
INTRODUCTION
Recent studies have uncovered that tumor types with high somatic mutation frequencies, such as non-small cell lung cancer (NSCLCs), melanoma, and colorectal cancer (CRC) [1], display the strongest responses to immune checkpoint inhibitors [2–7]. This link between mutational burden and immunotherapy efficacy is due to the generation of immunogenic, tumor-specific mutated peptides, termed neo-antigens. Neo-antigens have been linked to response to anti-PD-1 therapy in NSCLC [8] and anti-CTLA-4 therapy in melanoma [9]. Further, in muscle invasive bladder cancer, high neo-antigen load correlated with greater recurrence-free survival and low T cell receptor-β diversity, indicating oligoclonal T cell expansion [10]. Across multiple cancer types, predicted neo-antigen burden, but not overall mutational load, correlated with patient survival and CD8A and PD-1 expression [11], as well as immune cytolytic activity (GZMA and PRF1 expression) [12].
The clinical significance of neo-antigens is derived from their ability to drive a functional and specific anti-tumor immune response. Neo-antigen-specific T cells have been identified in chronic lymphocytic leukemia patients who demonstrated remission after allogeneic stem cell therapy [13], melanoma patients who were administered an IL-12-producing dendritic cell vaccine (after prior ipilimumab therapy) [14], as well as phase II melanoma patients who experienced tumor regression after adoptive transfer of autologous tumor infiltrating lymphocytes (TILs) [15]. In pre-clinical studies, multiple immunogenic mutated peptides capable of decreasing tumor growth in vivo were identified in the B16F10 melanoma cells line [16], and in a murine sarcoma model, tumor immunoediting led to clonal outgrowth of tumor cells lacking neo-antigen expression [17]. Further, immunization with neo-antigens has even proven therapeutically comparable to administration of immune checkpoint blockade in mice [18]. This therapeutic potential was translated to the clinical setting, as adoptive transfer of autologous tumor mutation-specific Th1 CD4 T cells from a patient with metastatic cholangiocarcinoma was able to induce tumor regression upon original administration as well as after tumor recurrence [19].
One potential driver of a hypermutable state capable of generating neo-antigens is the loss of DNA repair genes [20]. In CRC, mismatch repair (MMR) gene mutations differentiate microsatellite instable (MSI) from microsatellite stable (MSS) disease. Interestingly, MSI CRCs contain increased mutations, predicted neo-antigens, TILs (with a Th1 phenotype), and expression of immunosuppressive molecules, and MSI also confers a favorable prognosis [21–27]. Mutated MMR genes themselves have even been postulated as immunogenic antigens capable of driving the immune response in pancreatic cancer [28]. Importantly, in a phase 2 trial of metastatic CRC patients treated with the anti-PD-1 antibody, pembrolizumab, patients with MMR-deficient tumors had a objective response rate (ORR) of 40%, compared to 0% for those with MMR-proficient tumors. Similarly, patients with MMR-deficient non-CRC had a ORR of 71%. These MMR-deficient tumors contained significantly increased mutation and neo-antigen load, both of which were associated with increased progression-free survival [6]. Importantly, similar success has been replicated in MSI-high CRC [29] and across multiple other cancer types [30], leading to pembrolizumab being granted FDA approval for use in unresectable or metastatic MMR-deficient or MSI-high solid tumors. However, none of the above studies have demonstrated efficacy in MMR-deficient lung tumors, and immunotherapy clinical trials have only focused on the MMR DNA repair pathway.
In addition to mutations in MMR genes, deficiencies in other DNA repair genes have also been implicated in neo-antigen generation and impacting the immune response. BRCA1/2 and other homologous recombination (HR) gene mutations have been associated with increased neo-antigen load, CD3 and CD8 TILs, expression of cytotoxicity-related genes (in the TCR, IFNγ, and TNFR pathways), PD-1 and PD-L1 expression, and favorable overall survival (OS) in ovarian cancer [31, 32]. In endometrial cancer, samples with POLE mutations have shown increased neo-antigens, as well as PD-1 and PD-L1 expression [33]. Further, in NSCLC patients treated with anti-PD-1 blockade, responders with the highest mutational burden contained mutations in POLD1, POLE, and MSH2, and nonsynonymous mutational burden and neo-antigen load in these patients correlated with enhanced clinical response [8]. This effect of DNA repair loss to augment the anti-tumor immune response can explain the improved clinical outcomes and survival seen in patients with tumors harboring mutations in these critical genes [34–36].
In lung adenocarcinoma, nivolumab immunotherapy is approved for patients progressing on platinum-based chemotherapy, conferring a 51% 1 year OS rate compared to 39% with docetaxel [37]. Additionally, pembrolizumab is approved for PD-L1-positive metastatic NSCLC patients, with a reported 19.4% ORR and 12 month median OS (treated population was 81% non-squamous NSCLC) [5]. However, while neo-antigen and mutational burden have been linked to enhanced response to immunotherapies [8], biomarkers to aid in identifying these patients are lacking. In addition, the overall immunophenotype of the lung adenocarcinoma microenvironment still has not clearly been defined in a comprehensive manner. We hypothesize that mutations in DNA repair genes in lung adenocarcinoma are key players in determining total mutational burden, neo-antigen load, and, consequently, tumor microenvironment in increased TILs and immune infiltrate that can serve as future biomarkers for immune checkpoint efficacy.
