
INTRODUCTION
Cancer is a systems disease with a complicated pathogenesis. Tumorigenesis is characterized by abnormal regulation of cell growth and cell death. Mutations in proto-oncogenes and tumor suppressor genes underlie tumorigenesis by dysregulation of intracellular signaling pathways that are critical for the normal physiology of organisms [1]. The canonical Wnt/β-catenin signaling pathway is one such prominent pathway, being tightly regulated for maintenance of biological homeostasis. Frequently, this pathway is aberrantly activated in numerous cancers, including gastrointestinal, prostate, breast, and ovarian cancer [2–5]. Over the past several decades, studies of cancer genetics and genomics have demonstrated that multiple genes encoding key proteins in this signaling pathway are targets for recurrent mutational alterations. Among these proteins, β-catenin and Adenomatous polyposis coli (APC) are two key nodes that physically combine with each other in a complex. The tumor suppressor gene, APC, is one frequently mutated gene of Wnt signaling in human cancers [6]. As one of the central nodes, β-catenin contributes in transporting extracellular signals for nuclear programming [7]. β-catenin mutations may lead to constitutive activation of the Wnt/β-catenin signaling pathway and reprogramming of downstream nuclear transcriptional networks [8, 9]. In this review, we provide a focused overview of the integrated Wnt/β-catenin signaling pathway and the basic structure and biological roles of β-catenin. We also summarize the current understanding of β-catenin mutations in tumorigenesis and discuss their possible therapeutic implications for cancer.
OVERVIEW OF THE CANONICAL WNT/β-CATENIN SIGNALING PATHWAY
The Wnt gene was first discovered in 1982 by Nusse and Varmus while studying the transcription mechanisms for a tumor virus in a murine mammary tumor [10]. Initially identified as Int1, the gene was determined to encode proteins that transfer growth and development signals between cells. Further studies demonstrated that this gene could make Drosophila flies wingless in normal embryonic development, and it was renamed Wnt [11, 12]. The Wnt protein family is cysteine-rich secreted glycoprotein with both autocrine and paracrine functions [13]. There are currently 19 identified members, including wnt1, wnt3A, and wnt5A [14–16]. Wnt signaling has proven to contribute on the embryonic development. The three highly characterized Wnt signaling pathways are the noncanonical Wnt-Ca2+ pathway, noncanonical planar cell polarity pathway, and canonical Wnt/β-catenin signaling pathway [13, 17–19]. In general, these pathways can be placed in two categories according to the presence or absence of β-catenin: canonical or noncanonical, respectively. Several other signal transduction pathways involve Wnt, such as the Wnt/Rac, Wnt/cAMP, and Wnt/Rho pathways [18, 20, 21].
The complexity of the canonical Wnt/β-catenin pathway derives from the high number of ligands and receptors involved in signaling that can elicit a variety of intracellular responses [22, 23]. As the key intracellular transducer of this pathway, β-catenin plays important roles in the entire process (Figure 1). Activity of β-catenin is controlled by the destruction complex, consisting of APC, AXIN-1, AXIN-2, casein kinase-1α (CK-1), protein phosphatase 2A (PP2A), and glycogen synthase kinase (GSK)-3β [7, 24–26]. This pathway has two states dependent upon the presence or absence of Wnt ligands.
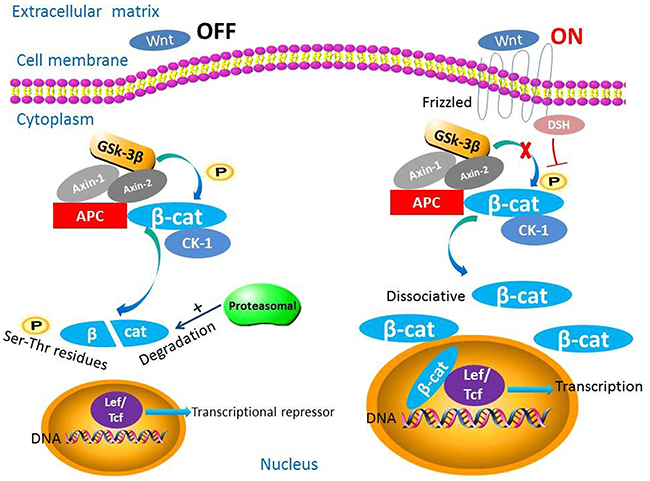
Figure 1: The biological roles of β-catenin in the Wnt/β-catenin signaling pathway. This pathway has two states dependent upon the presence or absence of Wnt ligands. When Wnt ligands are absent, β-catenin is phosphorylated by the destruction complex and degraded. When Wnt ligands are present, β-catenin is not degraded and translocates to the nucleus and functions as a transcription factor.
In the absence of Wnt ligands, cytoplasmic β-catenin is phosphorylated at N-terminal serine-threonine residues by the destruction complex and degraded by the proteasome via the ubiquitin-proteasome pathway [7, 27]. In general, the ubiquitin-proteasome pathway involves three parts: a ubiquitin-activating enzyme, a ubiquitin-conjugating enzyme, and a ubiquitin ligase [28]. Without nuclear accumulation of β-catenin, nuclear T-cell factor/lymphoid enhancer factor (TCF/LEF) transcription factors associate with co-repressor proteins via their high-mobility group domains and act as transcriptional repressors. Authors reported that the co-repressor proteins contain Groucho/transducin-like enhancer of split and CREB-binding protein (CBP) [29–31].
Alternatively, in the presence of Wnt, ligand binds to the cell-surface receptor Frizzled and acts on Dishevelled protein [32, 33]. Frizzled is a seven-pass transmembrane protein with a long amino terminal extension called a cysteine-rich domain. The cysteine-rich domain is a special structure where Wnt proteins bind directly [15, 16, 34]. In addition to Frizzled, a long single-pass transmembrane molecule named low-density lipoprotein receptor-related protein (LRP) is bound to Wnt ligands. The identity of this protein is LRP5 or LRP6 in vertebrates and in Drosophila, a similar protein is derived from the arrow gene [35]. The cytoplasmic tail of LRP may combine with Axin directly [36, 37]. Other single-pass transmembrane proteins, such as receptor-like tyrosine kinase and receptor tyrosine kinase-like orphan receptor-1/2, can function as co-receptors, influencing Wnt signaling [38–40]. Moreover, numerous studies have suggested that the R-Spondin proteins also play potential roles in Wnt signaling and could stabilize the levels of cytosolic β-catenin and dramatically synergize with wnt3A [41–43]. Following Wnt binding, the protein kinases (A, B, and C), phosphoinositide 3-kinase (PI3K)/Akt, and mitogen-activated protein kinase (MAPK) inhibit the phosphorylation of GSK-3β. The destruction complex then becomes inactive, preventing β-catenin phosphorylation and destruction. Hypophosphorylated β-catenin accumulates in the cytoplasm and eventually translocates to the nucleus to function as a transcriptional factor, despite lacking a nuclear localization signal [44–47]. Thus, the networks of a broad spectrum of Wnt downstream target genes are programmed [7]. These target genes, such as cyclin D1, insulin-like growth factor-1, and CD44, are critical to some hallmarks of cancer such as epithelial-to-mesenchymal transition and metastasis [48, 49].
OVERVIEW OF β-CATENIN AND ITS GENE MUTATIONS IN TUMORIGENESIS
The primary and three-dimensional structures of β-catenin
β-catenin is a multifunctional cytoplasmic protein composed of a polypeptide chain [50]. It is encoded by the gene CTNNB1, which maps to 3p21 [27] and with a size of 23.2 kb. CTNNB1 has 16 exons according to restricted mapping and partial sequence analysis [51]. The primary structure of β-catenin consists of three domains: a 550-amino-acid central repeat, an approximately 150-amino-acid N-terminal domain, and an approximately 100-amino-acid C-terminal domain on both sides (Figure 2) [52–56]. The central repeat domain is also known as armadillo repeats [57]. The armadillo repeat domain consists of 12 armadillo repeats [58], each of which contains approximately 42 residues that form three helices arranged in a triangular shape. A superhelix, formed by these 12 contiguous repeats, features a long, positively charged groove. The third helix of each repeat constitutes the floor of this groove [58]. Generally, the N-terminal domain is the phosphorylation site for GSK-3β and casein kinase-1 as well as the binding site for α-catenin with the C-terminal domain involved in combination with nuclear (TCF/ LEF) [59, 60]. The N- and C-terminal domains may combine with the armadillo repeat domain via a fold-back mechanism that may regulate the partner-binding properties of the armadillo repeat [61–64].
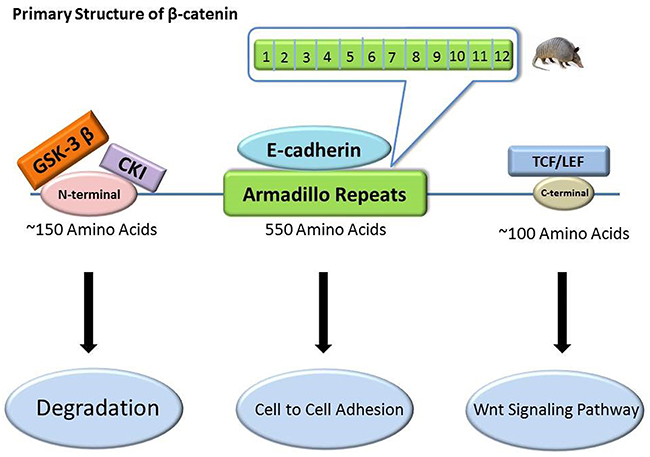
Figure 2: The primary structure of β-catenin and its relevant binding sites. β-catenin has three domains: a 550-amino-acid central repeat domain, an approximately 150-amino-acid N-terminal domain, and an approximately 100-amino-acid C-terminal domain. They are binding sites for E-cadherin, GSK-3β/CK-1, and TCF/LEF, respectively, and exert different roles.
