
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is among the most lethal malignancies in the US [1]. PDAC is currently the third most frequent cause of cancer deaths in the US and is expected to be the second deadliest cancer by 2030 [1, 2]. The 5-year survival rate in PDAC patients remains about 6% and the poor prognosis is attributed to several factors including late-stage diagnosis, an aggressive progression of the disease and high resistance to conventional therapies. Surgery remains the best option for the long-term survival of PDAC patients, however, only about 20% of patients are suitable for this procedure [3]. Furthermore, post-operative recurrence remains very common, and even after complete resection, the 5-year survival is only around 25% [1]. Therefore, improving systemic treatment strategies is highly desirable for PDAC patients. Gemcitabine (Gem) remained a standard therapy for PDAC for more than 16-years despite only a minimal clinical benefit [4]. FOLFIRINOX is a combination chemotherapy regimen that increased the median survival of unresectable PDAC patients to approximately 11 months. However, this regimen has a high toxicity potential, limiting its use to only patients with good performance status [5]. Nab-paclitaxel (NPT) is a next-generation taxane that has demonstrated significant antitumor efficacy in several solid tumors. Nab-paclitaxel is an approved treatment for breast cancer, NSCLC and PDAC [6]. Nab-paclitaxel combined with gemcitabine has recently become the standard treatment for unresectable PDAC after demonstrating a 1.8 months improvement in patient survival compared to gemcitabine monotherapy [7]. Due to the limited clinical efficacy of current cytotoxic chemotherapy regimens for PDAC patients, novel therapeutic approaches are urgently needed to further improve patient survival.
Activation of ERK signaling due to mutations in the RAS/RAF/MEK/ERK (MAPK) pathway has been implicated in several cancers [8]. In PDAC, activating KRAS mutations occur at a frequency of 90% [9], rendering this a potential therapeutic target of great interest. Developing drugs that directly target mutant KRAS protein remains challenging due to target specificity issues [10]. Therefore, alternative strategies focus on inhibition of downstream targets of the RAS-MAPK cascade. MEK is one such protein kinase located downstream of RAS/RAF, making it an attractive target for cancer therapy [11]. Trametinib (Tra, Supplementary Figure 1), a selective and reversible small molecule inhibitor of MEK1/2 kinase activity, has demonstrated antitumor efficacy in preclinical studies of several tumor types, with the largest effect in tumors harboring mutant BRAF or Ras [12]. Furthermore, trametinib is an FDA-approved treatment for V600E-mutant metastatic melanoma patients as a single agent or in combination with dabrafenib [13, 14]. In a preclinical study with patient-derived xenografts of PDAC, trametinib showed significant antitumor effects [15]. In addition, the first clinical study of trametinib monotherapy in PDAC demonstrated potential activity [16]. In a recent phase II clinical trial in PDAC patients, trametinib combination with gemcitabine demonstrated a 1.7 months increase in median overall survival (OS) compared with the gemcitabine alone. However, this difference in OS was evaluated as non-significant as the observed separation was not durable with a hazard ratio of 0.98 [17]. Furthermore, in a phase II clinical study in KRAS mutant NSCLC patients, selumetinib, another MEK inhibitor, plus docetaxel showed significant improvements in response rate and progression-free survival (PFS) [18], indicating differences in the synergy of MEK inhibitors with gemcitabine compared with taxanes. In this study, we report the antitumor efficacy of trametinib in combination with nab-paclitaxel-based chemotherapy regimens in preclinical models of pancreatic cancer.