RESULTS
The immunophenotype of lung adenocarcinoma is primarily infiltration by activated CD4 and CD8 T cells
To elucidate the immune microenvironment of TCGA lung adenocarcinoma samples, we utilized a pre-determined list of immune metagenes whose expression have been shown to accurately predict the infiltration of 28 immune cell populations [24]. Activated CD4 and activated CD8 T cells were the most prominent infiltrating cell types, present in 29.7% and 26.8% of samples, respectively. Infiltration by at least one T cell subtype was identified in 73% of samples (Figure 1A). Additionally, we analyzed the tendency for each pairing of immune cell types to infiltrate the same samples. Interestingly, infiltration by activated CD4 and CD8 T cells significantly co-occurred in the same tumors, and infiltration by both cell types also significantly co-occurred with myeloid dendritic cells (mDCs), effector memory CD4 T cells, and Th17 T cells, with both inversely correlating with activated B cells, immature B cells, regulatory T cells (Tregs) and γδ T cells (Figure 1B).
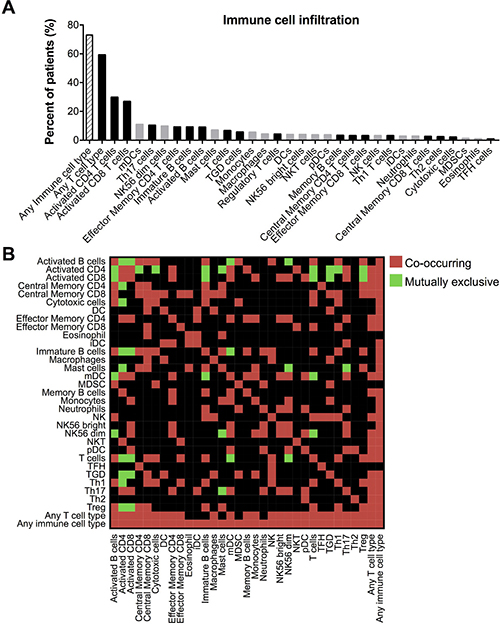
Figure 1: Comprehensive immunophenotype of the lung adenocarcinoma microenvironment. (A) Percent of tumors with positive infiltration by each adaptive (black bars) and innate (gray bars) immune cell type, as determined by immune metagene lists and GSEA analysis. Abbreviations: γδ T (TGD) cells, immature DCs (iDCs), myeloid derived suppressor cells (MDSCs), follicular helper T (TFH) cells. (B) Heat map indicating infiltration of immune cell types that either significantly (*P < 0.05) co-occur or are mutually exclusive within tumors.
Tumors with higher mutational burden contain increased TILs and a distinct molecular and histological phenotype
Mutational burden is known to impact tumor immunogenicity. To determine how the immunophenotype of lung adenocarcinoma is affected by increased mutational burden, we divided samples into those above or below the median mutation count. As expected, patients with tumors with high mutational burden were heavier smokers (Supplementary Figure 1A, 1B) and contained increased cytosine to adenine transversions (Supplementary Figure 1C), which has been previously linked to smoking [38–40]. Interestingly, the high mutation group also contained significantly higher infiltration by effector memory CD4 T cells, activated CD4 and CD8 T cells, mDCs, any T cell type, and any immune cell type (Figure 2A). Additionally, tumors with higher mutational burden were less likely to be oncogene-positive, as defined by containing an activating mutation in the RTK/RAS/RAF signaling pathway (Figure 2B). These tumors were also associated with a cluster 2 or 3 classification, defined by increased TP53 mutations, tumor ploidy, and copy number variations (CNVs) in 8q, 15q, and chromosome 7. High mutation tumors were less commonly cluster 4 samples, which are defined by low ploidy, 6q CNVs, CDKN2A methylation, and SETD2 mutations (Figure 2C). Finally, high mutation tumors were more often histologically proximal-inflammatory (squamoid; NF1 and TP53 mutations) or proximal proliferative (magnoid; KRAS mutation, STK11 inactivation), and significantly less likely to be terminal respiratory unit (bronchioid; EFGR mutations and kinase fusions) [38] (Figure 2D).
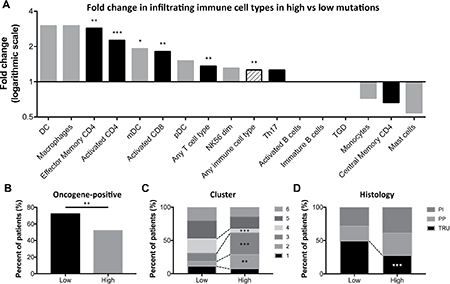
Figure 2: High mutation burden tumors contain increased TILS, immune infiltration, and a distinct molecular phenotype. (A) Fold change in the percent of patients with adaptive (black bars) and innate (gray bars) immune cell infiltration in those with high compared to low mutation burden, as defined by the median mutation count (excluding cell types with positive tumor infiltration in < 10 samples). Percent of high and low mutational burden tumors classified by (B) oncogene-positivity, (C) cluster type, and (D) histology. * P < 0.05, ** P < 0.01, *** P < 0.001.
DNA repair-deficient tumors contain increased mutation count, TILs
Mutations in DNA repair genes can consequently result in a hypermutable state with increased genomic instability. To test our hypothesis that mutations in these single DNA repair genes can serve as biomarkers for identifying NSCLC patients with higher mutational burden and increased TILs, we analyzed tumors specifically containing mutations in the HR pathway, the MMR pathway, or POLE. In each grouping, tumors with DNA repair deficiencies displayed significantly increased mutation counts (and a trending further increased mutation count with multiple HR and MMR genes mutated) (Figure 3A.i, 3B.i, 3C.i). Samples with HR mutations were significantly more infiltrated by activated CD4 T cells, neutrophils, natural killer T (NKT) cells, and were less likely to be infiltrated by activated B cells. Those with MMR mutations contained increased T cell infiltration, and POLE mutated samples contained increased infiltration by effector memory CD4 T cells, Th17 T cells, natural killer (NK)56 bright cells, T cells, plasmacytoid DCs (pDCs), Th1 T cells, and cytotoxic cells (Figure 3A.ii, 3B.ii, 3C.ii).