The basic biological function of β-catenin
β-catenin is crucial for two important developmental processes: 1) establishment and maintenance of cell-type-specific cell-to-cell adhesion and 2) regulation of target gene expression via the Wnt signaling pathway [52, 65]. It also regulates the stem cell transcriptome via the Wnt, Notch, Hippo, and Hedgehog pathways [66–69]. In most cases, β-catenin needs to associate with several other proteins to perform its functions. Under normal physiological conditions, β-catenin is mainly present in the cell membrane and can participate in a complex with E-cadherin via the armadillo repeats [70–72]. E-cadherin, also known as uvomorulin, is the major cadherin molecule expressed by epithelial cells [73–75]. This complex can mediate the connection of E-cadherin with the cytoskeleton and is involved in cell adhesion. The complex can function to maintain normal cell morphology and inhibit tumor cell invasion and metastasis. E-cadherin can also inhibit β-catenin’s transcriptional activities by recruiting β-catenin from its transcriptional complexes [76]. Furthermore, in adherens junctions, β-catenin combines with α-catenin. α-catenin can link adherens junctions with the actin cytoskeleton by associating with filamentous actin directly or indirectly; this cytoskeletal linkage is crucial for the adhesion of cells [52, 77]. In summary, β-catenin provides obvious connections among extracellular signals, cell-cycle management, and gene transcription [23, 78].
Hot-spot exon mutations of CTNNB1 can drive tumorigenesis
Exon 3 of CTNNB1 is a key exon encoding serine-threonine phosphorylation sites for GSK-3β that activates degradation of β-catenin [79]. The CTNNB1 mutations are frequently missense mutations [27]. Nearly all of them have been localized in this hot-spot exon 3, and most of them have occurred at S33, S37, S45, T41, D32, and G34 [6, 57, 80–82]. Of these, S33, S37, and T41 are the phosphorylation sites for GSK-3β; S45 is the phosphorylation site for casein kinase-1; and D32 and G34 are essential for the interaction of β-catenin with Fbw1.
Gene mutations leading to constitutive activation of the Wnt/β-catenin signaling pathway, especially the canonical one, are early events in the development of some cancer cases [83]. Moreover, a high level of β-catenin activity is required for cancer initiation [84]. Initial characterization of mutations of CTNNB1 and deregulation of the canonical Wnt pathway were in colorectal cancer cases [85, 86]. Since then, these mutations have been described and studied in several other types of malignancies [87–92]. Paul Polakis (2000) has summarized CTNNB1 mutation spots and rates in human cancers in detail [93]. For example, a mutation of β-catenin (S37F) activates Wnt signaling in several melanoma cell lines [87]. Such mutations have been shown to result in the accumulation of nuclear β-catenin and stabilization of the protein and tumorigenesis [94, 95]. These mutations stabilize β-catenin by abrogating the phosphorylation-dependent interaction of β-catenin with Fbw1 [57]. Fbw1 is a type of F-box protein that is a component of the ubiquitin ligase for β-catenin that associates with β-catenin phosphorylated by casein kinase-1α and GSK-3β resulting in ubiquitination and degradation of β-catenin [57, 96]. In addition, other relatively benign tumors, such as desmoid-type fibromatosis, also have CTNNB1 mutations and abnormal nuclear β-catenin expression [97, 98].
As described above, under normal physiological conditions, β-catenin is expressed mainly in the cell membrane. In contrast, the expression level of dissociative β-catenin in the cytoplasm is quite low owing to the combination with the destruction complex [7]. Whether the genes upstream from β-catenin are altered, or CTNNB1 mutates, once a threshold of β-catenin accumulates in the cytoplasm and nucleus, this status reflects abnormal expression of β-catenin [99]. When degradation of β-catenins inhibited the level of dissociated β-catenin in the cytoplasm increases, the protein will then accumulate at high levels in the nucleus. Of all the molecular alterations that lead to disruptions of β-catenin degradation, the most common are mutations that activate β-catenin or inactivate APC [94]. For example, a report suggests APC mutations can correlate with high expression levels of β-catenin whereas wild-type APC expression can reduce β-catenin levels in colorectal cancer cells [93, 100]. The accumulated β-catenin in the nucleus can then combine with TCF/LEF transcription factors [101–104]. The TCF/β-catenin complex can activate the transcription of proto-oncogenes in humans [105]. The result is activated transcription of downstream cell-proliferation related genes such as c-myc and cyclin D1. This process promotes cell proliferation [106]. Thus, abnormal expression of β-catenin in the cytoplasm and nucleus is regarded as an important indicator of malignancy [6, 79].
For some cancers, such as endometrioid endometrial carcinoma (EEC), studies have demonstrated that 40-60% of cases exhibited nuclear accumulation of β-catenin in tumor cells [107, 108]. Other studies identified an increase in CTNNB1 mutations in EEC compared to nonendometrioid endometrial carcinoma cases (NEEC) [27, 109, 110]. Detailed mutational rates are in Table 1. CTNNB1 mutations seemingly occurr in the early stages of endometrial carcinogenesis [110]. Additionally, authors have described nuclear accumulation of β-catenin and mutations in exon 3 of CTNNB1 in endometrioid ovarian carcinoma [111–116]. From these aspects, at least in part, both nuclear expression and mutations of β-catenin are characteristic of the endometrioid phenotype [9]. Liu and colleagues (2014) conducted a systematic, comprehensive study of CTNNB1 mutations in EEC [117]. They performed an integrated analysis including whole-exome sequencing, RNA sequencing, and reverse-phase protein array profiling. The clinical study population consisted of two groups: 271 EEC patients whose data were obtained from The Cancer Genome Atlas and a validation group of 184 EEC patients from The University of Texas MD Anderson Cancer Center. One of the four genes expression clusters in The Cancer Genome Atlas group, cluster II, was enriched with the highest proportion of CTNNB1 gene mutations. Eighty-seven percent of the tumors in this cluster carried CTNNB1 mutations. This cluster was associated with young, obese patients with grade 1 or 2 tumors that were early stage at diagnosis (stages I or II) and had decreased expression of estrogen and progesterone receptors. Importantly, this cluster was also associated with decreased overall survival. Table 1 lists the rates of CTNNB1 mutations in these cases.
Table 1: Frequency of CTNNB1 mutation in different types of EEC and EOC
Histology |
Mutation rate (%) |
Frequency (N) |
Reference |
|---|---|---|---|
EEC |
52 |
15/29 |
27 |
EEC |
19 |
88/454 |
109 |
EEC |
37 |
71/192 |
117 |
EEC |
19 |
35/183 |
117 |
NEEC |
0 |
0/14 |
27 |
AEH |
14 |
3/21 |
110 |
EOC |
50 |
3/6 |
111 |
EOC |
54 |
7/13 |
112 |
EOC |
16 |
10/63 |
114 |
EOC |
31 |
14/45 |
114 |
EOC |
26 |
6/23 |
115 |
EOC |
38 |
8/21 |
116 |
Although CTNNB1 mutations have been shown to induce the accumulation of nuclear β-catenin and activate the canonical Wnt pathway, some studies demonstrated that this accumulation can occur without CTNNB1 mutations [9, 106]. This may indicate that alterations of other molecules or signaling pathways, such as APC, AXIN1, AXIN2, γ-catenin, and the PI3K/AKT signaling pathway, can modulate the Wnt pathway. Mutations in AXIN1 and AXIN2 can activate the Wnt pathway and have been associated with the development of a subset of colon, hepatic, and ovarian carcinomas and medulloblastomas [114, 118–120]. Overexpression of γ-catenin has been demonstrated to stabilize and increase the nuclear localization of β-catenin [121]. In addition, activation of PI3K/AKT by different mechanisms, such as GSK-3β inhibition and Ras activation, has been associated with nuclear accumulation of β-catenin in some cancers [122–124]. In some cancer cell lines, PI3K/AKT signaling activated β-catenin–mediated transcription [125]. Inhibition of PI3K/AKT signaling reduced Wnt signaling in medulloblastoma cells [126]. Similarly, in colon cancers, hepatocyte growth factor and MAPK signaling pathway may be regulators of Wnt signaling to mediate tumor progression [124, 127–130].
There are also critical upstream regulators of β-catenin, specifically p53 and microRNAs (miRNAs), that impact canonical Wnt signaling. As a tumor suppressor, loss of p53 function can activate canonical Wnt pathway by its transcriptional activity [131]. miRNAs are small and non-coding RNAs that can interact with untranslated regions of mRNA targets; repressing gene expression post-transcriptionally [132, 133]. Studies in recent years have found that several miRNAs could regulate the canonical Wnt pathway. For example, miRNA-34 suppressed the transcriptional activity of β-catenin/TCF complexes by targeting the untranslated regions of Wnt pathway-regulated genes [131]. Additionally, miRNA-370-3p could inhibit downstream genes of Wnt pathway, like cyclin D1 and c-myc, by binding with the 3’-untranslated region of β-catenin mRNA directly [134]. Roman Anton and colleagues (2011) performed a systematic screen of 470 miRNAs and identified 38 miRNAs that either activate, or repress the canonical Wnt pathway [135]. miRNA-1 and miRNA-613 acted upstream of β-catenin while miRNA-25 functioned at the level of β-catenin. From these studies, we can conclude that other factors in addition to CTNNB1 mutation can activate the canonical Wnt pathway.
THE CONNECTION BETWEEN β-CATENIN EXPRESSION AND IMMUNOSUPPRESSION IN THE CANCER MICROENVIRONMENT
Many types of tumors, including hematological malignancies and melanoma have been treated with immunotherapy [136, 137]. However, this approach often has insufficient antitumor effects [138, 139]. One important obstacle of this treatment approach is immunoescape through localized immunosuppression and immunoresistance, which is one of the major malignant characteristics of cancer cells [140]. Cancer cells can activate some immunosuppressive cells such as regulatory dendritic cells (DCs), and regulatory T cells (Tregs) through production of many immunosuppressive molecules such as transforming growth factor (TGF)-β, interleukin (IL)-10 [140]. Researchers have found that β-catenin correlated to the infiltration of immune cells in the tumor microenvironment (Figure 3) [141, 142]. Tumor-intrinsic β-catenin signaling could inhibit T-cell infiltration in melanoma models and in patient-derived biopsies [141]. Melanoma gene microarray data have suggested that an activated Wnt/β-catenin signal in the cancer microenvironment can be correlated with a lack of an immune cell infiltration [143]. Additionally, overexpression and mutations of β-catenin can cause the production of IL-10 at high levels in melanoma cells [138]. Investigators have observed similar events in EC. In the immune microenvironment of EC, cancer cells can secrete many immunosuppressive molecules, such as IL-10, TGF-β, and indoleamine 2,3-dioxygenase [140, 144] that could suppress the differentiation, maturation, and function of DCs and effector T cells [145]. Other immunosuppressive cells, such as regulatory DCs and Tregs, can be induced not only by TGF-β, IL-10 and other immunosuppressive molecules, but also by EC directly [140, 146]. Regulatory DCs also can induce Tregs. In addition, Tregs can potently suppress T-cell-mediated immune responses [147–150]. According to a statistical analysis, CTNNB1 mutations were associated with TGF-β2, which contributes to tumor progression. These mutations also influenced the numbers of cytotoxic cells that can kill tumor cells, such as lymphocytes and macrophages [117]. From these data we may conclude that β-catenin has some connections with immunosuppresion in the cancer environment.
