
Cancer is no longer a problem of cancer cells alone since the development and metastasis of cancers clearly involves many aspects of the host. There is growing evidence for a direct mechanistic relationship between the changes induced by inflammation and epigenetic deregulation during tumor development and progression. The proclivity of tumor cells to undergo widespread dissemination to distant sites is the major cause of cancer mortality. Consequently, this process has been extensively studied, but the exact nature of the events that may contribute to, or are essential for, metastasis remain controversial. During malignant progression, it has been proposed that carcinoma cells undergo an epithelial-to-mesenchymal transition (EMT), in which they lose epithelial characteristics and acquire invasive properties and stem cell-like features (Fig. 1) [1]. Multiple signaling pathways including TGFβ, Wnt and Notch working together with transcription factors such as Slug, Snail, Twist, Zeb1 and 2 suppress E-cadherin and induce EMT resulting in loss of cell-cell adhesion and increased tumor progression and migration [2]. Moreover, gene expression patterns in human cancers indicate that de-differentiated cancer cells combine EMT properties with a stem-cell like phenotype to generate migrating cancer stem cells as the basis of metastasis [3]. Indeed, EMT activators such as Twist1, can co-induce EMT and stemness properties thereby linking EMT to cancer stem cells (CSCs) [4].
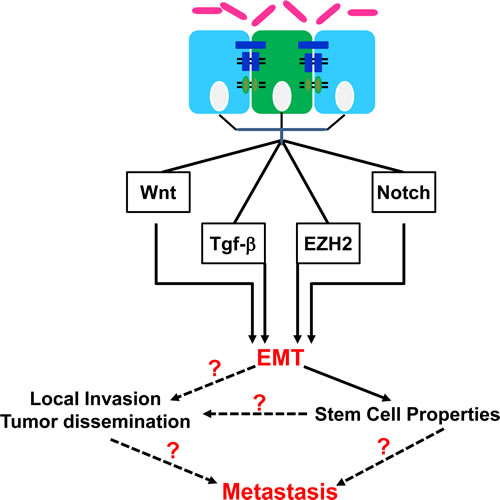
Figure 1: Epithelial-to-mesenchymal transition (EMT) pathways deregulated in cancer and the downstream effects. Citrobacter rodentium (CR) binding to the luminal surface of the large intestine in a susceptible host can influence the epigenetic signaling within the stem (green) and/or progenitor (blue) cells to promote inflammation and EMT. The diagram demonstrates a variety of effector pathways for EMT as well as the downstream consequences related to either local invasion or distant metastasis. Dotted lines and question marks represent the current focus in our laboratory.
Remarkably, several of the EMT-signaling pathways such as TGFβ, Wnt, Notch and NF-κB are also induced by microbial pathogens, therefore suggesting that pathogens may also be considered as EMT inducers [2]. It is noteworthy that bacterial or viral infections, and the resulting chronic inflammation, have been shown to predispose individuals to certain types of cancer. Recent studies also provide evidence of histone modification and chromatin remodeling as key regulators of eukaryotic transcription and therefore excellent targets for pathogenic infection [5]. Epigenetic remodeling including DNA methylation and posttranslational histone modifications, have recently been implicated in the regulation of EMT and tumor cell invasion [6]. Epigenetic dysregulation may contribute to inflammation-driven diseases including cancer, and can lead to the inappropriate silencing of genes necessary to inhibit cancer development. Growing evidence suggests a strong link of enhancer of zest homolog-2 (EZH2), a polycomb repressor complex-2 protein, to oncogenesis and to cancer-specific gene silencing. Overexpression of EZH2 has been found in many cancers and the expression level is correlated with tumor progression and poor prognosis [7].
Surprisingly, much of what we have learned about the process of EMT has come from elegant studies in tumor cell lines [8], while the characterization of the pathways leading to EMT in vivo is less clear. Rhim AD et al. [9] by doing lineage tracing in a pancreatic cancer model, recently demonstrated that cells acquire an invasive and metastatic phenotype before detectable tumor formation. Similarly, a recent study demonstrated expression of EMT markers during the pathogenesis of fistulae in Crohn’s disease [10]. Utilizing an in vivo model of Citrobacter rodentium (CR)-induced pre-neoplastic colonic crypt hyperplasia, we have recently shown that hyperplastic crypts when cultured in vitro, exhibit EMT-like changes that promotes tumorigenesis following a second hit [11]. We have also discovered that Dclk1 and Lgr5 as markers of quiescent and rapidly cycling stem cells [12], are differentially expressed during progression and regression phases of crypt hyperplasia [11]. In addition, the process of EMT was epigenetically regulated as CR-induced upregulation of EZH2 levels at peak crypt hyperplasia was associated with E-cadherin promoter leading to its downregulation which is the hallmark of EMT process. Thus, it is not unprecedented for either the hyperplastic crypts or intestinal adenomas such as those seen in mice mutant for Apc gene [13], to serve as precursors of malignant tumors before detectable local invasion or may be even metastasis. Interestingly, the CR-induced EMT was almost completely attenuated following blockade of the Wnt/β-catenin and Notch cross-talk in vivo [11]. Since these pathways are also implicated in epithelial stem cell/CSCs maintenance and proliferation and are generally thought as the cells of origin for a large proportion of CRCs, our studies provide an excellent platform to link enteric pathogens to the cancers of the large intestine and how they can promote either the onset or propagation of EMT signal that promotes metastasis and frank malignancy. Moreover, utilization of CR infection that models human EPEC and EHEC, to study pathogenesis of EZH2-mediated changes in Wnt signaling is clinically relevant and can lead to exciting data to treat diseases with infectious etiology.
