
INTRODUCTION
Nearly 68,000 adolescents and young adults (AYAs) aged 15 to 39 years were diagnosed with cancer in the US in 2002 [1], and females with breast, ovary, and uterine cancers constituted a large proportion of cases [2]. Since the prognosis for AYA patients with cancer has improved less than that for patients in the non-AYA age group [1, 3], better characterization of the properties of AYA cancers is urgently needed to facilitate understanding of the etiology of their early development and improve diagnosis and therapy.
The clinical and histopathological characteristics of tumors in AYA patients have been revealed by comparison with those of non-AYA patients in European/US populations [2]. Tumors of the breast in AYAs often lack expression of therapeutic targets, such as estrogen receptor (ER), progesterone receptor (PgR), and human epidermal growth factor receptor 2 (HER2) oncoprotein (i.e., they are of the triple-negative subtype), and have a poor prognosis [2, 4]. Malignant and borderline ovarian tumors are rare in adolescents compared with adults; however, they pose serious issues in that age group. Such ovarian tumors encompass a variety of subtypes; the most common are epithelial in origin; however, non-epithelial tumors, such as malignant germ cell and sex cord-stromal tumors, constitute a major fraction [5-8]. Cervical and endometrial carcinomas comprise a large proportion of AYA uterine tumors [9, 10].
Genome-wide mutation profiling of cancer genomes is a powerful method to identify actionable gene aberrations, and can facilitate elucidation of the mutagenic processes underlying the development of a variety of cancers [11-13]; however, few studies have focused on tumors in AYAs, and hence information on their associated gene aberrations is very limited. Approximately 2.5% of cancers in the Japanese population are diagnosed among the AYA age group, and, as in the US [2], breast, ovarian, and uterine cancers are the major types identified [14]. Here, we present gene aberration profiles of breast (N = 48), ovary (N = 22), and non-cervical uterine (N = 6) tumors from 76 AYA Japanese females at diagnosis.
RESULTS
Study cohort for genome-wide mutation profiling
The characteristics of the 76 sporadic AYA tumor cases studied here are presented in Table 1. The 48 breast tumors consisted of 47 carcinomas and one (2.1%) angiosarcoma. The histological and subtype distributions in the study cohort, including frequent luminal type carcinomas, were consistent with previous reports of breast tumors in Japanese AYAs [15]; however, they were different to the distributions among European/US patients, in whom both luminal and triple-negative tumors are common [16].
Table 1: Characteristics of the 76 female AYA tumor cases
Breast tumor (N=48) |
Ovarian tumor (N=22) |
Uterine tumor (N=6) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
N |
% |
N |
% |
N |
% |
||||
Age |
Mean (±SD) |
36.6 |
(±2.9) |
34.5 |
(±4.3) |
34.5 |
(±3.9) |
||
Stage |
0 |
2 |
4.2 |
- |
- |
0 |
0 |
||
I |
16 |
33.3 |
12 |
54.5 |
1 |
16.7 |
|||
II |
22 |
45.8 |
4 |
18.2 |
1 |
16.7 |
|||
III |
7 |
14.6 |
6 |
27.2 |
4 |
66.7 |
|||
IV |
0 |
0 |
0 |
0 |
0 |
0 |
|||
Unknown |
1 |
2.1 |
0 |
0 |
0 |
0 |
|||
Histology |
DCIS |
2 |
4.2 |
Serous (BM) |
4 (2) |
18.1 |
Endometrioid |
5 |
83.3 |
IDC |
43 |
89.6 |
Mucinous (BM) |
8 (2) |
36.3 |
Carcinosarcoma |
1 |
16.7 |
|
ILC |
1 |
2.1 |
Endometrioid (BM) |
3 (1) |
13.6 |
||||
Mucinous |
1 |
2.1 |
Clear cell |
4 |
18.1 |
||||
Angiosarcoma |
1 |
2.1 |
Carcinosarcoma |
1 |
4.5 |
||||
Immature teratoma |
1 |
4.5 |
|||||||
Primitive neuroectodermal tumor and adenosquamous cell carcinoma |
1 |
4.5 |
|||||||
Subtype |
Luminal |
36 |
75 |
||||||
Luminal HER2 |
3 |
6.3 |
|||||||
HER2 |
2 |
4.2 |
|||||||
Triple negative |
6 |
12.5 |
|||||||
Other |
1 |
2.1 |
|||||||
SD, standard deviation; DCIS, ductal carcinoma in situ; IDC, invasive ductal carcinoma; ILC, invasive lobular carcinoma; BM, borderline malignancy.