RESULTS
Nab-paclitaxel-based chemotherapy regimens and trametinib reduce tumor growth
In an AsPC-1 subcutaneous xenograft model, nab-paclitaxel alone, nab-paclitaxel plus gemcitabine and trametinib alone caused an inhibition in tumor growth while trametinib combination with chemotherapy regimens had a trend for additive effects (Figure 1A). Net tumor growth in different groups after 2-weeks of therapy was 432.6 mm3 in controls, 105.3 mm3 after NPT, 184 mm3 after Tra, 81 mm3 after NPT+Tra, 37.3 mm3 after NPT+Gem and –8.1 mm3 (tumor regression) after NPT+Gem+Tra (Figure 1B). Tumor weight at the completion of the experiment in different groups was 0.38 g in controls, 0.23 g in NPT, 0.31 g in Tra, 0.23 g in NPT+Tra, 0.15 g in NPT+Gem and 0.11 g in NPT+Gem+Tra (Figure 1C). In another Panc-1 subcutaneous xenograft model, nab-paclitaxel, nab-paclitaxel plus gemcitabine and trametinib therapy also caused an inhibition in tumor growth rates with a trend for additive effects in combination groups (Figure 2A). Net tumor growth in different groups was 274.1 mm3 in controls, 80.8 mm3 after NPT, 150.6 mm3 after Tra, 75.1 mm3 after NPT+Tra, 48.4 mm3 after NPT+Gem and 3.8 mm3 after NPT+Gem+Tra (Figure 2B). Tumor weight at the completion of the experiment in different groups was: 0.37 g in controls, 0.15 g in NPT, 0.18 g in Tra, 0.13 g in NPT+Tra, 0.09 g in NPT+Gem and 0.077 g in NPT+Gem+Tra (Figure 2C). Also, in these two subcutaneous xenograft experiments, no disenable therapy-related toxicity was observed during the therapy period and there was no significant change in the body weight of mice in all groups (Figures 1D, 2D).
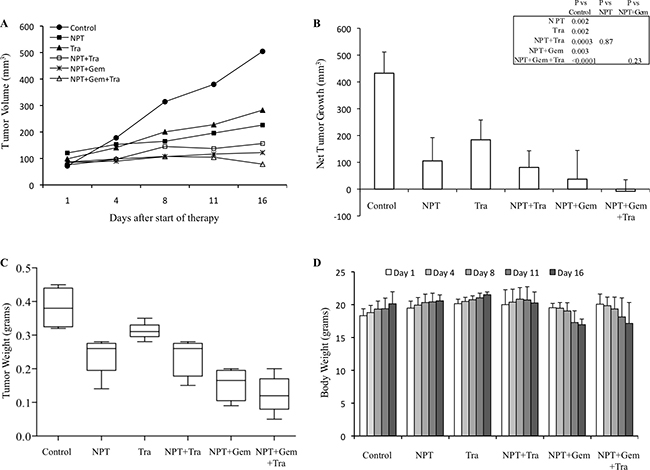
Figure 1: Antitumor activity of nab-paclitaxel-based chemotherapy regimens and trametinib in AsPC-1 tumor xenografts. AsPC-1 cells were subcutaneously injected into nude mice and treated with nab-paclitaxel-based chemotherapy regimens and trametinib for 2 weeks. (A) Tumor size as measured twice-weekly using calipers. (B) Net tumor growth, calculated by subtracting tumor volume on the first treatment day from that on the final day. (C) Mean tumor weight was calculated from final day tumor weights in each group, presented as a Box plot. (D) Mouse body weight was measured twice a week and presented as bar chart for the 2-week therapy period. Data are representative of mean values ± standard deviation from 5 mice per group.
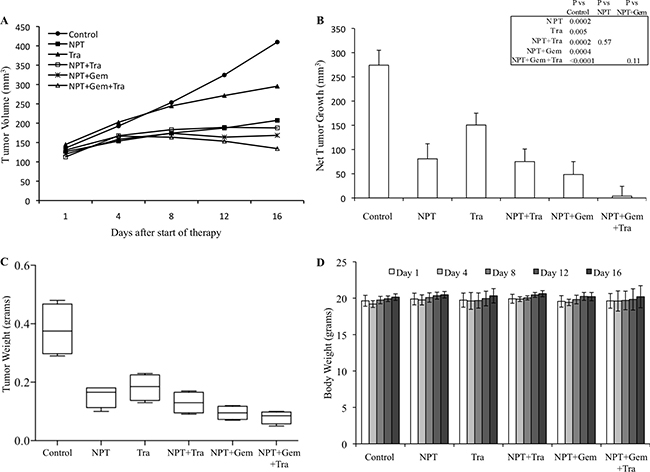
Figure 2: Antitumor activity of nab-paclitaxel-based chemotherapy regimens and trametinib in Panc-1 tumor xenografts. Panc-1 cells were subcutaneously injected in nude mice and treated with nab-paclitaxel-based chemotherapy regimens and trametinib for 2 weeks. (A) Tumor size as measured twice-weekly using calipers. (B) Net tumor growth, calculated by subtracting tumor volume on the first treatment day from that on the final day. (C) Mean tumor weight was calculated from final day tumor weights in each group and is presented as a Box plot. (D) Mouse body weight was measured twice a week and presented as bar chart for the 2-week therapy period. Data are representative of mean values ± standard deviation from 5 mice per group.