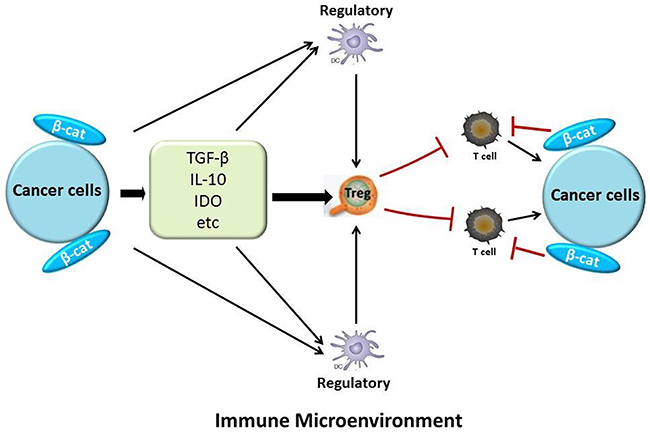
Figure 3: The relationship between β-catenin and immunosuppression in the tumor microenvironment. Cancer cells can induce relevant immunosuppressive molecules and cells, such as TGF-β, IL-10, indoleamine 2,3-dioxygenase (IDO), Tregs, and regulatory DCs. They can inhibit T cells, which have toxic effects on cancer cells. Also, cancer-intrinsic β-catenin signaling can decrease T-cell infiltration. β-catenin also helps cancer cells secrete immunosuppressive molecules.
THE CONNECTION BETWEEN β-CATENIN EXPRESSION AND METABOLIC REGULATION
Metabolic regulation is essential to tumorigenesis, and metabolic reprogramming is one of the hallmarks of tumor cells [151]. Metabolic reprogramming in cancer cells may contribute in targeting therapy resistance [152]. Via the Warburg effect, glucose metabolism can confer a powerful growth advantage to tumor cells from oxidative phosphorylation to glycolysis regardless of the oxygen supply [153–155]. Tumor cells need more glucose than normal cells to meet their elevated anabolic and energy demands, and are more sensitive to glucose deprivation, a phenomenon known as glucose addiction, [155, 156]. Tumor cells seem to prefer the glycolytic pathway to oxidative phosphorylation to produce ATP to support their anabolic production of biomass [157–159]. This preference may be a protective strategy against reactive oxygen species. Hence, glycolysis can protect genome integrity during DNA replication [160–162].
The Wnt/β-catenin signaling pathway can induce the Warburg effect and establish metabolic zonation (Figure 4) [153, 163–165]. However, once this signaling pathway is blocked, the free: bound NADH ratio and lactate production decrease. Decreases in both can indicate decreased glycolysis. In addition, the expression of two downstream target genes, the lactate transporter MCT-1 (SLC16A1) and pyruvate dehydrogenase kinase, isozyme 1, are also downregulated due to this blockage [166]. Pyruvate dehydrogenase kinase, isozyme 1 can phosphorylate and inhibit the pyruvate dehydrogenase complex in mitochondria, inhibiting the conversion of pyruvate to acetyl-CoA. This means that the next step, oxidative phosphorylation is blocked [167]. Therefore, pyruvate dehydrogenase kinase, isozyme 1 plays an important role in promotion of the glycolytic phenotype. Pyruvate kinase M2, one of the key enzymes in glucose metabolism, is highly expressed in human cancer cells and can stimulate glycolysis [168]. Authors have reported that nuclear pyruvate kinase M2 can activate β-catenin transactivation upon epidermal growth factor receptor activation [168].
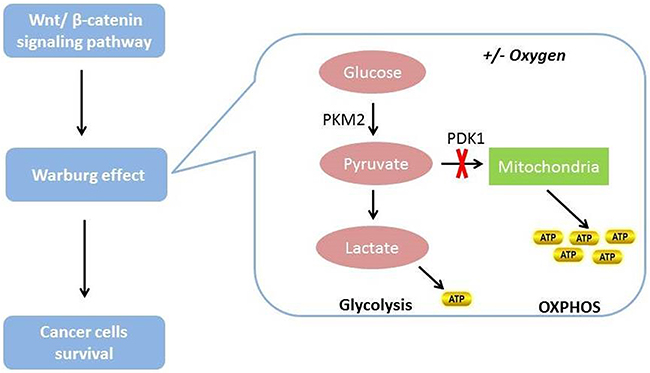
Figure 4: The relationship between Wnt/β-catenin signaling pathway and glucose metabolism in cancer cells. The Wnt/β-catenin signaling pathway can induce the Warburg effect and then promote cancer cell survival. Cancer cells prefer to use the Warburg effect to satisfy their energy demands regardless of the presence or absence of oxygen.
Metformin has been used to treat hyperglycemia and type 2 diabetes mellitus [169]. This drug mainly increases insulin sensitivity, suppresses hepatic glucose production, reduces glucose absorption from the intestines, and reduces the incidence of lipolysis in adipocytes [170, 171]. Metformin also has antineoplastic efficacy [172]. It can activate adenosine monophosphate-activated protein kinase (APMK), which maintains cellular energy homeostasis in response to various stimuli [173, 174]. In addition, sustained AMPK activation has been linked with apoptosis [175]. AMPK inhibits the expression of bone morphogenic protein and the activin membrane-bound inhibitor (Bambi), which can induce Wnt/β-catenin signaling pathway leading to cell proliferation and survival [173, 176]. Also, a positive feedback loop exists between Bambi and this pathway [173]. Taken together, these findings indicate that the Wnt/β-catenin signaling pathway has a relationship with the metabolicnetwork. Metabolic control can regulate the progression of tumors, which is accomplished by regulating the Wnt/β-catenin signaling pathway.
TARGETED APPLICATIONS OF β-CATENIN FOR CANCER TREATMENT
Researchers revealed that β-catenin was required for the development of triple-negative breast cancer that is estrogen-dependent [177]. In their study, they found that cell migration, colony formation, and epithelial-to-mesenchymal transition in vitro and tumorigenesis in vivo were regulated by β-catenin in triple-negative breast cancer. In addition, they proved that the transcriptional activity of β-catenin was quite important for the chemosensitivity of triple-negative breast cancer cells. So at least in part, β-catenin can be considered as one of therapeutic targets for this cancer.
Past studies have elaborated that antagonists of β-catenin or other proteins in the Wnt pathway could block this pathway and inhibit cell proliferation in many different tumor types [47, 95, 178–185]. Table 2 lists these antagonists and their possible mechanisms of action. For example, PKF115-584 is a small-molecule antagonist of the TCF/β-catenin complex [183]. It can decrease both cytoplasmic and nuclear β-catenin expression levels [186–188]. This antagonist downregulated the mRNA and protein expression levels for the critical proliferative genes of pancreatic neuroendocrine tumor cells [95]. Fiskus and colleagues (2015) used BC2059 to treat acute myeloid leukemia cells and found induction of apoptosis of the leukemia cells [47]. BC2059 is a small molecule that is considered to be an anthraquinone oxime analog. It can disrupt the binding of β-catenin to transducin β-like 1 and promote β-catenin degradation.
Table 2: The antagonists of β-catenin or other proteins in Wnt pathway and their possible mechanisms
Name |
Possible mechanisms |
|---|---|
PKF115-584183 |
Antagonist of the TCF/β-catenin complex Decreases both cytoplasmic and nuclear β-catenin expression |
BC205947 |
Anthraquinone oxime analog Disrupts the binding of β-catenin to transducin β-like 1; Promotes β-catenin degradation |
LF3180 |
Inhibits the β-catenin/TCF4 interaction Suppresses cell motility, cell-cycle progression, and the overexpression of Wnt target genes; Blocks the self-renewal capacity of cancer stem cells |
ICG-001181 |
Inhibits β-catenin/CREBBP interaction Downregulates Wnt target genes |
Salinomycin185 |
Small molecule inhibitor of LRP6 Downregulates Wnt target genes and cause cancer cell death |
Calphostin C182 |
Antagonist of the TCF/β-catenin complex Degrades β-catenin via a proteasome-dependent |
Xanthothricin182 |
Antagonist of the TCF/β-catenin complex Degrades β-catenin via a proteasome-dependent |
FH535184 |
Suppresses β-catenin /TCF-mediated transcription |
Besides the specific antagonists, hormones can influence the expression of β-catenin. For example, progesterone can counteract the accumulation of β-catenin in the nucleus and inhibit the Wnt/β-catenin signaling pathway [44, 108]. This hormone can also induce the expression of two inhibitors of the Wnt/β-catenin signaling pathway, DKK1 and FOXO1 [189–191]. DKK1 can inhibit Wnt/β-catenin signaling by binding to the Wnt co-receptors LRP5 and LRP6 [192]. For FOXO1, investigators found that it could inhibit this signaling by binding to β-catenin [191–193].
CONCLUSION
In summary, the important canonical Wnt/β-catenin signaling pathway exerts potent biological functions. As evidence, induction of tumorigenesis occurs when any step in this pathway is deregulated. β-catenin is the key intracellular transducer of this signaling pathway. Abnormal expression and mutations of β-catenin have been regarded as important signs of malignancy [6, 79]. Researchers have described and studied its mutations in many cancers, including colon cancer, melanoma, cervical carcinoma, endometrial cancer (EC), hepatoblastoma, and primitive neuroectodermal brain tumors. β-catenin mutations also can be considered to be the initiating factors for tumorigenesis and are positively related to poor survival, although the specific mechanism of this tumorigenesis deserves further study [8, 9, 112, 194]. Hence, we conclude that if we specifically inhibit the abnormal expression of β-catenin, cancers could be treated more effectively.