The 22 ovarian tumors consisted of 14 carcinomas, five borderline tumors, and three others, while the six uterine tumors consisted of five endometrioid carcinomas and one carcinosarcoma. All four major histological types of ovarian carcinoma were represented [17]. The histological distribution was consistent with that previously reported for ovarian and uterine tumors of Japanese patients [18, 19].
Germline mutations
Exome sequencing data generated from non-tumor DNA from 73/76 cases (three ovarian tumor cases without informed consent for germline mutation analysis were excluded) were analyzed to identify germline mutations in 25 known cancer susceptibility genes [20]. Germline mutations were identified in three cases: 2/48 cases with breast (4.2%) and 1/19 cases with ovarian (5.3%) cancer carried pathogenic deleterious germline mutations in the BRCA2 gene. The two cases with breast tumors also each carried an additional deleterious germline mutation: one in TP53 and the other in CHEK2. No other cases showed germline mutations in the 25 genes tested (Figure 1, Supplementary Table 1).
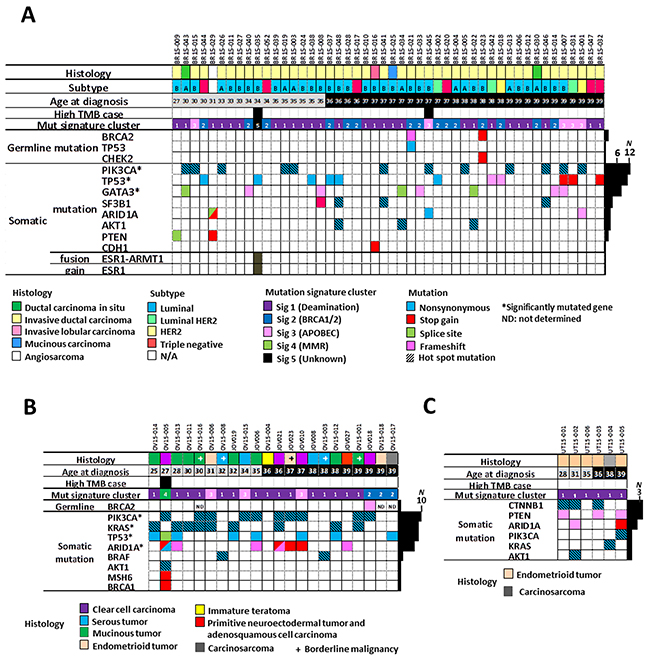
Figure 1: Gene aberration profiles of tumors of AYA Japanese females. (A) Forty-eight breast, (B) 22 ovarian, and (C) six uterine tumors. Clinical and histological factors, tumor mutation burden, mutational signature cluster groups (see Figure 2), germline mutations in 25 cancer susceptibility genes [20], and somatic mutations in representative cancer census genes known to be aberrant in female tumors are shown. High tumor burden cases were defined as those with > 10 single nucleotide variants per Mb [21]. A/B subtypes of luminal type tumors are indicated by the characters A and B. ND, not determined; N/A, not applicable; *, significantly mutated genes in breast and ovarian tumors defined by the MutSigCV program.
Somatic mutations
Next, we searched for somatic mutations by examining exome sequencing data from tumors from all 76 cases. A high tumor mutation burden (TMB), recently defined as > 10 SNVs/Mb [21], was found in two breast carcinomas (BR15-035T, TMB = 50.4 and BR15-045T, TMB = 16.4) and an ovarian carcinoma (OV15-005T, TMB = 250.9). Deleterious germline and somatic mutations, i.e., nonsense and frameshift insertion/deletion (indel) alterations, in six hypermutator genes consisting of four mismatch repair (MMR) genes (MLH1, MSH2, MSH6, and PMS2) and two DNA polymerase genes with proofreading function, POLD and POLE, were examined as potentially responsible for high TMB, since their aberration is established as associated with high TMB in a variety of human cancers [21-24]. The case with ovarian cancer had a deleterious somatic mutation in MSH6, a MMR gene, while the two cases with breast cancer lacked mutations in the six genes (Figure 1, Supplementary Table 1). The TMBs of the remaining breast and ovarian, and all six uterine, tumors were similar (medians 0.60, 0.64, and 0.59, respectively; P > 0.05 by Kruskal-Wallis test).