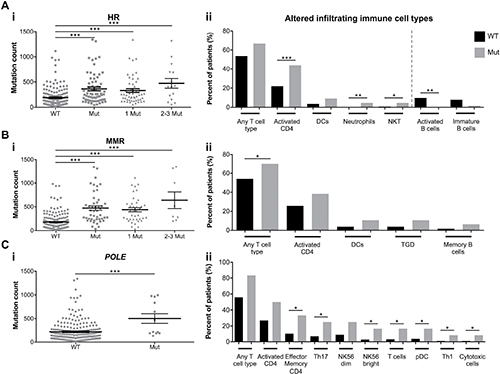
Figure 3: DNA repair mutations are biomarkers for total mutational burden and TILs. Total mutation count of tumors with mutations in (A.i) HR genes, (B.i) MMR genes, or (C.i) POLE. Results are presented as mean ± SEM. (A.ii), (B.ii), (C.ii) Percent of DNA repair wildtype (WT; black bars) or mutated (Mut; gray bars) tumors with immune cell infiltration (all immune cell types with P < 0.1 displayed. Red dotted line separates increased and decreased tumor infiltrating immune cell types). * P < 0.05, ** P < 0.01, *** P < 0.001.
We next analyzed the link between DNA repair gene mutations and mutations in type I interferon (IFN) genes, as tumor-derived type I IFNs are important for optimal immunosurveillance and anti-cancer therapy efficacy [41]. Samples with HR, MMR, and POLE mutations all contained increased frequencies of mutations in various type I IFN pathway genes. Notable, HR mutated tumors contained increased IFNβ (IFNB1) and IRF8 (type I IFN positive feedback loop) mutations, while MMR mutated tumors contained increased IFNα (IFNA5, IFNA14, IFNA21) and IFNAR2 (type I IFN receptor) mutations (Supplementary Figure 2A–2C), thereby indicating that while mutations in DNA repair genes are linked to increased tumor mutation count and TILs, they also may represent tumors with increased type I IFN gene mutations. Similarly, we also analyzed potential immune evasion as a result of mutations in class I HLA genes [42]. However, class I HLA mutations in the TCGA dataset we rare, with only 3, 0 , and 1 of the 230 samples containing mutations in HLA-A, HLA-B, and HLA-C, respectively. Of those, the 3 HLA-A mutated samples did not contain HR, MMR, or POLE mutations, while the single HLA-C mutated sample did contain a mutated HR pathway (data not shown).
Neo-antigen burden is linked to DNA repair mutations, increased TILs
The clinical and immunological relevance of increased mutational burden is that as more tumor mutations arise, there is a greater likelihood for somatic, non-synonymous mutations to result in the formation of immunogenic epitopes expressed only in cancerous cells. Therefore, we filtered total non-synonymous mutations into predicted neo-antigens, as determined by predicted immunogenicity (MHC binding affinity) (Supplementary Figure 3A), positive gene expression (Supplementary Figure 3B), and those whose non-mutated parental epitope were weak or non-MHC binders (Supplementary Figure 3C). Similar to total mutation count, neo-antigen burden was also significantly higher in samples with HR, MMR, or POLE mutations (Figure 4A–4C). Additionally, those samples with high neo-antigen load contained significantly increased infiltration by any immune cell type, overall T cells, activated CD8 T cells, and effector memory CD8 T cells (Figure 4D).
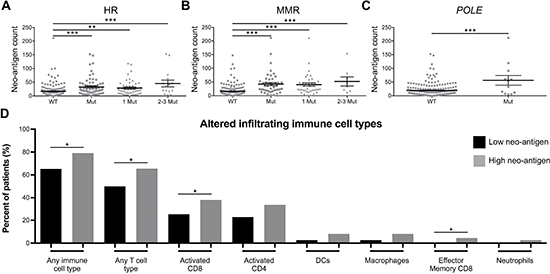
Figure 4: DNA repair mutations are biomarkers for high tumor neo-antigen load, which is associated with increased TILs. Neo-antigen count of tumors with (A) HR, (B) MMR, or (C) POLE mutations. Results are presented as mean ± SEM. (D) Percent of neo-antigen low (black bars) or high (gray bars) patients infiltrated by immune cell types (all immune cell types with P < 0.1 displayed). * P < 0.05, ** P < 0.01, *** P < 0.001.
Neo-antigens correlate with increased expression of multiple pro-inflammatory cytokines and immune-related genes, M1-polarized macrophage genes, PD-L1 and PD-1, and favorable survival outcome
While the link between neo-antigen load and TILs is now appreciated in many cancer types, the specific molecular causes and consequences of that association are not as well understood. We analyzed the correlations between neo-antigen burden and the expression of a comprehensive list of cytokines, chemokines, and immunomodulatory genes [24]. Significant positively or negatively correlated genes are reported in Figure 5A. Increasing neo-antigen load correlated with chemokines TNFRSF25, CCR1, and LTBR, immunomodulatory genes LAG3, PDCD1 (PD-1), GZMB, and FASLG, and multiple cytokines/cytokine receptors, including IFNγ, IL12RB2, and IL17RA. Additionally, when analyzing expression of gene markers to differentiate pro-inflammatory M1 from immunosuppressive M2 tumor-associated macrophages (TAMs) [43], tumors with more neo-antigens displayed significantly increased expression of M1 genes NOS2 and IL23A, and significantly (SOC2) or trending (CHIA, CHI3L1, CHI3L2, KLF4) decreased expression of multiple M2-associated genes (Figure 5B). Further, the top quartile of high neo-antigen load samples displayed increased PD-L1 (Figure 5C) and PD-1 (Figure 5D) expression. Finally, these high neo-antigen load patients trended toward increased OS (p = 0.12 when excluding the low neo-antigen outlier censored at 224 months) (Figure 5E).