Another noteworthy problem about β-catenin is that some studies demonstrated that β-catenin activation alone could not induce spontaneous development of hepatocellular carcinoma in mouse models [195]. β-catenin`s role of tumor promotion requires the participation of other proteins, such as constitutive androstane receptor, which is a primary regulator of drug metabolism and detoxification. As a multifunctional protein, β-catenin can participate in a variety of physiological activities. As we described above, β-catenin can enhance the survival of Tregs and induce the secretion of immunosuppressive cytokines such as IL-10 and TGF-β. Treatments focusing on immunosuppression in the cancer microenvironment resulting from β-catenin should deserve more attention. The Warburg effect is well known to be one of the hallmarks of tumor cells. These cells prefer to use glycolysis rather than oxidative phosphorylation for producing ATP regardless of the sufficiency of oxygen. Once the Wnt/β-catenin signaling pathway is blocked, the production of some downstream metabolites of glycolysis, such as lactate, will decrease. Given this, we can conclude that the progression of tumors can be limited by regulating their glucose metabolism. β-catenin, therefore, seems to be an ideal therapeutic target for cancer. Besides the antagonists of β-catenin, hormones such as progesterone can also be of worth for cancer treatments. Certainly, more effective antagonists of β-catenin should be developed and applied in the clinic for cancer treatment. In the near future, early carcinogenesis may be initially inhibited by controlling abnormal β-catenin activity.
Abbreviations
APC, adenomatosis polyposis coli; CK-1, casein kinase-1α; PP2A, protein phosphatase 2A; GSK-3β, glycogen synthase kinase-3β; TCF/LEF, T-cell factor/lymphoid enhancer factor; CBP, CREB-binding protein; LRP, low-density lipoprotein receptor-related protein; PI3K, phosphoinositide 3-kinase; MAPK, mitogen-activated protein kinase; EEC, endometrioid endometrial carcinoma; NEEC, nonendometrioid endometrial carcinoma; miRNAs, microRNAs; DCs, dendritic cells; Tregs, regulatory T cells; TGF, transforming growth factor; IL, interleukin; APMK, adenosine monophosphate-activated protein kinase; Bambi, bone morphogenic protein and the activin membrane-bound inhibitor; EC, endometrial cancer; AEH, atypical endometrial hyperplasia; EOC, endometrioid ovarian cancer
Author contributions
All authors have read and approved the final version of the manuscript. CG drafted the manuscript and generated tables and figures. YMW, RB, LHS and FXX contributed to the writing and editing of the manuscript. WZ contributed to the writing of the manuscript and supervised the project.
ACKNOWLEDGMENTS
We would like to thank the Department of Scientific Publications at MD Anderson Cancer Center and M. Elizabeth Forbes and Mac Robinson from the Wake Forest Baptist Comprehensive Cancer Center for the English editing of this manuscript.
CONFLICTs OF INTEREST
The authors declare that they have no competing interest.
FUNDING
This work was supported in part by the NIH SPORE in Uterine Cancer NIH 2P50 CA098258. CG is supported by a fellowship from the China Education Council.
REFERENCES
1. Morin PJ. Beta-catenin signaling and cancer. Bioessays. 1999; 21:1021–30. https://doi.org/10.1002/(SICI)1521-1878(199912)22:1<1021::AID-BIES6>3.0.CO;2-P.
2. White BD, Chien AJ, Dawson DW. Dysregulation of Wnt/beta-catenin signaling in gastrointestinal cancers. Gastroenterology. 2012; 142:219–32. https://doi.org/10.1053/j.gastro.2011.12.001.
3. Robinson DR, Zylstra CR, Williams BO. Wnt signaling and prostate cancer. Curr Drug Targets. 2008; 9:571–80.
4. Mukherjee N, Bhattacharya N, Alam N, Roy A, Roychoudhury S, Panda CK. Subtype-specific alterations of the Wnt signaling pathway in breast cancer: clinical and prognostic significance. Cancer Sci. 2012; 103:210–20. https://doi.org/10.1111/j.1349-7006.2011.02131.x.
5. Gatcliffe TA, Monk BJ, Planutis K, Holcombe RF. Wnt signaling in ovarian tumorigenesis. Int J Gynecol Cancer. 2008; 18:954–62. https://doi.org/10.1111/j.1525-1438.2007.01127.x.
6. Polakis P. Wnt signaling in cancer. Cold Spring Harb Perspect Biol. 2012. https://doi.org/10.1101/cshperspect.a008052.
7. Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006; 127:469–80. https://doi.org/10.1016/j.cell.2006.10.018.
8. Saegusa M, Hashimura M, Yoshida T, Okayasu I. beta-Catenin mutations and aberrant nuclear expression during endometrial tumorigenesis. Br J Cancer. 2001; 84:209–17. https://doi.org/10.1054/bjoc.2000.1581.
9. Moreno-Bueno G, Hardisson D, Sanchez C, Sarrio D, Cassia R, Garcia-Rostan G, Prat J, Guo M, Herman JG, Matias-Guiu X, Esteller M, Palacios J. Abnormalities of the APC/beta-catenin pathway in endometrial cancer. Oncogene. 2002; 21:7981–90. https://doi.org/10.1038/sj.onc.1205924.
10. Nusse R, Varmus HE. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell. 1982; 31:99–109.
11. Cadigan KM, Nusse R. Wnt signaling: a common theme in animal development. Genes Dev. 1997; 11:3286–305.
12. Kuhl M, Sheldahl LC, Park M, Miller JR, Moon RT. The Wnt/Ca2+ pathway: a new vertebrate Wnt signaling pathway takes shape. Trends Genet. 2000; 16:279–83.
13. MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009; 17:9–26. https://doi.org/10.1016/j.devcel.2009.06.016.
14. Willert K, Brown JD, Danenberg E, Duncan AW, Weissman IL, Reya T, Yates JR 3rd, Nusse R. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature. 2003; 423:448–52. https://doi.org/10.1038/nature01611.
15. Hsieh JC, Rattner A, Smallwood PM, Nathans J. Biochemical characterization of Wnt-frizzled interactions using a soluble, biologically active vertebrate Wnt protein. Proc Natl Acad Sci U S A. 1999; 96:3546–51.
16. Dann CE, Hsieh JC, Rattner A, Sharma D, Nathans J, Leahy DJ. Insights into Wnt binding and signalling from the structures of two Frizzled cysteine-rich domains. Nature. 2001; 412:86–90. https://doi.org/10.1038/35083601.
17. Arend RC, Londono-Joshi AI, Straughn JM Jr, Buchsbaum DJ. The Wnt/beta-catenin pathway in ovarian cancer: a review. Gynecol Oncol. 2013; 131:772–9. https://doi.org/10.1016/j.ygyno.2013.09.034.
18. Semenov MV, Habas R, Macdonald BT, He X. SnapShot: noncanonical Wnt signaling pathways. Cell. 2007; 131:1378. https://doi.org/10.1016/j.cell.2007.12.011.
19. Rao TP, Kuhl M. An updated overview on Wnt signaling pathways: a prelude for more. Circ Res. 2010; 106:1798–806. https://doi.org/10.1161/CIRCRESAHA.110.219840.
20. Schulte G. International Union of Basic and Clinical Pharmacology. LXXX. The class Frizzled receptors. Pharmacol Rev. 2010; 62:632–67. https://doi.org/10.1124/pr.110.002931.
21. Chiurillo MA. Role of the Wnt/beta-catenin pathway in gastric cancer: an in-depth literature review. World J Exp Med. 2015; 5:84–102. https://doi.org/10.5493/wjem.v5.i2.84.
22. Kikuchi A, Yamamoto H, Sato A. Selective activation mechanisms of Wnt signaling pathways. Trends Cell Biol. 2009; 19:119–29. https://doi.org/10.1016/j.tcb.2009.01.003.
23. van Amerongen R, Nusse R. Towards an integrated view of Wnt signaling in development. Development. 2009; 136:3205–14. https://doi.org/10.1242/dev.033910.
24. Hart MJ, de los Santos R, Albert IN, Rubinfeld B, Polakis P. Downregulation of beta-catenin by human Axin and its association with the APC tumor suppressor, beta-catenin and GSK3 beta. Curr Biol. 1998; 8:573–81.
25. Behrens J, Jerchow BA, Wurtele M, Grimm J, Asbrand C, Wirtz R, Kuhl M, Wedlich D, Birchmeier W. Functional interaction of an axin homolog, conductin, with beta-catenin, APC, and GSK3beta. Science. 1998; 280:596–9.
26. Hsu W, Zeng L, Costantini F. Identification of a domain of Axin that binds to the serine/threonine protein phosphatase 2A and a self-binding domain. J Biol Chem. 1999; 274:3439–45.
27. Machin P, Catasus L, Pons C, Munoz J, Matias-Guiu X, Prat J. CTNNB1 mutations and beta-catenin expression in endometrial carcinomas. Hum Pathol. 2002; 33:206–12.
28. Ciechanover A. The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J. 1998; 17:7151–60. https://doi.org/10.1093/emboj/17.24.7151.
29. Zhao DM, Yu S, Zhou X, Haring JS, Held W, Badovinac VP, Harty JT, Xue HH. Constitutive activation of Wnt signaling favors generation of memory CD8 T cells. J Immunol. 2010; 184:1191–9. https://doi.org/10.4049/jimmunol.0901199.
30. Verbeek S, Izon D, Hofhuis F, Robanus-Maandag E, te Riele H, van de Wetering M, Oosterwegel M, Wilson A, MacDonald HR, Clevers H. An HMG-box-containing T-cell factor required for thymocyte differentiation. Nature. 1995; 374:70–4. https://doi.org/10.1038/374070a0.
31. Hurlstone A, Clevers H. T-cell factors: turn-ons and turn-offs. EMBO J. 2002; 21:2303–11. https://doi.org/10.1093/emboj/21.10.2303.
32. Dale TC. Signal transduction by the Wnt family of ligands. Biochem J. 1998; 329:209–23.
33. Howell BW, Herz J. The LDL receptor gene family: signaling functions during development. Curr Opin Neurobiol. 2001; 11:74–81.
34. Bhanot P, Brink M, Samos CH, Hsieh JC, Wang Y, Macke JP, Andrew D, Nathans J, Nusse R. A new member of the frizzled family from Drosophila functions as a Wingless receptor. Nature. 1996; 382:225–30. https://doi.org/10.1038/382225a0.
35. Wehrli M, Dougan ST, Caldwell K, O’Keefe L, Schwartz S, Vaizel-Ohayon D, Schejter E, Tomlinson A, DiNardo S. arrow encodes an LDL-receptor-related protein essential for Wingless signalling. Nature. 2000; 407:527–30. https://doi.org/10.1038/35035110.
36. Tolwinski NS, Wehrli M, Rives A, Erdeniz N, DiNardo S, Wieschaus E. Wg/Wnt signal can be transmitted through arrow/LRP5,6 and Axin independently of Zw3/Gsk3beta activity. Dev Cell. 2003; 4:407–18.