The 76 tumors exhibited five mutational signatures, four of which showed high cosine similarity (≥ 0.9) with 30 known signatures deposited in the Catalogue of Somatic Mutations in Cancer (COSMIC) database (http://cancer.sanger.ac.uk/cosmic/signatures) and had been detected in breast, ovarian, and uterine cancer genomes in previous studies [12, 25] (Supplementary Figure 1). Hierarchical cluster analysis of mutational signatures revealed that the 76 cases could be divided into five groups (Figure 2). The largest group (N = 48; 63%) comprised cases in which COSMIC-signature 1 (resulting from spontaneous deamination of 5-methylcytosine) was predominant. In the second largest group (N = 18; 24%) COSMIC-signature 3 (associated with BRCA1 and BRCA2 mutations) was predominant; all three cases with germline BRCA2 mutations were included in this group. In the third largest group (N = 8; 11%) COSMIC-signature 2 (attributed to activity of members of the APOBEC cytidine deaminase family) was predominant; however, expression levels of APOBEC genes were similar between these cases and those in other COSMIC signature groups (Supplementary Figure 1E). The remaining two groups each contained a single high TMB case. Case OV15-005T with the somatic MSH6 mutation showed strong identity with the COSMIC-signature 6 cluster associated with MMR deficiency. BR15-035T showed strong identity with an unknown signature pattern enriched in CpC to CpA mutations.
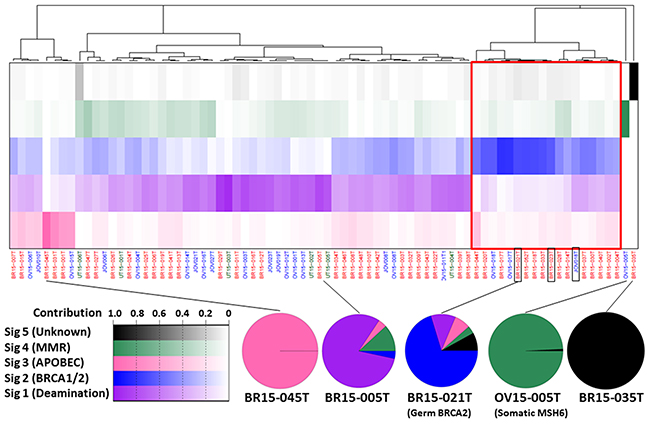
Figure 2: The spectrum of mutational signatures within 76 AYA female tumors. Samples are ordered according to hierarchical clustering performed on signatures. The IDs of the three cases with germline BRCA2 mutations are boxed in black. The top panel shows the proportion of each signature in each sample (increasing with color intensity as shown bottom left). The proportion of each signature in representative cases is represented bottom right.
Profiles of aberrations in cancer gene census genes
MutSigCV analysis identified several Cancer Gene Census (CGC) genes (http://cancer.sanger.ac.uk/census/) as having significant roles in the development/progression of AYA tumors; PIK3CA and TP53 were prominent in breast tumors, and PIK3CA, KRAS, TP53, and ARID1A in ovarian tumors (Figure 1), all of which are also frequently mutated in non-AYA breast and ovarian tumors [12, 16, 26-28]. A deleterious mutation in CDH1 was identified in the invasive lobular carcinoma (ILC), BR15-016T, consistent with a previous study showing frequent CDH1 mutation in ILC [29]. Among the ovarian tumors, KRAS mutations were more frequent in mucinous tumors than in other types (7/8 vs. 2/14; P = 0.0015 by Fisher’s exact test). PIK3CA and ARID1A mutations were more frequent in clear cell or endometrioid tumors than in other types (6/7 vs. 4/15 and 4/7 vs. 3/15; P = 0.016 and 0.11, respectively), consistent with previous studies of all-age-group ovarian tumors [13, 15, 27]. Mutations in CTNNB1, PTEN, and ARID1A were recurrent among the six uterine tumors (50%, 50%, and 33%, respectively), which is also consistent with previous reports from all-age-group uterine tumors [30]. These findings indicate that aberrations in the same sets of genes contribute to breast, ovarian, and uterine tumorigenesis in both AYA and non-AYA individuals.