Nab-paclitaxel-based chemotherapy regimens and trametinib increase animal survival
In an AsPC-1 pancreatic cancer peritoneal dissemination model, animal survival in different groups, compared with controls (20 days) was increased after therapy with NPT (33 days, a 65% increase), Tra (31 days, a 55% increase), NPT+Tra (37 days, a 85% increase), NPT+Gem (39 days, a 95% increase) and NPT+Gem+Tra (49 days, a 145% increase) (Figure 3A, 3B). No significant change in the body weight of mice was observed during 2-week therapy period, indicating that there was no discernable therapy-associated toxicity in all therapy groups.
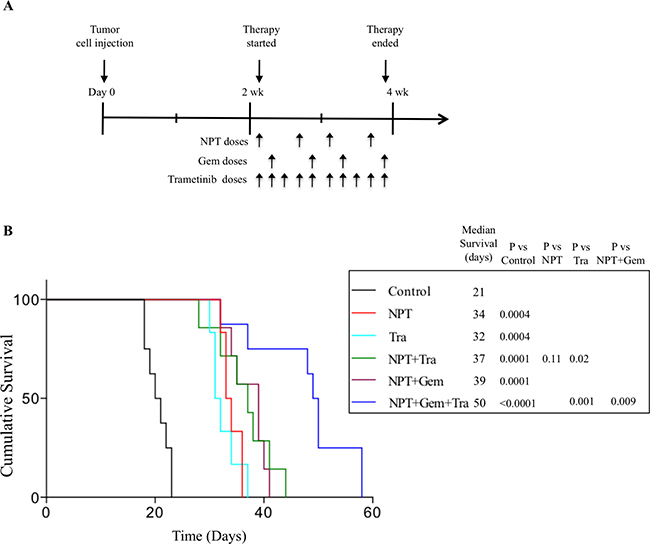
Figure 3: Prolongation of animal survival by nab-paclitaxel-based chemotherapy regimens and trametinib. (A) Schematic representation of experimental procedure. AsPC-1 cells (0.75 × 106) were injected intraperitoneally in NOD/SCID mice and treatment was started after 2 weeks with nab-paclitaxel, gemcitabine and trametinib for 2 weeks. (B) The curve represents the animal survival time from the beginning of therapy. Statistical group differences in survival time were calculated using logrank testing.
Nab-paclitaxel-based chemotherapy and trametinib: tumor cell proliferation and apoptosis
The antitumor mechanisms of nab-paclitaxel-based chemotherapy regimens and trametinib were investigated by IHC analysis of subcutaneous tumor tissues.
Ki67 staining demonstrated that nab-paclitaxel, nab-paclitaxel plus gemcitabine and trametinib decreased tumor cell proliferation and combination therapy groups demonstrated a trend for an additive effect. Intratumoral proliferative index, the percentage of Ki67-positive cells over the total number of cells per high-power field, in different therapy groups was: controls (45.4), NPT (21.8), Tra (26.6), NPT+Tra (15.1), NPT+Gem (13.3) and NPT+Gem+Tra (8.2) (Figure 4A).
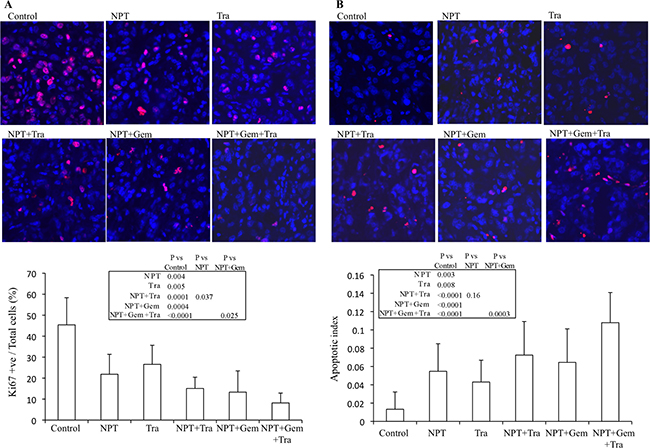
Figure 4: Mechanisms of antitumor activity of nab-paclitaxel-based chemotherapy regimens and trametinib. Nude mice were subcutaneously injected with AsPC-1 cells and treated with nab-paclitaxel, gemcitabine and trametinib for 2 weeks. (A) Intratumoral proliferation was measured by immunostaining tissue sections for Ki67 nuclear antigen. Ki67-positive cells were counted in five different high power fields. (B) Intratumoral apoptosis was measured by staining tumor tissue section with TUNEL procedure. TUNEL-positive apoptotic cells were counted in five different high power fields. For both immunostaining experiments, slides were photographed under a fluorescent microscope and the data are expressed as the mean ± standard deviation.