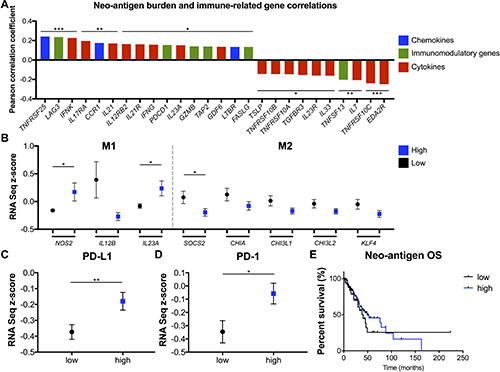
Figure 5: Neo-antigen load correlates with multiple immune-related genes and cytokines, M1-polarized TAMs, PD-L1, PD-1, and favorable OS. (A) Pearson correlation coefficients between neo-antigen count and RNA-seq expression levels of multiple chemokines, immunomodulatory genes, and cytokines (only significantly correlating genes reported). (B) Expression of M1 TAM- and M2 TAM-defining genes in high and low neo-antigen burden tumors (all genes with P < 0.2 displayed). (C) PD-L1 and (D) PD-1 expression in high (top quartile) and low neo-antigen load tumors. Results are presented as mean ± SEM. (E) OS of neo-antigen load high (top quartile) and low tumors. * P < 0.05, ** P < 0.01, *** P < 0.001.
DISCUSSION
Recent scientific and clinical advances in cancer immunotherapy have highlighted the importance of understanding neo-antigens and the immunological tumor microenvironment. Specifically in lung cancer, immune checkpoint blockade has shown great promise. However, we are still lacking a comprehensive understanding of the immunophenotype of these tumors. Additionally, while deficiencies in DNA repair genes, mostly in the MMR pathway, are known to accelerate mutation rates and affect prognosis, their relevance in immunotherapy in lung cancer has been unclear.
We first analyzed immune cell infiltration, utilizing a comprehensive list of “immune metagenes” to accurately predict the presence of up to 28 intra-tumoral immune cell types in a large cohort of lung adenocarcinoma patients. Interestingly, activated CD4 and CD8 T cells were the most commonly infiltrating cell types, followed by mDCs, all of which are desired for an optimal anti-tumor response. 73% of the samples were infiltrated by at least one of the T cell subtypes (Figure 1A). Further, activated CD4 and CD8 T cells were low in tumors infiltrated by activated B cells or immature B cells (Figure 1B), in agreement with a study demonstrating increased CD8 tumor infiltration and decreased tumor growth after B cell depletion [44]. Activated CD8 and CD4 infiltrated tumors also contained decreased infiltration by Tregs but increased Th17 T cell infiltration. Differentiation of T cells into either of these cell types inhibits the generation of the other, yet while Tregs are known to promote immunosuppression and tumor growth [45], intra-tumoral Th17 T cells can increase IFNγ production and tumor infiltration by CD8 TILs, NK cells, and DCs [46]. Also of note, tumors infiltrated by activated CD4 and CD8 T cells also contained increased infiltration by γδ T cells, which can also promote the anti-tumor immune response [47].
We next sought to determine the significance of mutational burden on the immunological response in lung adenocarcinoma. As hypothesized, tumors with high mutational count displayed increased infiltration by activated CD4 and CD8 T cells, effector memory CD4 T cells, and mDCs (Figure 2A). Importantly, tumors containing mutations in HR genes, MMR genes, or POLE contained significantly higher mutational load than DNA repair wildtype tumors, and similarly contained significantly increased infiltration by many of the T cell types and other immune cells desirable for anti-tumor activity. Further, HR-deficient tumors also contained decreased infiltration by B cells, a potentially immunosuppressive and tumor-promoting cell type [48] (Figure 3). Therefore, detecting mutations in the single POLE gene or the small HR or MMR gene lists can accurately be used as biomarkers for identifying patients whose tumors likely contain high mutational burden and, consequently, increased TILs.
To clarify the clinical relevance of these findings, we refined overall mutational burden to specifically consider at predicted immunogenic neo-antigens. HR-, MMR-, and POLE-deficient tumors again contained increased neo-antigens and T cell infiltration (Figure 4). Interestingly, increasing neo-antigen load strongly positively correlated with expression of many immune-related genes. Notably, neo-antigens increased with increasing expression of IFNγ and IL12RB2, indicating a Th1 skewed signature, as is desirable in an anti-tumor response. Neo-antigen load also correlated with increased expression of IL23A, IL21, and IL17RA, all of which are players in the Th17 T cell pathway, which is implicated in both tumorigenesis as well as the anti-tumor immune response [46]. Also correlating were GZMB and FASLG, both necessary for anti-tumor CD8 T cell cytotoxic functionality [49], PD-1 and LAG3, representing T cell activation and exhaustion [50], and TAP2, necessary in MHC I antigen presentation [51]. Cytokines inversely correlating with neo-antigen burden included TSLP and IL33, involved in the Th2 response [52], IL7, implicated with pro- and anti-tumorigenic properties [53], EDA2R, which is involved in p53 signaling [53], and multiple genes in the TRAIL apoptosis pathway. Additionally, neo-antigen load correlated with increased expression of the chemokines TNFRSF25, CCR1, and LTBR, which may serve as valuable targets in future tumor immunology studies (Figure 5A).