RNA sequencing detected a novel in-frame fusion gene, ESR1-ARMT1, between ESR1, encoding estrogen receptor 1, and ARMT1, encoding acidic residue methyltransferase 1, in an ER+ luminal type invasive ductal carcinoma, case BR15-035T (Supplementary Figure 2). The increased genome copy numbers of both loci in this case, together with the location of these two genes neighboring CCDC170 on chromosome 6q25, suggest that this fusion was generated by tandem duplication of the ARMT1-CCDC170-ESR1 locus, as identified in breast cancers bearing the recurrent ESR1-CCDC170 fusion [31].
Proportion of cases with actionable gene aberrations
Hot spot activating mutations in the PIK3CA, KRAS, BRAF, and AKT1 genes, copy number gains in HER2, and deleterious BRCA1, BRCA2, PTEN, and ARID1A mutations were considered actionable gene aberrations (Figure 3), since drugs targeting the molecules encoded by these loci are available or being developed in clinical trials. In addition, high TMB was deemed an actionable aberration, as this feature is associated with response to immune checkpoint inhibitory therapy [21, 32]. Based on these criteria, 24 breast (50%), 17 ovarian (77%), and five uterine (83%) tumor cases were judged to have actionable gene aberrations.
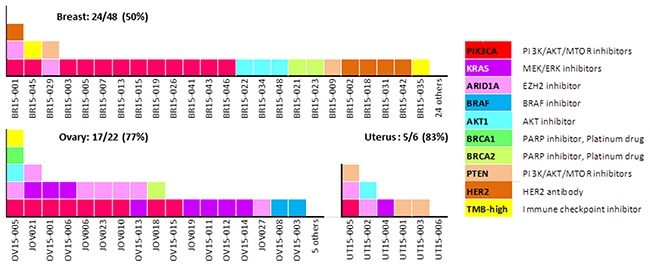
Figure 3: Fractions of AYA female tumors with potentially actionable gene aberrations. Gene aberrations and their corresponding therapeutic agents are shown for each case.
DISCUSSION
Here, the genome-wide profiles of 76 sporadic tumors of AYA Japanese females were investigated to determine the underlying mutagenic processes and elucidate actionable gene aberrations. Germline mutations in 25 genes established as involved in hereditary tumors [20] were detected only in a small fraction of patients: 4.2% of cases with breast tumors and 5.3% of those with ovarian tumors. The frequencies among breast and ovarian tumor cases were considerably lower than those found in sporadic cases among AYAs in the US (Supplementary Table 2). Thus, germline mutations in those susceptibility genes may contribute to the development of a smaller subset of sporadic AYA tumors in females in Japan than in those in the US. It was noted that two individuals with breast cancer with germline BRCA2 mutation also carried another deleterious germline mutation, respectively. Double germline mutations have been observed in several US patients (Supplementary Table 2), therefore, their pathogenic and clinical significance in AYA-tumors should be further investigated in larger sets of samples.
Mutational signature analysis revealed that our cases had the same signatures as those observed in all-age-group studies of breast, ovarian, and uterine tumors [11-13]. The prevalence of COSMIC-signatures 1 and 2 in female AYA tumors was in common with that in a variety of tumors [11], indicating that the mutagenic mechanisms are similar [11]. By contrast, a substantial proportion of the cases (24%) bore a signature of mutagenesis associated with BRCA1 and BRCA2 deficiency, which is exclusively observed in breast, ovarian, and pancreatic tumors [11]. This mutational signature profile of AYA tumors is quite similar to that of sporadic breast and ovarian cancers in general [12, 13]. Therefore, the mutation burden during tumorigenesis is likely to be similar between tumors in AYA and non-AYA females. The breast cancer case, BR-035T, showed a signature highly enriched in CpC to CpA mutations. To the best of our knowledge, this patient had not suffered from specific carcinogen exposure; therefore, the underlying process that initiated mutagenesis remains unclear.