37. Mao J, Wang J, Liu B, Pan W, Farr GH 3rd, Flynn C, Yuan H, Takada S, Kimelman D, Li L, Wu D. Low-density lipoprotein receptor-related protein-5 binds to Axin and regulates the canonical Wnt signaling pathway. Mol Cell. 2001; 7:801–9.
38. Hendrickx M, Leyns L. Non-conventional Frizzled ligands and Wnt receptors. Dev Growth Differ. 2008; 50:229–43. https://doi.org/10.1111/j.1440-169X.2008.01016.x.
39. Kikuchi A, Yamamoto H, Sato A, Matsumoto S. New insights into the mechanism of Wnt signaling pathway activation. Int Rev Cell Mol Biol. 2011; 291:21–71. https://doi.org/10.1016/B978-0-12-386035-4.00002-1.
40. Niehrs C. The complex world of WNT receptor signalling. Nat Rev Mol Cell Biol. 2012; 13:767–79. https://doi.org/10.1038/nrm3470.
41. Kazanskaya O, Glinka A, del Barco Barrantes I, Stannek P, Niehrs C, Wu W. R-Spondin2 is a secreted activator of Wnt/beta-catenin signaling and is required for Xenopus myogenesis. Dev Cell. 2004; 7:525–34. https://doi.org/10.1016/j.devcel.2004.07.019.
42. Binnerts ME, Kim KA, Bright JM, Patel SM, Tran K, Zhou M, Leung JM, Liu Y, Lomas WE 3rd, Dixon M, Hazell SA, Wagle M, Nie WS, et al. R-Spondin1 regulates Wnt signaling by inhibiting internalization of LRP6. Proc Natl Acad Sci U S A. 2007; 104:14700–5. https://doi.org/10.1073/pnas.0702305104.
43. Wei Q, Yokota C, Semenov MV, Doble B, Woodgett J, He X. R-spondin1 is a high affinity ligand for LRP6 and induces LRP6 phosphorylation and beta-catenin signaling. J Biol Chem. 2007; 282:15903–11. https://doi.org/10.1074/jbc.M701927200.
44. Wang Y, Hanifi-Moghaddam P, Hanekamp EE, Kloosterboer HJ, Franken P, Veldscholte J, van Doorn HC, Ewing PC, Kim JJ, Grootegoed JA, Burger CW, Fodde R, Blok LJ. Progesterone inhibition of Wnt/beta-catenin signaling in normal endometrium and endometrial cancer. Clin Cancer Res. 2009; 15:5784–93. https://doi.org/10.1158/1078-0432.CCR-09-0814.
45. van der Zee M, Jia Y, Wang Y, Heijmans-Antonissen C, Ewing PC, Franken P, DeMayo FJ, Lydon JP, Burger CW, Fodde R, Blok LJ. Alterations in Wnt-beta-catenin and Pten signalling play distinct roles in endometrial cancer initiation and progression. J Pathol. 2013; 230:48–58. https://doi.org/10.1002/path.4160.
46. Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004; 20:781–810. https://doi.org/10.1146/annurev.cellbio.20.010403.113126.
47. Fiskus W, Sharma S, Saha S, Shah B, Devaraj SG, Sun B, Horrigan S, Leveque C, Zu Y, Iyer S, Bhalla KN. Pre-clinical efficacy of combined therapy with novel beta-catenin antagonist BC2059 and histone deacetylase inhibitor against AML cells. Leukemia. 2015; 29:1267–78. https://doi.org/10.1038/leu.2014.340.
48. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646–74. https://doi.org/10.1016/j.cell.2011.02.013.
49. Thiery JP. Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003; 15:740–6.
50. Xing Y, Takemaru K, Liu J, Berndt JD, Zheng JJ, Moon RT, Xu W. Crystal structure of a full-length beta-catenin. Structure. 2008; 16:478–87. https://doi.org/10.1016/j.str.2007.12.021.
51. Nollet F, Berx G, Molemans F, van Roy F. Genomic organization of the human beta-catenin gene (CTNNB1). Genomics. 1996; 32:413–24. https://doi.org/10.1006/geno.1996.0136.
52. Daniels DL, Eklof Spink K, Weis WI. Beta-catenin: molecular plasticity and drug design. Trends Biochem Sci. 2001; 26:672–8.
53. Peifer M, Berg S, Reynolds AB. A repeating amino acid motif shared by proteins with diverse cellular roles. Cell. 1994; 76:789–91.
54. Jiang J, Struhl G. Regulation of the Hedgehog and Wingless signalling pathways by the F-box/WD40-repeat protein Slimb. Nature. 1998; 391:493–6. https://doi.org/10.1038/35154.
55. Rimm DL, Koslov ER, Kebriaei P, Cianci CD, Morrow JS. Alpha 1(E)-catenin is an actin-binding and -bundling protein mediating the attachment of F-actin to the membrane adhesion complex. Proc Natl Acad Sci U S A. 1995; 92:8813–7.
56. Stadeli R, Hoffmans R, Basler K. Transcription under the control of nuclear Arm/beta-catenin. Curr Biol. 2006; 16: R378–85. https://doi.org/10.1016/j.cub.2006.04.019.
57. Kikuchi A. Tumor formation by genetic mutations in the components of the Wnt signaling pathway. Cancer Sci. 2003; 94:225–9.
58. Xu W, Kimelman D. Mechanistic insights from structural studies of beta-catenin and its binding partners. J Cell Sci. 2007; 120:3337–44. https://doi.org/10.1242/jcs.013771.
59. Kishida S, Yamamoto H, Ikeda S, Kishida M, Sakamoto I, Koyama S, Kikuchi A. Axin, a negative regulator of the wnt signaling pathway, directly interacts with adenomatous polyposis coli and regulates the stabilization of beta-catenin. J Biol Chem. 1998; 273:10823–6.
60. Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. Beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997; 16:3797–804. https://doi.org/10.1093/emboj/16.13.3797.
61. Castano J, Raurell I, Piedra JA, Miravet S, Dunach M, Garcia de Herreros A. Beta-catenin N- and C-terminal tails modulate the coordinated binding of adherens junction proteins to beta-catenin. J Biol Chem. 2002; 277:31541–50. https://doi.org/10.1074/jbc.M204376200.
62. Cox RT, Pai LM, Kirkpatrick C, Stein J, Peifer M. Roles of the C terminus of armadillo in wingless signaling in drosophila. Genetics. 1999; 153:319–32.
63. Piedra J, Martinez D, Castano J, Miravet S, Dunach M, de Herreros AG. Regulation of beta-catenin structure and activity by tyrosine phosphorylation. J Biol Chem. 2001; 276:20436–43. https://doi.org/10.1074/jbc.M100194200.
64. Solanas G, Miravet S, Casagolda D, Castano J, Raurell I, Corrionero A, de Herreros AG, Dunach M. beta-Catenin and plakoglobin N- and C-tails determine ligand specificity. J Biol Chem. 2004; 279:49849–56. https://doi.org/10.1074/jbc.M408685200.
65. Baum B, Georgiou M. Dynamics of adherens junctions in epithelial establishment, maintenance, and remodeling. J Cell Biol. 2011; 192:907–17. https://doi.org/10.1083/jcb.201009141.
66. Noubissi FK, Goswami S, Sanek NA, Kawakami K, Minamoto T, Moser A, Grinblat Y, Spiegelman VS. Wnt signaling stimulates transcriptional outcome of the Hedgehog pathway by stabilizing GLI1 mRNA. Cancer Res. 2009; 69:8572–8. https://doi.org/10.1158/0008-5472.CAN-09-1500.
67. Rosenbluh J, Nijhawan D, Cox AG, Li X, Neal JT, Schafer EJ, Zack TI, Wang X, Tsherniak A, Schinzel AC, Shao DD, Schumacher SE, Weir BA, et al. beta-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell. 2012; 151:1457–73. https://doi.org/10.1016/j.cell.2012.11.026.
68. Shimizu T, Kagawa T, Inoue T, Nonaka A, Takada S, Aburatani H, Taga T. Stabilized beta-catenin functions through TCF/LEF proteins and the Notch/RBP-Jkappa complex to promote proliferation and suppress differentiation of neural precursor cells. Mol Cell Biol. 2008; 28:7427–41. https://doi.org/10.1128/MCB.01962-07.
69. Tsai BP, Hoverter NP, Waterman ML. Blending hippo and WNT: sharing messengers and regulation. Cell. 2012; 151:1401–3. https://doi.org/10.1016/j.cell.2012.12.007.
70. Wijnhoven BP, Dinjens WN, Pignatelli M. E-cadherin-catenin cell-cell adhesion complex and human cancer. Br J Surg. 2000; 87:992–1005. https://doi.org/10.1046/j.1365-2168.2000.01513.x.
71. Conacci-Sorrell M, Zhurinsky J, Ben-Ze’ev A. The cadherin-catenin adhesion system in signaling and cancer. J Clin Invest. 2002; 109:987–91. https://doi.org/10.1172/JCI15429.
72. Kemler R. From cadherins to catenins: cytoplasmic protein interactions and regulation of cell adhesion. Trends Genet. 1993; 9:317–21.
73. Takeichi M. Cadherin cell adhesion receptors as a morphogenetic regulator. Science. 1991; 251:1451–5.
74. Fujimori T, Takeichi M. Disruption of epithelial cell-cell adhesion by exogenous expression of a mutated nonfunctional N-cadherin. Mol Biol Cell. 1993; 4:37–47.
75. Kintner C. Regulation of embryonic cell adhesion by the cadherin cytoplasmic domain. Cell. 1992; 69:225–36.
76. Howng SL, Wu CH, Cheng TS, Sy WD, Lin PC, Wang C, Hong YR. Differential expression of Wnt genes, beta-catenin and E-cadherin in human brain tumors. Cancer Lett. 2002; 183:95–101.
77. Yap AS, Brieher WM, Gumbiner BM. Molecular and functional analysis of cadherin-based adherens junctions. Annu Rev Cell Dev Biol. 1997; 13:119–46. https://doi.org/10.1146/annurev.cellbio.13.1.119.
78. Beyer C, Schramm A, Akhmetshina A, Dees C, Kireva T, Gelse K, Sonnylal S, de Crombrugghe B, Taketo MM, Distler O, Schett G, Distler JH. beta-catenin is a central mediator of pro-fibrotic Wnt signaling in systemic sclerosis. Ann Rheum Dis. 2012; 71:761–7. https://doi.org/10.1136/annrheumdis-2011-200568.