Outside of T cells, TAMs have emerged an important focus of research and therapeutic targets. Specifically, M1-polarized macrophages have been shown to promote anti-tumor activity and decrease tumor growth while M2 macrophages suppress TILs and promote tumorigenesis [54]. We found that high neo-antigen load correlated with increased expression of multiple M1 genes and decreased expression of multiple M2 genes. This finding supports a novel link between neo-antigens and M1 TAM polarization. This may be due to the interplay between an increased activated adaptive T cell response with a Th1/Th17 signature and the infiltrating innate cells. Finally, tumors with increased neo-antigens contained increased expression of PD-L1 (Figure 5C), which is induced by IFNγ [55], and PD-1 (Figure 5D), as well as a trending increase in OS (Figure 5E). While past studies have reported a more dramatic gap in survival between high and low neo-antigen groups, our analysis was limited because for the TCGA data the treatments and disease stage were not controlled between groups, many patients were censored at early time points, and many patients had resectable early stage disease as opposed to late stage disease important for survival analysis.
Overall, we comprehensively analyzed the immunophenotype of the lung adenocarcinoma microenvironment. This study demonstrates that lung tumors with DNA repair deficiencies, either in the MMR pathway, HR pathway, or POLE, can be utilized as biomarkers for total mutational burden, neo-antigen load, and TILs, which will be of utmost value as immunotherapies continue to grow and expand clinically. These results provide a foundation for further studies exploring the clinical implications of using DNA repair gene mutations as a predictive marker for immunotherapy response.
MATERIALS AND METHODS
Clinical data, immune infiltration, DNA repair
This study utilized cBioPortal [56, 57] to obtain data from the lung adenocarcinoma cohort of The Cancer Genome Atlas (TCGA) [38] to analyze RNA-sequencing (RNA-seq) gene expression and clinical data (n = 515) and DNA mutations (n = 230). Predicted infiltration by 28 distinct immune cell types was performed as previously described [24], utilizing 812 “immune metagenes” derived from 813 microarrays over 36 studies. Expression of these genes was used as input in Gene Set Enrichment Analysis (GSEA), and any immune cell types with a false discovery rate (q-value) of ≤ 10% were considered as positively infiltrating into that tumor sample. In the DNA repair genes analysis, the homologous recombination pathway gene list included ATR, ATM, CHEK1, CHEK2, BRCA1, BRCA2, BAP1, BARD1, FANCD2, FANCE, FANCC, FANCA, RAD50, RAD51, and PALB2, and the mismatch repair pathway gene list included MLH1, MLH3, MSH2, MSH3, MSH4, MSH5, MSH6, PMS1, PMS2, PMS2L3, PCNA, EXO1, POLD1, RFC1, RFC2, RFC3, RFC4, and RFC5 [58]. In the type I IFN signaling pathway analysis, the gene list included all genes with available TCGA data from the gene ontology (GO) accession number 0060337 [59, 60].
Neo-antigen prediction
HLA typing, neo-antigen identification, and HLA-peptide affinity prediction were performed using HLAminer [61],Variant Effect Predictor Tool [62], NetMHCpan [63], and the UCSC browser [64] (http://www.genome.ucsc.edu/), with most computations performed on the National Cancer Institute (NCI) Cancer Genomics Cloud (CGC) in the Seven Bridges Genomics implementation. The neo-antigen affinity scores were generated between all possible 9 amino acid length peptides containing a mutated site with the 6 predicted HLA types, using the CloudNeo pipeline [65]. A control analysis was performed with the homologous non-mutated 9 amino acid length sequences. Neo-antigens were defined as mutated peptides with a binding score IC50 < 500 nM, positive gene expression, and whose non-mutated wildtype peptide was a weak or non-MHC binder IC50 > 500 nM.
Statistical analysis
As appropriate, z-score between two population proportions, unpaired two-tailed Student’s or Welch’s t-test, Log-rank test for survival, and the Pearson correlation coefficient were used for statistical assessment. Results are presented as percentages, fold change (logarithmic scale), mean ± standard error of the mean (SEM), Pearson correlation coefficient, RNA-seq z-score, or percent survival, as indicated. * P < 0.05, ** P < 0.01, *** P < 0.001.
Abbreviations
NSCLC, non-small cell lung cancer; CRC, colorectal cancer; TIL, tumor infiltrating lymphocyte; MMR, mismatch repair; MSI, microsatellite instable; MSS, microsatellite stable; ORR, objective response rate; HR, homologous recombination; OS, overall survival; mDC, myeloid dendritic cell; Treg, regulatory T cells; CNV, copy number variations; TAM, tumor-associated macrophage; TCGA, The Cancer Genome Atlas; GSEA, Gene Set Enrichment Analysis; CGC, Cancer Genomics Cloud; SEM, standard error of the mean; TGD, γδ T cell; NKT, natural killer T cell; pDC, plasmacytoid DC; NK, natural killer cell; iDC, immature DC; MDSC, myeloid derived suppressor cell; TFH, follicular helper T cell; WT, wildtype; Mut, mutated; RNA-seq, RNA-sequencing.
