innate immune responses and insulin resistance. Endocrinology. 2010; 151:5624–3.
38. Fredenburgh LE, Liang OD, Macias AA, Polte TR, Liu X, Riascos DF, Chung SW, Schissel SL, Ingber DE, Mitsialis SA, Kourembanas S, Perrella MA. Absence of cyclooxygenase-2 exacerbates hypoxia-induced pulmonary hypertension and enhances contractility of vascular smooth muscle cells. Circulation. 2008; 117:2114–22.
39. Fouty BW, Grimison B, Fagan KA, Le Cras TD, Harral JW, Hoedt-Miller M, Sclafani RA, Rodman DM. p27(Kip1) is important in modulating pulmonary artery smooth muscle cell proliferation. Am J Respir Cell Mol Biol. 2001; 25:652–658.