TUNEL assay detected that nab-paclitaxel and trametinib monotherapies increased tumor cell apoptosis and the combination therapy groups demonstrated a trend for an additive effect. The apoptotic index readings, by therapy group, were: controls (0.013), NPT (0.055), Tra (0.043), NPT+Tra (0.072), NPT+Gem (0.064) and NPT+Gem+Tra (0.108) (Figure 4B).
Further investigation of the tumor growth inhibitory action of nab-paclitaxel-based chemotherapy regimens and trametinib exhibited that trametinib significantly decreased ERK phosphorylation and concomitantly increased the expression of apoptosis-associated proteins cleaved caspase-3 and cleaved PARP-1 (Figure 5).
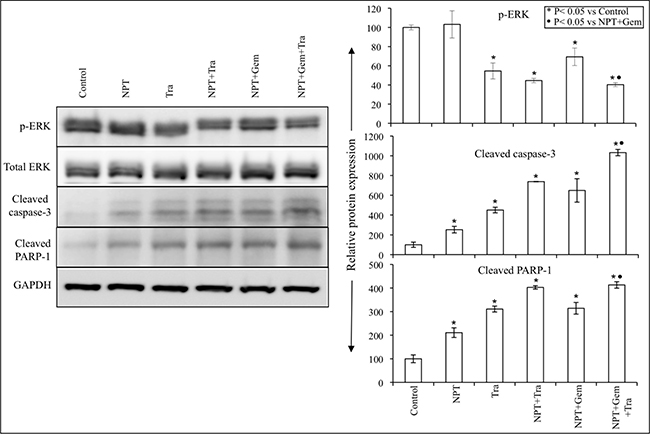
Figure 5: Nab-paclitaxel-based chemotherapy regimens and trametinib effects on their molecular targets in vivo. Tumor lysates were prepared from tumor tissues obtained from AsPC-1 tumor-bearing mice and were analyzed by immunoblotting. Data are representative of pooled lysates obtained from tumor sections of at least 5 mice in each therapy group. The intensity of bands was quantitated by densitometry and is represented in the bar graph after normalizing values with corresponding total protein expression or GAPDH expression.
Nab-paclitaxel-based chemotherapy and trametinib: In vitro cell viability and cellular targets
In vitro cell viability examination of human PDAC epithelial cells with different mutations [19], indicated that both, nab-paclitaxel plus gemcitabine and trametinib, suppressed cell proliferation. In the combination therapy groups, at high dose (10 μM), a statistically significant additive response on cell proliferation inhibition was observed in N+G+Tra compared with N+G or Tra groups for all the four cell lines tested (Figure 6). Nab-paclitaxel plus gemcitabine treatment dose-dependently inhibited cell proliferation, and at 10 μM concentration of each drug inhibition in proliferation was 47.6% (AsPC-1), 66.6% (Panc-1), 51.7% (Mia PaCa-2) and 80% (CFPAC) (Figure 6). Addition of trametinib (10 μM) to the nab-paclitaxel plus gemcitabine treatment group caused an additive effect and inhibition in cell proliferation in this combination group was 75% (AsPC-1), 82.6% (Panc-1), 65.6% (Mia PaCa-2) and 89.4% (CFPAC) (Figure 6). Immunoblot analysis determined that trametinib blocked the expression of phospho-ERK and concomitantly increased the expression of cleaved caspase-3 and cleaved PARP-1 proteins (Figure 7).