Author contributions
JFA and YKC formulated hypotheses and designed experiments, JFA performed all data collection, analyses, and manuscript preparation and review. PB and SN performed neo-antigen predictions. FJG aided in manuscript review. JHC designed experiments, performed neo-antigen predictions, and aided in manuscript review.
ACKNOWLEDGMENTS
The authors thank Gaurav Kaushik for assistance with computational implementations on the Cancer Genomics Cloud.
CONFLICTS OF INTEREST
All authors certify that they have no conflicts of interest to disclose.
FUNDING
JHC was supported by the NCI of the National Institutes of Health under award R21CA191848 and supplement R21CA191848-01A1S1. Research was also partially supported by the NCI under award P30CA034196.
REFERENCES
1. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, Boyault S, Burkhardt B, Butler AP, et al. Signatures of mutational processes in human cancer. Nature. 2013; 500:415–21. https://doi.org/10.1038/nature12477.
2. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, Dronca R, Gangadhar TC, Patnaik A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013; 369:134–44. https://doi.org/10.1056/NEJMoa1305133.
3. Lynch TJ, Bondarenko I, Luft A, Serwatowski P, Barlesi F, Chacko R, Sebastian M, Neal J, Lu H, Cuillerot JM, Reck M. Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: results from a randomized, double-blind, multicenter phase II study. J Clin Oncol. 2012; 30:2046–54. https://doi.org/10.1200/JCO.2011.38.4032.
4. Phan GQ, Yang JC, Sherry RM, Hwu P, Topalian SL, Schwartzentruber DJ, Restifo NP, Haworth LR, Seipp CA, Freezer LJ, Morton KE, Mavroukakis SA, Duray PH, et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci U S A. 2003; 100:8372–7. https://doi.org/10.1073/pnas.1533209100.
5. Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, Carcereny E, Ahn MJ, Felip E, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015; 372:2018–28. https://doi.org/10.1056/NEJMoa1501824.
6. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, Biedrzycki B, Donehower RC, Zaheer A, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015; 372:2509–20. https://doi.org/10.1056/NEJMoa1500596.
7. Champiat S, Ferte C, Lebel-Binay S, Eggermont A, Soria JC. Exomics and immunogenics: Bridging mutational load and immune checkpoints efficacy. Oncoimmunology. 2014; 3:e27817. https://doi.org/10.4161/onci.27817.
8. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015; 348:124–8. https://doi.org/10.1126/science.aaa1348.
9. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, Hollmann TJ, Bruggeman C, Kannan K, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014; 371:2189–99. https://doi.org/10.1056/NEJMoa1406498.
10. Choudhury NJ, Kiyotani K, Yap KL, Campanile A, Antic T, Yew PY, Steinberg G, Park JH, Nakamura Y, O’Donnell PH. Low T-cell Receptor Diversity, High Somatic Mutation Burden, and High Neoantigen Load as Predictors of Clinical Outcome in Muscle-invasive Bladder Cancer. Eur Urol. 2015; 2:445–452. https://doi.org/10.1016/j.euf.2015.09.007.
11. Brown SD, Warren RL, Gibb EA, Martin SD, Spinelli JJ, Nelson BH, Holt RA. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. 2014; 24:743–50. https://doi.org/10.1101/gr.165985.113.
12. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015; 160:48–61. https://doi.org/10.1016/j.cell.2014.12.033.
13. Rajasagi M, Shukla SA, Fritsch EF, Keskin DB, DeLuca D, Carmona E, Zhang W, Sougnez C, Cibulskis K, Sidney J, Stevenson K, Ritz J, Neuberg D, et al. Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemia. Blood. 2014; 124:453–62. https://doi.org/10.1182/blood-2014-04-567933.
14. Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Ly A, Lie WR, Hildebrand WH, Mardis ER, Linette GP. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. 2015; 348:803–8. https://doi.org/10.1126/science.aaa3828.
15. Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, Lin JC, Teer JK, Cliften P, Tycksen E, Samuels Y, Rosenberg SA. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med. 2013; 19:747–52. https://doi.org/10.1038/nm.3161.
16. Castle JC, Kreiter S, Diekmann J, Lower M, van de Roemer N, de Graaf J, Selmi A, Diken M, Boegel S, Paret C, Koslowski M, Kuhn AN, Britten CM, et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 2012; 72:1081–91. https://doi.org/10.1158/0008-5472.CAN-11-3722.
17. Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, Arthur CD, White JM, Chen YS, Shea LK, Hundal J, Wendl MC, Demeter R, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. 2012; 482:400–4. https://doi.org/10.1038/nature10755.
18. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber WJ, Mulder GE, Toebes M, Vesely MD, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014; 515:577–81. https://doi.org/10.1038/nature13988.
19. Tran E, Turcotte S, Gros A, Robbins PF, Lu YC, Dudley ME, Wunderlich JR, Somerville RP, Hogan K, Hinrichs CS, Parkhurst MR, Yang JC, Rosenberg SA. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science. 2014; 344:641–5. https://doi.org/10.1126/science.1251102.
20. Loeb LA. A mutator phenotype in cancer. Cancer Res. 2001; 61:3230–9.
21. Tougeron D, Fauquembergue E, Rouquette A, Le Pessot F, Sesboue R, Laurent M, Berthet P, Mauillon J, Di Fiore F, Sabourin JC, Michel P, Tosi M, Frebourg T, et al. Tumor-infiltrating lymphocytes in colorectal cancers with microsatellite instability are correlated with the number and spectrum of frameshift mutations. Mod Pathol. 2009; 22:1186–95. https://doi.org/10.1038/modpathol.2009.80.