Consistent with the mutational signature data, AYA tumors showed mutations in the same set of genes as non-AYA tumors. Deleterious mutations in GATA3, an activating SF3B1 mutation (K700E), and an activating AKT1 mutation (E17K) were observed in eight (16.7%), three (6.3%), and three (6.3%) breast tumors, respectively. These frequencies are higher than those in overall breast cancers [12, 16, 33]. Thus, these gene aberrations could be preferentially involved in the development of tumors in AYA females. In the present study, significant fractions of breast (50%), ovarian (77%), and uterine (83%) tumors had actionable gene mutations and gains (Figure 3), while actionable oncogene fusions, as frequently observed in AYA lung tumors [34, 35], were not discovered. Recently, it was reported that a mutational “BRCAness” signature is a predictor for functional BRCA1/BRCA2 deficiency [36]; therefore, the 15 BRCA1/2 mutation-negative breast/ovarian tumors with a predominant COSMIC-signature 3 could be responsive to PARP inhibitors and platinum agents due to deficiency in DNA double strand break repair. If that were the case, the fractions of breast and ovarian tumors with actionable gene aberrations in the current study would increase to 74% and 86%, respectively (Supplementary Figure 3).
An ESR1-ARMT1 fusion was detected in a case of breast carcinoma. Several types of gene fusions including ESR1 have been reported in ER+ breast cancers [37]. All known ESR fusion proteins have a common structure, where the ligand-binding domain of the ESR1 protein is absent, but the hormone-independent transactivation domain and DNA-binding domain are retained, suggesting their significance in resistance to endocrine therapy. The ESR1-ARMT1 fusion protein identified in the current study retains the ligand-binding and transactivation domain, but lacks the DNA-binding domain (Supplementary Figure 2A), and patient BR15-035T, from whom it was isolated, had not received endocrine therapy. Thus, the effect of ESR1-ARMT1 fusion on resistance to endocrine therapy is unknown.
Overall, our results from gene profiling of tumors from 76 female AYAs lead us to conclude that the mutational processes in these malignancies, as well as their aberrant genes, are largely shared with non-AYA tumors. High frequencies of actionable gene aberrations, including high TMB and the “BRCAness” mutational signature, indicate that the efficacy of molecular targeting and immune checkpoint inhibitory therapies should be studied in AYA patients, along with non-AYA patients. In addition, more extensive study of germline mutations in genes other than the 25 examined in this study will facilitate our understanding of hereditary factors involved in AYA tumor development.
MATERIALS AND METHODS
Patients
The AYA tumors analyzed in the present study were obtained from consecutive cases aged 15 to 39 years, who were diagnosed with breast, ovarian, or uterine tumors and underwent surgery at the National Cancer Center Hospital (NCCH), Tokyo, Japan, or at the Jikei University Hospital (JUH), Tokyo, Japan, and for whom snap-frozen tumor and non-tumor tissues were available in the NCCH and JUH-Gynecology Biobanks. None of the 76 patients had received any pre-surgical treatment, and there was no obvious family history of cancers. Seventy-three patients provided informed consent for both somatic and germline gene aberration analysis. The remaining three, OV15-016, OV15-017, and OV15-018, only provided informed consent for somatic mutation analysis, in which non-tumor DNA is used as a reference to identify somatic mutations in tumor DNA; these three patients did not give consent for germline mutation analysis, where non-tumor tissue DNA is used for detection of germline mutations. The institutional review boards of the National Cancer Center and Jikei University approved the study. Clinical data for each patient, including their age, and tumor stage, histology, and subtype, were collected retrospectively. Breast tumors were diagnosed according to the Union for International Cancer Control TNM classification (7th edition). Subtyping of breast tumors was performed based on the status of ER, PgR, HER2, and Ki67, which were examined by immunohistochemistry, as previously described [19]. Ovarian and uterine tumors were diagnosed in accordance with the International Federation of Gynecology and Obstetrics (FIGO) system and classified according to the World Health Organization (WHO) classification of tumors [38].
Whole exome and RNA sequencing
Exome sequencing was conducted using 200 ng of genomic DNA isolated from snap-frozen tumor and non-tumor tissues obtained from 76 patients. Exome capture was performed using the Agilent SureSelect Human V5 platform, according to the manufacturer’s instructions. The median sequencing depths of tumor and non-tumor DNA were 203 (range, 128–228) and 104 (79–136), respectively. Somatic single nucleotide variants (SNVs) were called using the MuTect program for variants present in bi-directional reads [39]. Somatic insertion/deletion (indel) mutations were called using the GATK Somatic Indel Detector, while germline SNVs and indels were called using the GATK program (https://www.broadinstitute.org/gatk/). Significantly mutated genes were defined by a q value of < 0.10, using the MutSigCV program [28]. Pathogenic germline mutations in 25 known cancer susceptibility genes [20] were defined as “pathogenic variants” deposited in the ClinVar database (http://www.ncbi.nlm.nih.gov/clinvar/), and as deleterious variations, i.e., nonsense SNVs and frameshift indel variants. The 25 genes examined consisted of 12 breast cancer susceptibility genes (ATM, BARD1, BRCA1, BRCA2, BRIP1, CDH1, CHEK2, NBN, PALB2, PTEN, STK11, and TP53) and 13 cancer susceptibility genes (APC, BMPR1A, CDK4, CDKN2A, EPCAM, MLH1, MSH2, MSH6, MUTYH, PMS2, RAD51C, RAD51D, and SMAD4). These genes were selected because they are well-documented moderate- and high-risk genes for female tumors [20].