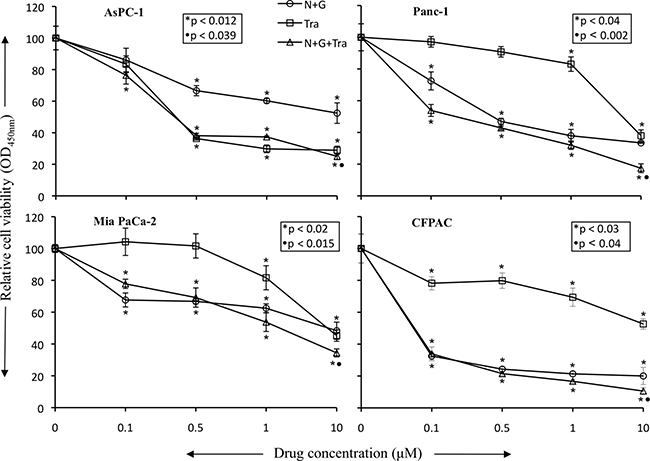
Figure 6: Nab-paclitaxel plus gemcitabine and trametinib inhibit in vitro PDAC cell proliferation. PDAC cells (AsPC-1, Panc-1, Mia PaCa-2, CFPAC) were plated on 96-well plates and treated with 100 nM to 10 μM concentrations of nab-paclitaxel, gemcitabine and trametinib. After 72 hours, 10 μl WST-1 reagent was added in each well and incubated for 2 additional hours. The absorbance at 450 nm was measured using a microplate reader. The resulting number of viable cells was calculated by measuring absorbance of color produced in each well. Data are the mean ± SD of triplicate determinations. The p values represent significance of the difference between the control and treated groups.
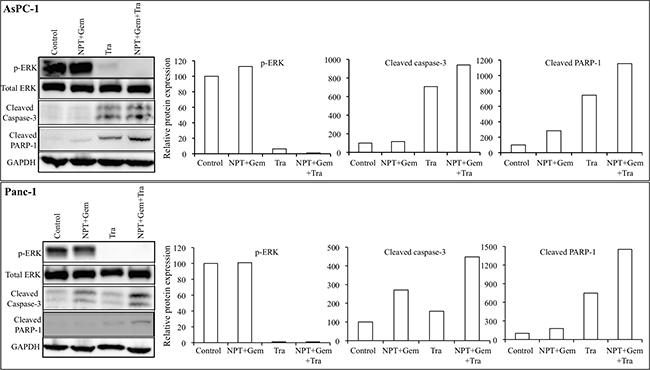
Figure 7: Nab-paclitaxel-based chemotherapy regimens and trametinib effects on their molecular targets in vitro. A sub-confluent monolayer of human PDAC cells AsPC-1 and Panc-1 was treated with nab-paclitaxel (10 μM), gemcitabine (10 μM) and trametinib (10 μM), either alone or in combination for 16 hours. Total cell extracts were analyzed by immunoblotting. The intensity of bands was quantitated by densitometry and is represented in the bar graph after normalizing values against corresponding total protein expression or GAPDH expression. Data are representative of two independent experiments with similar results.
DISCUSSION
Pancreatic cancer is frequently refractory to cytotoxic and targeted therapies, mainly due to the marked genetic heterogeneity and resultant complexity in molecular signaling [20]. Specifically, PDACs exhibit a high frequency of activating mutations in the KRAS oncogene, inactivating mutations in tumor suppressor genes p16/CDKN2A and TP53 and inactivation of the SMAD4 gene [21–23]. Activating mutation in the KRAS oncogene is a hallmark of PDAC as it is crucial for PDAC growth and progression and occurs in approximately 95% of patients [24]. Further, the importance of the KRAS oncogene in PDAC development has been demonstrated in genetically engineered mouse models [25, 26]. Therefore, investigating strategies for effective inhibition of KRAS signaling is critically important in pancreatic cancer research [25, 27]. Small molecule inhibitors of downstream targets of the RAS-RAF-MEK-ERK pathway are in clinical development and have shown promising efficacy in RAS/RAF-driven tumors [28, 29].
Trametinib has been shown to prevent RAS-dependent MEK phosphorylation and cause prolonged inhibition of ERK signaling [12]. Trametinib is highly selective for their targets, which may lead to fewer off-target effects [30]. In PDAC, trametinib augmented combined treatment with EGFR and HER2 inhibitors in patient-derived xenograft models [15, 31]. Early clinical studies in PDAC with trametinib monotherapy or its combination with gemcitabine showed promising results [16, 32]. Although a phase II clinical trial in untreated metastatic PDAC patients could not demonstrate significant clinical efficacy of the combination of gemcitabine and trametinib [17], another study in NSCLC with selumetinib as a MEK inhibitor demonstrated significant clinical benefits in combination with docetaxel, indicating the possibility of some synergy potential of MEK inhibitors with taxanes compared with gemcitabine [18]. Therefore, we explored the antitumor benefits of combining trametinib with the current standard of care chemotherapy regimen containing both the next-generation taxane nab-paclitaxel and gemcitabine in preclinical models of pancreatic cancer.