22. Smyrk TC, Watson P, Kaul K, Lynch HT. Tumor-infiltrating lymphocytes are a marker for microsatellite instability in colorectal carcinoma. Cancer. 2001; 91:2417–22.
23. Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, Blosser RL, Fan H, Wang H, Luber BS, Zhang M, Papadopoulos N, Kinzler KW, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015; 5:43–51. https://doi.org/10.1158/2159-8290.CD-14-0863.
24. Angelova M, Charoentong P, Hackl H, Fischer ML, Snajder R, Krogsdam AM, Waldner MJ, Bindea G, Mlecnik B, Galon J, Trajanoski Z. Characterization of the immunophenotypes and antigenomes of colorectal cancers reveals distinct tumor escape mechanisms and novel targets for immunotherapy. Genome Biol. 2015; 16:64. https://doi.org/10.1186/s13059-015-0620-6.
25. Ishikawa T, Fujita T, Suzuki Y, Okabe S, Yuasa Y, Iwai T, Kawakami Y. Tumor-specific immunological recognition of frameshift-mutated peptides in colon cancer with microsatellite instability. Cancer Res. 2003; 63:5564–72.
26. Gatalica Z, Snyder C, Maney T, Ghazalpour A, Holterman DA, Xiao N, Overberg P, Rose I, Basu GD, Vranic S, Lynch HT, Von Hoff DD, Hamid O. Programmed cell death 1 (PD-1) and its ligand (PD-L1) in common cancers and their correlation with molecular cancer type. Cancer Epidemiol Biomarkers Prev. 2014; 23:2965–70. https://doi.org/10.1158/1055-9965.EPI-14-0654.
27. Dolcetti R, Viel A, Doglioni C, Russo A, Guidoboni M, Capozzi E, Vecchiato N, Macri E, Fornasarig M, Boiocchi M. High prevalence of activated intraepithelial cytotoxic T lymphocytes and increased neoplastic cell apoptosis in colorectal carcinomas with microsatellite instability. Am J Pathol. 1999; 154:1805–13. https://doi.org/10.1016/S0002-944065436-3.
28. Okada T, Noji S, Goto Y, Iwata T, Fujita T, Okada T, Matsuzaki Y, Kuwana M, Hirakata M, Horii A, Matsuno S, Sunamura M, Kawakami Y. Immune responses to DNA mismatch repair enzymes hMSH2 and hPMS1 in patients with pancreatic cancer, dermatomyositis and polymyositis. Int J Cancer. 2005; 116:925–33. https://doi.org/10.1002/ijc.21118.
29. Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, Desai J, Hill A, Axelson M, Moss RA, Goldberg MV, Cao ZA, Ledeine JM, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017; 18:1182–1191. https://doi.org/10.1016/S1470-204530422-9.
30. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, Wong F, Azad NS, Rucki AA, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017; 357:409–13. https://doi.org/10.1126/science.aan6733.
31. Clarke B, Tinker AV, Lee CH, Subramanian S, van de Rijn M, Turbin D, Kalloger S, Han G, Ceballos K, Cadungog MG, Huntsman DG, Coukos G, Gilks CB. Intraepithelial T cells and prognosis in ovarian carcinoma: novel associations with stage, tumor type, and BRCA1 loss. Mod Pathol. 2009; 22:393–402. https://doi.org/10.1038/modpathol.2008.191.
32. Strickland KC, Howitt BE, Shukla SA, Rodig S, Ritterhouse LL, Liu JF, Garber JE, Chowdhury D, Wu CJ, D’Andrea AD, Matulonis UA, Konstantinopoulos PA. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget. 2016; 7:13587–98. https://doi.org/10.18632/oncotarget.7277.
33. Howitt BE, Shukla SA, Sholl LM, Ritterhouse LL, Watkins JC, Rodig S, Stover E, Strickland KC, D’Andrea AD, Wu CJ, Matulonis UA, Konstantinopoulos PA. Association of Polymerase e-Mutated and Microsatellite-Instable Endometrial Cancers With Neoantigen Load, Number of Tumor-Infiltrating Lymphocytes, and Expression of PD-1 and PD-L1. JAMA Oncol. 2015; 1:1319–23. https://doi.org/10.1001/jamaoncol.2015.2151.
34. Yap KL, Kiyotani K, Tamura K, Antic T, Jang M, Montoya M, Campanile A, Yew PY, Ganshert C, Fujioka T, Steinberg GD, O’Donnell PH, Nakamura Y. Whole-exome sequencing of muscle-invasive bladder cancer identifies recurrent mutations of UNC5C and prognostic importance of DNA repair gene mutations on survival. Clin Cancer Res. 2014; 20:6605–17. https://doi.org/10.1158/1078-0432.CCR-14-0257.
35. Yamamoto H, Itoh F, Nakamura H, Fukushima H, Sasaki S, Perucho M, Imai K. Genetic and clinical features of human pancreatic ductal adenocarcinomas with widespread microsatellite instability. Cancer Res. 2001; 61:3139–44.
36. Benatti P, Gafa R, Barana D, Marino M, Scarselli A, Pedroni M, Maestri I, Guerzoni L, Roncucci L, Menigatti M, Roncari B, Maffei S, Rossi G, et al. Microsatellite instability and colorectal cancer prognosis. Clin Cancer Res. 2005; 11:8332–40. https://doi.org/10.1158/1078-0432.CCR-05-1030.
37. Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E, Barlesi F, Kohlhaufl M, Arrieta O, et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med. 2015; 373:1627–39. https://doi.org/10.1056/NEJMoa1507643.
38. Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014; 511:543–50. https://doi.org/10.1038/nature13385.
39. Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, Sougnez C, Auclair D, Lawrence MS, et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012; 150:1107–20. https://doi.org/10.1016/j.cell.2012.08.029.
40. Govindan R, Ding L, Griffith M, Subramanian J, Dees ND, Kanchi KL, Maher CA, Fulton R, Fulton L, Wallis J, Chen K, Walker J, McDonald S, et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell. 2012; 150:1121–34. https://doi.org/10.1016/j.cell.2012.08.024.
41. Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol. 2015; 15:405–14. https://doi.org/10.1038/nri3845.
42. Shukla SA, Rooney MS, Rajasagi M, Tiao G, Dixon PM, Lawrence MS, Stevens J, Lane WJ, Dellagatta JL, Steelman S, Sougnez C, Cibulskis K, Kiezun A, et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat Biotechnol. 2015; 33:1152–8. https://doi.org/10.1038/nbt.3344.
43. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011; 11:723–37. https://doi.org/10.1038/nri3073.
44. Affara NI, Ruffell B, Medler TR, Gunderson AJ, Johansson M, Bornstein S, Bergsland E, Steinhoff M, Li Y, Gong Q, Ma Y, Wiesen JF, Wong MH, et al. B cells regulate macrophage phenotype and response to chemotherapy in squamous carcinomas. Cancer Cell. 2014; 25:809–21. https://doi.org/10.1016/j.ccr.2014.04.026.
45. Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017; 27:109–18. https://doi.org/10.1038/cr.2016.151.
46. Ye J, Livergood RS, Peng G. The role and regulation of human Th17 cells in tumor immunity. Am J Pathol. 2013; 182:10–20. https://doi.org/10.1016/j.ajpath.2012.08.041.
47. Silva-Santos B, Serre K, Norell H. gammadelta T cells in cancer. Nat Rev Immunol. 2015; 15:683–91. https://doi.org/10.1038/nri3904.
48. Shalapour S, Font-Burgada J, Di Caro G, Zhong Z, Sanchez-Lopez E, Dhar D, Willimsky G, Ammirante M, Strasner A, Hansel DE, Jamieson C, Kane CJ, Klatte T, et al. Immunosuppressive plasma cells impede T-cell-dependent immunogenic chemotherapy. Nature. 2015; 521:94–8. https://doi.org/10.1038/nature14395.
49. Andersen MH, Schrama D, Thor Straten P, Becker JC. Cytotoxic T cells. J Invest Dermatol. 2006; 126:32–41. https://doi.org/10.1038/sj.jid.5700001.
50. Grosso JF, Goldberg MV, Getnet D, Bruno TC, Yen HR, Pyle KJ, Hipkiss E, Vignali DA, Pardoll DM, Drake CG. Functionally distinct LAG-3 and PD-1 subsets on activated and chronically stimulated CD8 T cells. J Immunol. 2009; 182:6659–69. https://doi.org/10.4049/jimmunol.0804211.
51. Lankat-Buttgereit B, Tampe R. The transporter associated with antigen processing: function and implications in human diseases. Physiol Rev. 2002; 82:187–204. https://doi.org/10.1152/physrev.00025.2001.
52. Bartemes KR, Kita H. Dynamic role of epithelium-derived cytokines in asthma. Clin Immunol. 2012; 143:222–35. https://doi.org/10.1016/j.clim.2012.03.001.
53. Lin J, Zhu Z, Xiao H, Wakefield MR, Ding VA, Bai Q, Fang Y. The role of IL-7 in Immunity and Cancer. Anticancer Res. 2017; 37:963–7. https://doi.org/10.21873/anticanres.11405.
54. Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014; 41:49–61. https://doi.org/10.1016/j.immuni.2014.06.010.
55. Blank C, Brown I, Peterson AC, Spiotto M, Iwai Y, Honjo T, Gajewski TF. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. 2004; 64:1140–5.
56. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013; 6:pl1. https://doi.org/10.1126/scisignal.2004088.
57. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2:401–4. https://doi.org/10.1158/2159-8290.CD-12-0095.
58. Chae YK, Anker JF, Carneiro BA, Chandra S, Kaplan J, Kalyan A, Santa-Maria CA, Platanias LC, Giles FJ. Genomic landscape of DNA repair genes in cancer. Oncotarget. 2016; 7:23312–21. https://doi.org/10.18632/oncotarget.8196.
59. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000; 25:25–9. https://doi.org/10.1038/75556.
60. The Gene Ontology Consortium. Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 2017; 45:D331-D8. https://doi.org/10.1093/nar/gkw1108.
61. Warren RL, Choe G, Freeman DJ, Castellarin M, Munro S, Moore R, Holt RA. Derivation of HLA types from shotgun sequence datasets. Genome Med. 2012; 4:95. https://doi.org/10.1186/gm396.
62. McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P, Cunningham F. The Ensembl Variant Effect Predictor. Genome Biol. 2016; 17:122. https://doi.org/10.1186/s13059-016-0974-4.
63. Andreatta M, Nielsen M. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics. 2016; 32:511–7. https://doi.org/10.1093/bioinformatics/btv639.
64. Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002; 12:996–1006. https://doi.org/10.1101/gr.229102.
65. Bais P, Namburi S, Gatti DM, Zhang X, Chuang JH. CloudNeo: A cloud pipeline for identifying patient-specific tumor neoantigens. Bioinformatics. 2017; 33: 3110–3112. https://doi.org/10.1093/bioinformatics/btx375.