Microorganisms are causally implicated in slightly over 20% of all human cancers. Viruses, bacteria and parasites are among the most important pathogens associated with carcinogenesis. It is therefore imperative to focus on understanding the mechanistic basis of malignant transformation initiated by pathogens, an area that promises exciting prophylactic, diagnostic, and therapeutic applications. In conclusion, even though we still lack novel approaches to overcome therapeutic resistance, a combined regimen of chemotherapy and radiation therapy remains the central strategy to fight cancer. Results of the current study [11] suggest that the acquisition of EMT phenotype triggered by an enteric pathogen could be associated with an early onset metastatic spread in genetically predisposed individuals. Since pathogens associated with either chronic pathologies or those that interfere with innate immunity have been described to induce EMT, targeting EMT and CSC molecular pathways activated by pathogens, may represent a unique therapeutic alternative to conventional anti-neoplastic strategy to mitigate early stage metastasis and/or frank malignancy.
References
1. Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nature Rev Cancer 2009; 9: 265-273.
2. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009; 139: 871-890.
3. Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Opinion: migrating cancer stem cells - an integrated concept of malignant tumour progression. Nat Rev Cancer. 2005; 5: 744-9.
4. Li CW, Xia W, Huo L, Lim SO, Wu Y, Hsu JL, Chao CH, Yamaguchi H, Yang NK, Ding Q, Wang Y, Lai YJ, LaBaff AM, Wu TJ, Lin BR, Yang MH, Hortobagyi GN, Hung MC. Epithelial-mesenchymal transition induced by TNF-α requires NF-kB-mediated transcriptional upregulation of Twist1. Cancer Res. 2012; 72: 1290-300.
5. Hamon MA, Cossart P. Histone modifications and chromatin remodeling during bacterial infections. Cell Host Microbe 2008; 4: 100-109.
6. Wu MZ, Tsai YP, Yang MH, Huang CH, Chang SY, Chang CC, Teng SC, Wu KJ. Interplay between HDAC3 and WDR5 is essential for hypoxia-induced epithelial-mesenchymal transition. Mol Cell. 2011; 43: 811-822.
7. Crea F, Fornaro L, Bocci G, Sun L, Farrar WL, Falcone A, Danesi R. EZH2 inhibition: targeting the crossroad of tumor invasion and angiogenesis. Cancer Metastasis Rev. 2012; 31: 753-761.
8. Hofman P, Vouret-Craviari V. Microbes-induced EMT at the crossroad of inflammation and cancer. Gut Microbes 2012; 3: 176-185.
9. Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, Reichert M, Beatty GL, Rustgi AK, Vonderheide RH, Leach SD, Stanger BZ. EMT and dissemination precede pancreatic tumor formation. Cell 2012; 148: 349-361.
10. Bataille F, Rohrmeier C, Bates R, Weber A, Rieder F, Brenmoehl J, Strauch U, Farkas S, Fürst A, Hofstädter F, Schölmerich J, Herfarth H, Rogler G. Evidence for a role of epithelial mesenchymal transition during pathogenesis of fistulae in Crohn’s disease. Inflamm Bowel Dis. 2008; 14: 1514-1527.
11. Chandrakesan P, Roy B, Jakkula LU, Ahmed I, Ramamoorthy P, Tawfik O, Papineni R, Houchen C, Anant S, Umar S. Utility of a bacterial infection model to study epithelial-mesenchymal transition, mesenchymal-epithelial transition or tumorigenesis. Oncogene 2014; 33: 2639-2654.
12. May R, Sureban SM, Hoang N, Riehl TE, Lightfoot SA, Ramanujam R, Wyche JH, Anant S, Houchen CW. Doublecortin and CaM kinase-like-1 and leucine-rich-repeat-containing G-protein-coupled receptor mark quiescent and cycling intestinal stem cells, respectively. Stem Cells. 2009; 27: 2571-9.
13. Ahmed I, Roy B, Chandrakesan P, Venugopal A, Xia L, Jensen R, Anant S, Umar S. Evidence of functional cross talk between the Notch and NF-kB pathways in non-neoplastic hyperproliferating colonic epithelium. Am J Physiol Gastrointest Liver Physiol. 2013; 304: G356-370.