RNA samples (200 ng) extracted from snap-frozen tissues using TRizol reagent (Thermo Fisher Scientific) were subjected to RNA sequencing using the TruSeq RNA Sample Prep Kit (Illumina). Fusion transcripts were detected using the TopHat-Fusion algorithm [40].
Mutational signature analysis
Mutational signatures were analyzed by non-negative matrix factorization (NMF), which was applied to the 96 possible mutations occurring in a trinucleotide context, as previously described [11, 25]. NMF was performed with various numbers of signatures, from one to ten, in this study. Obtained signatures were compared with those in the COSMIC database (http://cancer.sanger.ac.uk/cosmic/siganatures). The similarity was quantified using cosine similarity as previously described [11, 25].
Statistical analyses
Statistical analyses of differences in clinico-pathological factors and genetic aberrations were tested by Mann-Whitney U, Kruskal-Wallis, Pearson’s chi2, and Fisher’s exact tests.
Further information
See Supplementary Materials.
Abbreviations
AYA, adolescent and young adult; CGC, Cancer Gene Census; COSMIC, Catalogue of Somatic Mutations in Cancer; ER, estrogen receptor; HER2, human epidermal growth factor receptor 2; indel, insertion/deletion; ILC, invasive lobular carcinoma; JUH, Jikei University Hospital; MMR, mismatch repair; NCCH, National Cancer Center Hospital; NMF, non-negative matrix factorization; PgR, progesterone receptor; SNV, single nucleotide variant; TMB, tumor mutation burden.
Author contributions
TKo had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. KT, AO, TKi, TKa and TKo contributed to the study concept and design. YK, AS, MS, TH, KS, YS, RW, HY, MY, CS, KTa, HO, SH and KK contributed to the acquisition of the data. YK, HT, TH and TKo contributed to the analysis and interpretation of the data. YK drafted the manuscript. TKo critically revised the manuscript for important intellectual content. YK, TH and TKo were responsible for the statistical analysis. KT, AO, TKi and TKa provided administrative and material support. All authors read and approved the final version of the manuscript.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to declare.
FUNDING
This work was supported in part by grants-in-aid from the Japan Agency for Medical Research and Development (AMED; 17ck0106168h0003; T. Kohno), the Ministry of Education, Culture, Sports, Science, and Technology of Japan (KAKENHI; 17K11310; T. Kato), and the National Cancer Center Research and Development Fund (NCC Biobank).
REFERENCES
1. Schmidt C. Lack of progress in teen and young adult cancers concerns researchers, prompts study. J Natl Cancer Inst. 2006; 98: 1760-1763.
2. Bleyer A, Barr R, Hayes-Lattin B, Thomas D, Ellis C, Anderson B; Biology and Clinical Trials Subgroups of the US National Cancer Institute Progress Review Group in Adolescent and Young Adult Oncology. The distinctive biology of cancer in adolescents and young adults. Nat Rev Cancer. 2008; 8: 288-298.
3. Bleyer A. Young adult oncology: the patients and their survival challenges. CA Cancer J Clin. 2007; 57: 242-255.
4. Cleator S, Heller W, Coombes RC. Triple-negative breast cancer: therapeutic options. Lancet Oncol. 2007; 8: 235-244.
5. Heo SH, Kim JW, Shin SS, Jeong SI, Lim HS, Choi YD, Lee KH, Kang WD, Jeong YY, Kang HK. Review of ovarian tumors in children and adolescents: radiologic-pathologic correlation. Radiographics. 2014; 34: 2039-2055.