79. Xia J, Urabe K, Moroi Y, Koga T, Duan H, Li Y, Furue M. beta-Catenin mutation and its nuclear localization are confirmed to be frequent causes of Wnt signaling pathway activation in pilomatricomas. J Dermatol Sci. 2006; 41:67–75. https://doi.org/10.1016/j.jdermsci.2005.09.005.
80. Liu C, Li Y, Semenov M, Han C, Baeg GH, Tan Y, Zhang Z, Lin X, He X. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell. 2002; 108:837–47.
81. Yanagawa S, Matsuda Y, Lee JS, Matsubayashi H, Sese S, Kadowaki T, Ishimoto A. Casein kinase I phosphorylates the Armadillo protein and induces its degradation in Drosophila. EMBO J. 2002; 21:1733–42. https://doi.org/10.1093/emboj/21.7.1733.
82. Amit S, Hatzubai A, Birman Y, Andersen JS, Ben-Shushan E, Mann M, Ben-Neriah Y, Alkalay I. Axin-mediated CKI phosphorylation of beta-catenin at Ser 45: a molecular switch for the Wnt pathway. Genes Dev. 2002; 16:1066–76. https://doi.org/10.1101/gad.230302.
83. Lax SF. Molecular genetic pathways in various types of endometrial carcinoma: from a phenotypical to a molecular-based classification. Virchows Arch. 2004; 444:213–23. https://doi.org/10.1007/s00428-003-0947-3.
84. Dajani R, Fraser E, Roe SM, Yeo M, Good VM, Thompson V, Dale TC, Pearl LH. Structural basis for recruitment of glycogen synthase kinase 3beta to the axin-APC scaffold complex. EMBO J. 2003; 22:494–501. https://doi.org/10.1093/emboj/cdg068.
85. Rubinfeld B, Souza B, Albert I, Muller O, Chamberlain SH, Masiarz FR, Munemitsu S, Polakis P. Association of the APC gene product with beta-catenin. Science. 1993; 262:1731–4.
86. Wang Z, Vogelstein B, Kinzler KW. Phosphorylation of beta-catenin at S33, S37, or T41 can occur in the absence of phosphorylation at T45 in colon cancer cells. Cancer Res. 2003; 63:5234–5.
87. Rubinfeld B, Robbins P, El-Gamil M, Albert I, Porfiri E, Polakis P. Stabilization of beta-catenin by genetic defects in melanoma cell lines. Science. 1997; 275:1790–2.
88. Su TH, Chang JG, Yeh KT, Lin TH, Lee TP, Chen JC, Lin CC. Mutation analysis of CTNNB1 (beta-catenin) and AXIN1, the components of Wnt pathway, in cervical carcinomas. Oncol Rep. 2003; 10:1195–200.
89. Udatsu Y, Kusafuka T, Kuroda S, Miao J, Okada A. High frequency of beta-catenin mutations in hepatoblastoma. Pediatr Surg Int. 2001; 17:508–12.
90. Koch A, Waha A, Tonn JC, Sorensen N, Berthold F, Wolter M, Reifenberger J, Hartmann W, Friedl W, Reifenberger G, Wiestler OD, Pietsch T. Somatic mutations of WNT/wingless signaling pathway components in primitive neuroectodermal tumors. Int J Cancer. 2001; 93:445–9.
91. Semba S, Han SY, Ikeda H, Horii A. Frequent nuclear accumulation of beta-catenin in pituitary adenoma. Cancer. 2001; 91:42–8.
92. Lee CI, Hsu MY, Chou CH, Wang C, Lo YS, Loh JK, Howng SL, Hong YR. CTNNB1 (beta-catenin) mutation is rare in brain tumours but involved as a sporadic event in a brain metastasis. Acta Neurochir (Wien). 2009; 151:1107–11. https://doi.org/10.1007/s00701-009-0242-4.
93. Polakis P. Wnt signaling and cancer. Genes Dev. 2000; 14:1837–51.
94. Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science. 1997; 275:1787–90.
95. Jiang X, Cao Y, Li F, Su Y, Li Y, Peng Y, Cheng Y, Zhang C, Wang W, Ning G. Targeting beta-catenin signaling for therapeutic intervention in MEN1-deficient pancreatic neuroendocrine tumours. Nat Commun. 2014; 5:5809. https://doi.org/10.1038/ncomms6809.
96. Kitagawa M, Hatakeyama S, Shirane M, Matsumoto M, Ishida N, Hattori K, Nakamichi I, Kikuchi A, Nakayama K, Nakayama K. An F-box protein, FWD1, mediates ubiquitin-dependent proteolysis of beta-catenin. EMBO J. 1999; 18:2401–10. https://doi.org/10.1093/emboj/18.9.2401.
97. Enzo MV, Rastrelli M, Rossi CR, Hladnik U, Segat D. The Wnt/beta-catenin pathway in human fibrotic-like diseases and its eligibility as a therapeutic target. Mol Cell Ther. 2015; 3:1. https://doi.org/10.1186/s40591-015-0038-2.
98. Lam AP, Gottardi CJ. Beta-catenin signaling: a novel mediator of fibrosis and potential therapeutic target. Curr Opin Rheumatol. 2011; 23:562–7. https://doi.org/10.1097/BOR.0b013e32834b3309.
99. Hugh TJ, Dillon SA, Taylor BA, Pignatelli M, Poston GJ, Kinsella AR. Cadherin-catenin expression in primary colorectal cancer: a survival analysis. Br J Cancer. 1999; 80:1046–51. https://doi.org/10.1038/sj.bjc.6690461.
100. Fodde R, Smits R, Clevers H. APC, signal transduction and genetic instability in colorectal cancer. Nat Rev Cancer. 2001; 1:55–67. https://doi.org/10.1038/35094067.
101. Molenaar M, van de Wetering M, Oosterwegel M, Peterson-Maduro J, Godsave S, Korinek V, Roose J, Destree O, Clevers H. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell. 1996; 86:391–9.
102. Huber O, Korn R, McLaughlin J, Ohsugi M, Herrmann BG, Kemler R. Nuclear localization of beta-catenin by interaction with transcription factor LEF-1. Mech Dev. 1996; 59:3–10.
103. Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 1996; 382:638–42. https://doi.org/10.1038/382638a0.
104. Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008; 8:387–98. https://doi.org/10.1038/nrc2389.
105. Barker N, Morin PJ, Clevers H. The Yin-Yang of TCF/beta-catenin signaling. Adv Cancer Res. 2000; 77:1–24.
106. Fukuchi T, Sakamoto M, Tsuda H, Maruyama K, Nozawa S, Hirohashi S. Beta-catenin mutation in carcinoma of the uterine endometrium. Cancer Res. 1998; 58:3526–8.
107. Amant F, Moerman P, Neven P, Timmerman D, Van Limbergen E, Vergote I. Endometrial cancer. Lancet. 2005; 366:491–505. https://doi.org/10.1016/S0140-6736(05)67063-8.
108. Nei H, Saito T, Yamasaki H, Mizumoto H, Ito E, Kudo R. Nuclear localization of beta-catenin in normal and carcinogenic endometrium. Mol Carcinog. 1999; 25:207–18.
109. Byron SA, Gartside M, Powell MA, Wellens CL, Gao F, Mutch DG, Goodfellow PJ, Pollock PM. FGFR2 point mutations in 466 endometrioid endometrial tumors: relationship with MSI, KRAS, PIK3CA, CTNNB1 mutations and clinicopathological features. PLoS One. 2012; 7: e30801. https://doi.org/10.1371/journal.pone.0030801.
110. Moreno-Bueno G, Hardisson D, Sarrio D, Sanchez C, Cassia R, Prat J, Herman JG, Esteller M, Matias-Guiu X, Palacios J. Abnormalities of E- and P-cadherin and catenin (beta-, gamma-catenin, and p120ctn) expression in endometrial cancer and endometrial atypical hyperplasia. J Pathol. 2003; 199:471–8. https://doi.org/10.1002/path.1310.
111. Palacios J, Gamallo C. Mutations in the beta-catenin gene (CTNNB1) in endometrioid ovarian carcinomas. Cancer Res. 1998; 58:1344–7.
112. Gamallo C, Palacios J, Moreno G, Calvo de Mora J, Suarez A, Armas A. Beta-catenin expression pattern in stage I and II ovarian carcinomas : relationship with beta-catenin gene mutations, clinicopathological features, and clinical outcome. Am J Pathol. 1999; 155:527–36.
113. Wright K, Wilson P, Morland S, Campbell I, Walsh M, Hurst T, Ward B, Cummings M, Chenevix-Trench G. Beta-catenin mutation and expression analysis in ovarian cancer: exon 3 mutations and nuclear translocation in 16% of endometrioid tumours. Int J Cancer. 1999; 82:625–9.
114. Wu R, Zhai Y, Fearon ER, Cho KR. Diverse mechanisms of beta-catenin deregulation in ovarian endometrioid adenocarcinomas. Cancer Res. 2001; 61:8247–55.
115. Saegusa M, Okayasu I. Frequent nuclear beta-catenin accumulation and associated mutations in endometrioid-type endometrial and ovarian carcinomas with squamous differentiation. J Pathol. 2001; 194:59–67. https://doi.org/10.1002/path.856.
116. Moreno-Bueno G, Gamallo C, Perez-Gallego L, de Mora JC, Suarez A, Palacios J. Beta-Catenin expression pattern, beta-catenin gene mutations, and microsatellite instability in endometrioid ovarian carcinomas and synchronous endometrial carcinomas. Diagn Mol Pathol. 2001; 10:116–22.
117. Liu Y, Patel L, Mills GB, Lu KH, Sood AK, Ding L, Kucherlapati R, Mardis ER, Levine DA, Shmulevich I, Broaddus RR, Zhang W. Clinical significance of CTNNB1 mutation and Wnt pathway activation in endometrioid endometrial carcinoma. J Natl Cancer Inst. 2014. https://doi.org/10.1093/jnci/dju245.
118. Liu W, Dong X, Mai M, Seelan RS, Taniguchi K, Krishnadath KK, Halling KC, Cunningham JM, Boardman LA, Qian C, Christensen E, Schmidt SS, Roche PC, et al. Mutations in AXIN2 cause colorectal cancer with defective mismatch repair by activating beta-catenin/TCF signalling. Nat Genet. 2000; 26:146–7. https://doi.org/10.1038/79859.
119. Satoh S, Daigo Y, Furukawa Y, Kato T, Miwa N, Nishiwaki T, Kawasoe T, Ishiguro H, Fujita M, Tokino T, Sasaki Y, Imaoka S, Murata M, et al. AXIN1 mutations in hepatocellular carcinomas, and growth suppression in cancer cells by virus-mediated transfer of AXIN1. Nat Genet. 2000; 24:245–50. https://doi.org/10.1038/73448.
120. Dahmen RP, Koch A, Denkhaus D, Tonn JC, Sorensen N, Berthold F, Behrens J, Birchmeier W, Wiestler OD, Pietsch T. Deletions of AXIN1, a component of the WNT/wingless pathway, in sporadic medulloblastomas. Cancer Res. 2001; 61:7039–43.
121. Morgan RG, Pearn L, Liddiard K, Pumford SL, Burnett AK, Tonks A, Darley RL. Gamma-Catenin is overexpressed in acute myeloid leukemia and promotes the stabilization and nuclear localization of beta-catenin. Leukemia. 2013; 27:336–43. https://doi.org/10.1038/leu.2012.221.
122. Desbois-Mouthon C, Cadoret A, Blivet-Van Eggelpoel MJ, Bertrand F, Cherqui G, Perret C, Capeau J. Insulin and IGF-1 stimulate the beta-catenin pathway through two signalling cascades involving GSK-3beta inhibition and Ras activation. Oncogene. 2001; 20:252–9. https://doi.org/10.1038/sj.onc.1204064.
123. Satyamoorthy K, Li G, Vaidya B, Patel D, Herlyn M. Insulin-like growth factor-1 induces survival and growth of biologically early melanoma cells through both the mitogen-activated protein kinase and beta-catenin pathways. Cancer Res. 2001; 61:7318–24.
124. Ormanns S, Neumann J, Horst D, Kirchner T, Jung A. WNT signaling and distant metastasis in colon cancer through transcriptional activity of nuclear beta-Catenin depend on active PI3K signaling. Oncotarget. 2014; 5:2999–3011. https://doi.org/10.18632/oncotarget.1626.
125. Fang D, Hawke D, Zheng Y, Xia Y, Meisenhelder J, Nika H, Mills GB, Kobayashi R, Hunter T, Lu Z. Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J Biol Chem. 2007; 282:11221–9. https://doi.org/10.1074/jbc.M611871200.
126. Baryawno N, Sveinbjornsson B, Eksborg S, Chen CS, Kogner P, Johnsen JI. Small-molecule inhibitors of phosphatidylinositol 3-kinase/Akt signaling inhibit Wnt/beta-catenin pathway cross-talk and suppress medulloblastoma growth. Cancer Res. 2010; 70:266–76. https://doi.org/10.1158/0008-5472.CAN-09-0578.
127. Vermeulen L, De Sousa EM, van der Heijden M, Cameron K, de Jong JH, Borovski T, Tuynman JB, Todaro M, Merz C, Rodermond H, Sprick MR, Kemper K, Richel DJ, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2010; 12:468–76. https://doi.org/10.1038/ncb2048.
128. Phelps RA, Chidester S, Dehghanizadeh S, Phelps J, Sandoval IT, Rai K, Broadbent T, Sarkar S, Burt RW, Jones DA. A two-step model for colon adenoma initiation and progression caused by APC loss. Cell. 2009; 137:623–34. https://doi.org/10.1016/j.cell.2009.02.037.
129. Horst D, Chen J, Morikawa T, Ogino S, Kirchner T, Shivdasani RA. Differential WNT activity in colorectal cancer confers limited tumorigenic potential and is regulated by MAPK signaling. Cancer Res. 2012; 72:1547–56. https://doi.org/10.1158/0008-5472.CAN-11-3222.
130. Janssen KP, Alberici P, Fsihi H, Gaspar C, Breukel C, Franken P, Rosty C, Abal M, El Marjou F, Smits R, Louvard D, Fodde R, Robine S. APC and oncogenic KRAS are synergistic in enhancing Wnt signaling in intestinal tumor formation and progression. Gastroenterology. 2006; 131:1096–109. https://doi.org/10.1053/j.gastro.2006.08.011.
131. Kim NH, Kim HS, Kim NG, Lee I, Choi HS, Li XY, Kang SE, Cha SY, Ryu JK, Na JM, Park C, Kim K, Lee S, et al. p53 and microRNA-34 are suppressors of canonical Wnt signaling. Sci Signal. 2011; 4:ra71. https://doi.org/10.1126/scisignal.2001744.
132. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004; 116:281–97.
133. Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol. 2005; 6:376–85. https://doi.org/10.1038/nrm1644.
134. Peng Z, Wu T, Li Y, Xu Z, Zhang S, Liu B, Chen Q, Tian D. MicroRNA-370-3p inhibits human glioma cell proliferation and induces cell cycle arrest by directly targeting beta-catenin. Brain Res. 2016; 1644:53–61. https://doi.org/10.1016/j.brainres.2016.04.066.
135. Anton R, Chatterjee SS, Simundza J, Cowin P, Dasgupta R. A systematic screen for micro-RNAs regulating the canonical Wnt pathway. PLoS One. 2011; 6:e26257. https://doi.org/10.1371/journal.pone.0026257.
136. Fujiwara H. [Adoptive immunotherapy using gene-modified T cells to express target-specific receptors against hematological malignancy]. [Article in Japanese]. Rinsho Ketsueki. 2014; 55:2162–74.
137. Hao MZ, Zhou WY, Du XL, Chen KX, Wang GW, Yang Y, Yang JL. Novel anti-melanoma treatment: focus on immunotherapy. Chin J Cancer. 2014; 33:458–65. https://doi.org/10.5732/cjc.014.10118.
138. Yaguchi T, Goto Y, Kido K, Mochimaru H, Sakurai T, Tsukamoto N, Kudo-Saito C, Fujita T, Sumimoto H, Kawakami Y. Immune suppression and resistance mediated by constitutive activation of Wnt/beta-catenin signaling in human melanoma cells. J Immunol. 2012; 189:2110–7. https://doi.org/10.4049/jimmunol.1102282.
139. Cormier JN, Salgaller ML, Prevette T, Barracchini KC, Rivoltini L, Restifo NP, Rosenberg SA, Marincola FM. Enhancement of cellular immunity in melanoma patients immunized with a peptide from MART-1/Melan A. Cancer J Sci Am. 1997; 3:37–44.
140. Yaguchi T, Sumimoto H, Kudo-Saito C, Tsukamoto N, Ueda R, Iwata-Kajihara T, Nishio H, Kawamura N, Kawakami Y. The mechanisms of cancer immunoescape and development of overcoming strategies. Int J Hematol. 2011; 93:294–300. https://doi.org/10.1007/s12185-011-0799-6.
141. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. 2015; 523:231–5. https://doi.org/10.1038/nature14404.
142. Ding Y, Shen S, Lino AC, Curotto de Lafaille MA, Lafaille JJ. Beta-catenin stabilization extends regulatory T cell survival and induces anergy in nonregulatory T cells. Nat Med. 2008; 14:162–9. https://doi.org/10.1038/nm1707.
143. Gajewski TF, Fuertes M, Spaapen R, Zheng Y, Kline J. Molecular profiling to identify relevant immune resistance mechanisms in the tumor microenvironment. Curr Opin Immunol. 2011; 23:286–92. https://doi.org/10.1016/j.coi.2010.11.013.
144. Vanderstraeten A, Luyten C, Verbist G, Tuyaerts S, Amant F. Mapping the immunosuppressive environment in uterine tumors: implications for immunotherapy. Cancer Immunol Immunother. 2014; 63:545–57. https://doi.org/10.1007/s00262-014-1537-8.
145. Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005; 5:263–74. https://doi.org/10.1038/nrc1586.
146. Yamagami W, Susumu N, Tanaka H, Hirasawa A, Banno K, Suzuki N, Tsuda H, Tsukazaki K, Aoki D. Immunofluorescence-detected infiltration of CD4+FOXP3+ regulatory T cells is relevant to the prognosis of patients with endometrial cancer. Int J Gynecol Cancer. 2011; 21:1628–34. https://doi.org/10.1097/IGC.0b013e31822c271f.
147. Curotto de Lafaille MA, Lafaille JJ. CD4(+) regulatory T cells in autoimmunity and allergy. Curr Opin Immunol. 2002; 14:771–8.
148. Gavin M, Rudensky A. Control of immune homeostasis by naturally arising regulatory CD4+ T cells. Curr Opin Immunol. 2003; 15:690–6.
149. Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol. 2005; 6:345–52. https://doi.org/10.1038/ni1178.
150. Bluestone JA, Abbas AK. Natural versus adaptive regulatory T cells. Nat Rev Immunol. 2003; 3:253–7. https://doi.org/10.1038/nri1032.
151. Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer’s Achilles’ heel. Cancer Cell. 2008; 13:472–82. https://doi.org/10.1016/j.ccr.2008.05.005.
152. Masui K, Tanaka K, Ikegami S, Villa GR, Yang H, Yong WH, Cloughesy TF, Yamagata K, Arai N, Cavenee WK, Mischel PS. Glucose-dependent acetylation of Rictor promotes targeted cancer therapy resistance. Proc Natl Acad Sci U S A. 2015; 112:9406–11. https://doi.org/10.1073/pnas.1511759112.
153. Chafey P, Finzi L, Boisgard R, Cauzac M, Clary G, Broussard C, Pegorier JP, Guillonneau F, Mayeux P, Camoin L, Tavitian B, Colnot S, Perret C. Proteomic analysis of beta-catenin activation in mouse liver by DIGE analysis identifies glucose metabolism as a new target of the Wnt pathway. Proteomics. 2009; 9:3889–900. https://doi.org/10.1002/pmic.200800609.
154. Warburg O. On the origin of cancer cells. Science. 1956; 123:309–14.
155. Shaw RJ. Glucose metabolism and cancer. Curr Opin Cell Biol. 2006; 18:598–608. https://doi.org/10.1016/j.ceb.2006.10.005.
156. Gillies RJ, Robey I, Gatenby RA. Causes and consequences of increased glucose metabolism of cancers. J Nucl Med. 2008; 49: 24S–42S. https://doi.org/10.2967/jnumed.107.047258.
157. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008; 7:11–20. https://doi.org/10.1016/j.cmet.2007.10.002.
158. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009; 324:1029–33. https://doi.org/10.1126/science.1160809.