6. You W, Dainty LA, Rose GS, Krivak T, McHale MT, Olsen CH, Elkas JC. Gynecologic malignancies in women aged less than 25 years. Obstet Gynecol. 2005; 105: 1405-1409.
7. Heravi-Moussavi A, Anglesio MS, Cheng SW, Senz J, Yang W, Prentice L, Fejes AP, Chow C, Tone A, Kalloger SE, Hamel N, Roth A, Ha G, et al. Recurrent somatic DICER1 mutations in nonepithelial ovarian cancers. N Engl J Med. 2012; 366: 234-242.
8. Quirk JT, Natarajan N. Ovarian cancer incidence in the United States, 1992-1999. Gynecol Oncol. 2005; 97: 519-523.
9. Benshushan A. Endometrial adenocarcinoma in young patients: evaluation and fertility-preserving treatment. Eur J Obstet Gynecol Reprod Biol. 2004; 117: 132-137.
10. Zhang C, Hu W, Jia N, Li Q, Hua K, Tao X, Wang L, Feng W. Uterine carcinosarcoma and high-risk endometrial carcinomas: a clinicopathological comparison. Int J Gynecol Cancer. 2015; 25: 629-636.
11. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, Boyault S, Burkhardt B, Butler AP, et al. Signatures of mutational processes in human cancer. Nature. 2013; 500: 415-421.
12. Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, Martincorena I, Alexandrov LB, Martin S, Wedge DC, Van Loo P, Ju YS, Smid M, et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature. 2016; 534: 47-54.
13. Wang YK, Bashashati A, Anglesio MS, Cochrane DR, Grewal DS, Ha G, McPherson A, Horlings HM, Senz J, Prentice LM, Karnezis AN, Lai D, Aniba MR, et al. Genomic consequences of aberrant DNA repair mechanisms stratify ovarian cancer histotypes. Nat Genet. 2017; 49: 856-865.
14. Katanoda K, Shibata A, Matsuda T, Hori M, Nakata K, Narita Y, Ogawa C, Munakata W, Kawai A, Nishimoto H. Childhood, adolescent and young adult cancer incidence in Japan in 2009-2011. Jpn J Clin Oncol. 2017; 47: 762-771.
15. Yoshida M, Shimizu C, Fukutomi T, Tsuda H, Kinoshita T, Akashi-Tanaka S, Ando M, Hojo T, Fujiwara Y. Prognostic factors in young Japanese women with breast cancer: prognostic value of age at diagnosis. Jpn J Clin Oncol. 2011; 41: 180-189.
16. Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, Pugh M, Jones L, Russell R, Sammut SJ, Tsui DW, Liu B, Dawson SJ, et al. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun. 2016; 7: 11479.
17. Heintz AP, Odicino F, Maisonneuve P, Quinn MA, Benedet JL, Creasman WT, Ngan HY, Pecorelli S, Beller U. Carcinoma of the ovary. FIGO 26th Annual Report on the Results of Treatment in Gynecological Cancer. Int J Gynaecol Obstet. 2006; 95: S161-192.
18. Haruta S, Furukawa N, Yoshizawa Y, Tsunemi T, Nagai A, Kawaguchi R, Tanase Y, Yoshida S, Kobayashi H. Molecular genetics and epidemiology of epithelial ovarian cancer (Review). Oncol Rep. 2011; 26: 1347-1356.
19. Akahira J, Tokunaga H, Toyoshima M, Takano T, Nagase S, Yoshinaga K, Tase T, Wada Y, Ito K, Niikura H, Yamada H, Sato A, Sasano H, Yaegashi N. Prognoses and prognostic factors of carcinosarcoma, endometrial stromal sarcoma and uterine leiomyosarcoma: a comparison with uterine endometrial adenocarcinoma. Oncology. 2006; 71: 333-340.
20. Tung N, Lin NU, Kidd J, Allen BA, Singh N, Wenstrup RJ, Hartman AR, Winer EP, Garber JE. Frequency of germline mutations in 25 cancer susceptibility genes in a sequential series of patients with breast cancer. J Clin Oncol. 2016; 34: 1460-1468.
21. Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, Schrock A, Campbell B, Shlien A, Chmielecki J, Huang F, He Y, Sun J, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017; 9: 34.