159. Zhang S, Yang C, Yang Z, Zhang D, Ma X, Mills G, Liu Z. Homeostasis of redox status derived from glucose metabolic pathway could be the key to understanding the Warburg effect. Am J Cancer Res. 2015; 5:1265–80.
160. Brand KA, Hermfisse U. Aerobic glycolysis by proliferating cells: a protective strategy against reactive oxygen species. FASEB J. 1997; 11:388–95.
161. Chen Z, Odstrcil EA, Tu BP, McKnight SL. Restriction of DNA replication to the reductive phase of the metabolic cycle protects genome integrity. Science. 2007; 316:1916–9. https://doi.org/10.1126/science.1140958.
162. Zhang S, Yang C, Yang Z, Zhang D, Ma X, Mills G, Liu Z. Homeostasis of redox status derived from glucose metabolic pathway could be the key to understanding the Warburg effect. Am J Cancer Res. 2015; 5:928–44.
163. Benhamouche S, Decaens T, Godard C, Chambrey R, Rickman DS, Moinard C, Vasseur-Cognet M, Kuo CJ, Kahn A, Perret C, Colnot S. Apc tumor suppressor gene is the ‘zonation-keeper’ of mouse liver. Dev Cell. 2006; 10:759–70. https://doi.org/10.1016/j.devcel.2006.03.015.
164. Lee SY, Jeon HM, Ju MK, Kim CH, Yoon G, Han SI, Park HG, Kang HS. Wnt/Snail signaling regulates cytochrome C oxidase and glucose metabolism. Cancer Res. 2012; 72:3607–17. https://doi.org/10.1158/0008-5472.CAN-12-0006.
165. Esen E, Chen J, Karner CM, Okunade AL, Patterson BW, Long F. WNT-LRP5 signaling induces Warburg effect through mTORC2 activation during osteoblast differentiation. Cell Metab. 2013; 17:745–55. https://doi.org/10.1016/j.cmet.2013.03.017.
166. Pate KT, Stringari C, Sprowl-Tanio S, Wang K, TeSlaa T, Hoverter NP, McQuade MM, Garner C, Digman MA, Teitell MA, Edwards RA, Gratton E, Waterman ML. Wnt signaling directs a metabolic program of glycolysis and angiogenesis in colon cancer. EMBO J. 2014; 33:1454–73. https://doi.org/10.15252/embj.201488598.
167. Roche TE, Baker JC, Yan X, Hiromasa Y, Gong X, Peng T, Dong J, Turkan A, Kasten SA. Distinct regulatory properties of pyruvate dehydrogenase kinase and phosphatase isoforms. Prog Nucleic Acid Res Mol Biol. 2001; 70:33–75.
168. Yang W, Xia Y, Ji H, Zheng Y, Liang J, Huang W, Gao X, Aldape K, Lu Z. Nuclear PKM2 regulates beta-catenin transactivation upon EGFR activation. Nature. 2011; 480:118–22. https://doi.org/10.1038/nature10598.
169. Pierotti MA, Berrino F, Gariboldi M, Melani C, Mogavero A, Negri T, Pasanisi P, Pilotti S. Targeting metabolism for cancer treatment and prevention: metformin, an old drug with multi-faceted effects. Oncogene. 2013; 32:1475–87. https://doi.org/10.1038/onc.2012.181.
170. Giannarelli R, Aragona M, Coppelli A, Del Prato S. Reducing insulin resistance with metformin: the evidence today. Diabetes Metab. 2003; 29: 6S28–35.
171. Detaille D, Guigas B, Leverve X, Wiernsperger N, Devos P. Obligatory role of membrane events in the regulatory effect of metformin on the respiratory chain function. Biochem Pharmacol. 2002; 63:1259–72.
172. Giovannucci E, Harlan DM, Archer MC, Bergenstal RM, Gapstur SM, Habel LA, Pollak M, Regensteiner JG, Yee D. Diabetes and cancer: a consensus report. Diabetes Care. 2010; 33:1674–85. https://doi.org/10.2337/dc10-0666.
173. Subramaniam N, Sherman MH, Rao R, Wilson C, Coulter S, Atkins AR, Evans RM, Liddle C, Downes M. Metformin-mediated Bambi expression in hepatic stellate cells induces prosurvival Wnt/beta-catenin signaling. Cancer Prev Res (Phila). 2012; 5:553–61. https://doi.org/10.1158/1940-6207.CAPR-12-0053.
174. Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 2011; 25:1895–908. https://doi.org/10.1101/gad.17420111.
175. Canto C, Auwerx J. AMP-activated protein kinase and its downstream transcriptional pathways. Cell Mol Life Sci. 2010; 67:3407–23. https://doi.org/10.1007/s00018-010-0454-z.
176. Shin HW, Park SY, Lee KB, Jang JJ. Down-regulation of Wnt signaling during apoptosis of human hepatic stellate cells. Hepatogastroenterology. 2009; 56:208–12.
177. Xu J, Prosperi JR, Choudhury N, Olopade OI, Goss KH. Beta-Catenin is required for the tumorigenic behavior of triple-negative breast cancer cells. PLoS One. 2015; 10:e0117097. https://doi.org/10.1371/journal.pone.0117097.
178. Ashihara E, Takada T, Maekawa T. Targeting the canonical Wnt/beta-catenin pathway in hematological malignancies. Cancer Sci. 2015; 106:665–71. https://doi.org/10.1111/cas.12655.
179. Sebio A, Kahn M, Lenz HJ. The potential of targeting Wnt/beta-catenin in colon cancer. Expert Opin Ther Targets. 2014; 18:611–5. https://doi.org/10.1517/14728222.2014.906580.
180. Fang L, Zhu Q, Neuenschwander M, Specker E, Wulf-Goldenberg A, Weis WI, von Kries JP, Birchmeier W. A small-molecule antagonist of the beta-catenin/TCF4 interaction blocks the self-renewal of cancer stem cells and suppresses tumorigenesis. Cancer Res. 2016; 76:891–901. https://doi.org/10.1158/0008-5472.CAN-15-1519.
181. Gaddis M, Gerrard D, Frietze S, Farnham PJ. Altering cancer transcriptomes using epigenomic inhibitors. Epigenetics Chromatin. 2015; 8:9. https://doi.org/10.1186/1756-8935-8-9.
182. Zeller J, Turbiak AJ, Powelson IA, Lee S, Sun D, Showalter HD, Fearon ER. Investigation of 3-aryl-pyrimido[5,4-e][1, 2, 4]triazine-5,7-diones as small molecule antagonists of beta-catenin/TCF transcription. Bioorg Med Chem Lett. 2013; 23:5814–20. https://doi.org/10.1016/j.bmcl.2013.08.111.
183. Lepourcelet M, Chen YN, France DS, Wang H, Crews P, Petersen F, Bruseo C, Wood AW, Shivdasani RA. Small-molecule antagonists of the oncogenic Tcf/beta-catenin protein complex. Cancer Cell. 2004; 5:91–102.
184. Handeli S, Simon JA. A small-molecule inhibitor of Tcf/beta-catenin signaling down-regulates PPARgamma and PPARdelta activities. Mol Cancer Ther. 2008; 7:521–9. https://doi.org/10.1158/1535-7163.MCT-07-2063.
185. Lu W, Li Y. Salinomycin suppresses LRP6 expression and inhibits both Wnt/beta-catenin and mTORC1 signaling in breast and prostate cancer cells. J Cell Biochem. 2014; 115:1799–807. https://doi.org/10.1002/jcb.24850.
186. Jansen SR, Van Ziel AM, Baarsma HA, Gosens R. {beta}-Catenin regulates airway smooth muscle contraction. Am J Physiol Lung Cell Mol Physiol. 2010; 299:L204–14. https://doi.org/10.1152/ajplung.00020.2010.
187. Gandhirajan RK, Staib PA, Minke K, Gehrke I, Plickert G, Schlosser A, Schmitt EK, Hallek M, Kreuzer KA. Small molecule inhibitors of Wnt/beta-catenin/lef-1 signaling induces apoptosis in chronic lymphocytic leukemia cells in vitro and in vivo. Neoplasia. 2010; 12:326–35.
188. Sinnberg T, Menzel M, Ewerth D, Sauer B, Schwarz M, Schaller M, Garbe C, Schittek B. Beta-catenin signaling increases during melanoma progression and promotes tumor cell survival and chemoresistance. PLoS One. 2011; 6:e23429. https://doi.org/10.1371/journal.pone.0023429.
189. Niehrs C. Function and biological roles of the Dickkopf family of Wnt modulators. Oncogene. 2006; 25:7469–81. https://doi.org/10.1038/sj.onc.1210054.
190. Takano M, Lu Z, Goto T, Fusi L, Higham J, Francis J, Withey A, Hardt J, Cloke B, Stavropoulou AV, Ishihara O, Lam EW, Unterman TG, et al. Transcriptional cross talk between the forkhead transcription factor forkhead box O1A and the progesterone receptor coordinates cell cycle regulation and differentiation in human endometrial stromal cells. Mol Endocrinol. 2007; 21:2334–49. https://doi.org/10.1210/me.2007-0058.
191. Almeida M, Han L, Martin-Millan M, O’Brien CA, Manolagas SC. Oxidative stress antagonizes Wnt signaling in osteoblast precursors by diverting beta-catenin from T cell factor- to forkhead box O-mediated transcription. J Biol Chem. 2007; 282:27298–305. https://doi.org/10.1074/jbc.M702811200.
192. Wang K, Zhang Y, Li X, Chen L, Wang H, Wu J, Zheng J, Wu D. Characterization of the Kremen-binding site on Dkk1 and elucidation of the role of Kremen in Dkk-mediated Wnt antagonism. J Biol Chem. 2008; 283:23371–5. https://doi.org/10.1074/jbc.M802376200.
193. Essers MA, de Vries-Smits LM, Barker N, Polderman PE, Burgering BM, Korswagen HC. Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science. 2005; 308:1181–4. https://doi.org/10.1126/science.1109083.
194. Ikeda T, Yoshinaga K, Semba S, Kondo E, Ohmori H, Horii A. Mutational analysis of the CTNNB1 (beta-catenin) gene in human endometrial cancer: frequent mutations at codon 34 that cause nuclear accumulation. Oncol Rep. 2000; 7:323–6.
195. Dong B, Lee JS, Park YY, Yang F, Xu G, Huang W, Finegold MJ, Moore DD. Activating CAR and beta-catenin induces uncontrolled liver growth and tumorigenesis. Nat Commun. 2015; 6:5944. https://doi.org/10.1038/ncomms6944.