22. Howitt BE, Shukla SA, Sholl LM, Ritterhouse LL, Watkins JC, Rodig S, Stover E, Strickland KC, D’Andrea AD, Wu CJ, Matulonis UA, Konstantinopoulos PA. Association of polymerase e-mutated and microsatellite-instable endometrial cancers with neoantigen load, number of tumor-infiltrating lymphocytes, and expression of PD-1 and PD-L1. JAMA Oncol. 2015; 1: 1319-1323.
23. Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, Nakagawa H, Sotamaa K, Prior TW, Westman J, Panescu J, Fix D, Lockman J, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med. 2005; 352: 1851-1860.
24. Esteban-Jurado C, Gimenez-Zaragoza D, Munoz J, Franch-Expósito S, Álvarez-Barona M, Ocaña T, Cuatrecasas M, Carballal S, López-Cerón M, Marti-Solano M, Díaz-Gay M, van Wezel T, Castells A, et al. POLE and POLD1 screening in 155 patients with multiple polyps and early-onset colorectal cancer. Oncotarget. 2017; 8: 26732-26743. https://doi.org/10.18632/oncotarget.15810.
25. Alexandrov LB, Nik-Zainal S, Wedge DC, Campbell PJ, Stratton MR. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013; 3: 246-259.
26. Jones S, Wang TL, Shih IM, Mao TL, Nakayama K, Roden R, Glas R, Slamon D, Diaz LA Jr, Vogelstein B, Kinzler KW, Velculescu VE, Papadopoulos N. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010; 330: 228-231.
27. Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, Senz J, McConechy MK, Anglesio MS, Kalloger SE, Yang W, Heravi-Moussavi A, Giuliany R, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010; 363: 1532-1543.
28. Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, Kiezun A, Hammerman PS, McKenna A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013; 499: 214-218.
29. Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, Zhang H, McLellan M, Yau C, Kandoth C, Bowlby R, Shen H, Hayat S, et al. Comprehensive molecular portraits of invasive lobular breast cancer. Cell. 2015; 163: 506-519.
30. Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, Benz CC, Yau C, Laird PW, Ding L, et al; Cancer Genome Atlas Research Network. Integrated genomic characterization of endometrial carcinoma. Nature. 2013; 497: 67-73.
31. Veeraraghavan J, Tan Y, Cao XX, Kim JA, Wang X, Chamness GC, Maiti SN, Cooper LJ, Edwards DP, Contreras A, Hilsenbeck SG, Chang EC, Schiff R, Wang XS. Recurrent ESR1-CCDC170 rearrangements in an aggressive subset of oestrogen receptor-positive breast cancers. Nat Commun. 2014; 5: 4577.
32. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015; 348: 124-128.
33. Maguire SL, Leonidou A, Wai P, Marchio C, Ng CK, Sapino A, Salomon AV, Reis-Filho J, Weigelt B, Natrajan RC. SF3B1 mutations constitute a novel therapeutic target in breast cancer. J Pathol. 2015; 235: 571-580.
34. Tanaka K, Hida T, Oya Y, Yoshida T, Shimizu J, Mizuno T, Kuroda H, Sakakura N, Yoshimura K, Horio Y, Sakao Y, Yatabe Y. Unique prevalence of oncogenic genetic alterations in young patients with lung adenocarcinoma. Cancer. 2017; 123: 1731-1740.
35. Kohno T, Ichikawa H, Totoki Y, Yasuda K, Hiramoto M, Nammo T, Sakamoto H, Tsuta K, Furuta K, Shimada Y, Iwakawa R, Ogiwara H, Oike T, et al. KIF5B-RET fusions in lung adenocarcinoma. Nat Med. 2012; 18: 375-377.
36. Davies H, Glodzik D, Morganella S, Yates LR, Staaf J, Zou X, Ramakrishna M, Martin S, Boyault S, Sieuwerts AM, Simpson PT, King TA, Raine K, et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med. 2017; 23: 517-525.
37. Veeraraghavan J, Ma J, Hu Y, Wang XS. Recurrent and pathological gene fusions in breast cancer: current advances in genomic discovery and clinical implications. Breast Cancer Res Treat. 2016; 158: 219-232.
38. Kurman RJ, Carcangiu ML, Herrington CS, Young RH. World Health Organization Classification of Tumours Pathology & Genetics Tumours of Female Reproductive Organs. Lyon: IARC Press. 2014.
39. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013; 31: 213-219.
40. Kim D, Salzberg SL. TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome Biol. 2011; 12: R72.