We detected a significant expression of phospho-MEK and phospho-ERK (Supplementary Figure 2) in all seven PDAC cells tested, confirming the prevalence of the MAPK signaling cascade in PDAC cellular activation and progression. In subcutaneous xenografts of two aggressive KRAS-mutant cell lines, AsPC-1 and Panc-1, we determined that trametinib not only inhibited tumor growth as a monotherapy, but it also demonstrated a trend for an additive effect in combination with chemotherapy regimens, although it did not reach statistical significance (Figures 1, 2). The trend for additive effect was more pronounced in the NPT+Gem group compared to the NPT group. Tumor cell proliferation and apoptosis rate evaluated in tumor tissue correlated with the tumor growth experiment results, supporting a trend for an additive effect on decreased proliferation and increased apoptosis by addition of trametinib (Figure 4). Furthermore, the addition of trametinib to chemotherapy regimens caused an obvious and statistically significant increase in animal survival in our peritoneal dissemination model (Figure 3). Smaller additive effects of trametinib in the subcutaneous model suggest the possibility of the local tumor microenvironment factors making MEK inhibition less relevant to PDAC growth compared with the more clinically relevant peritoneal dissemination model where MEK inhibition caused a significant improvement in the chemotherapy response. Target protein determination in protein lysates from subcutaneous xenografts showed that trametinib significantly decreased phosphorylated ERK and increased the expression of apoptosis-associated proteins, suggesting that the trametinib therapy is sufficiently affecting its target within the local tumor setting. Early MEK inhibitors such as PD0325901, selumetinib and cobimetinib blocked ERK activation, but concomitant interference with ERK-dependent negative feedback resulted in the accumulation of phospho-MEK and rebound in ERK signaling [33, 34]. Newer MEK inhibitors including trametinib cause minimal to no increase in phospho-MEK or recovery in ERK activity, resulting in lasting inhibition of MAPK pathway and superior antitumor activity [34].
Previous studies in our laboratory have employed an established peritoneal dissemination model of PDAC to study animal survival using KRAS-mutant AsPC-1 cells that resembles the human disease in its aggressiveness and metastatic progression [35, 36]. With this approach, we observed that animal survival was improved by trametinib with and without nab-paclitaxel or nab-paclitaxel plus gemcitabine. More importantly, trametinib augmented nab-paclitaxel-based chemotherapy effects for the greatest improvement in animal survival. These results are consistent with a recent study demonstrating that trametinib in combination with gemcitabine had enhanced antitumor efficacy in a patient-derived xenograft model of PDAC in inhibiting growth rate of liver metastases and increasing PFS [37]. This observation is through our data extended to nab-paclitaxel, which appears to be specifically suited for in vivo therapy benefits when combined with trametinib. Although the appropriate sequence for the administration of MEK inhibitor and chemotherapy agents has been shown to impact clinical outcomes [17, 38], we have not evaluated the effects of varied dosing schedule of these drugs. In this study, nab-paclitaxel effects on enhanced tissue distribution and tumor penetration of drugs might be partially responsible for enhanced antitumor response in combination therapy groups as nab-paclitaxel therapy was started together with trametinib and a day before gemcitabine [39].
In vitro cell viability analysis revealed that trametinib inhibited proliferation of KRAS-mutant PDAC cells while the combination of trametinib with nab-paclitaxel plus gemcitabine had additive effects on inhibition in cell proliferation at high dose level (10 μM). Among these cells, AsPC-1 cells were most sensitive to trametinib and a significant decrease in cell viability was observed at all dose levels, and a very little additive benefit on cell viability inhibition was observed by addition of trametinib to chemotherapy. Other PDAC cells tested showed a small effect on cell viability at low dose levels and the effect was significant at high dose levels. The differential sensitivity of pancreatic cancer cells to MEK inhibition can be attributed to the KRAS mutational subtype, copy number and the presence of PIK3CA co-mutation [40, 41]. In a phase 1 study, the maximum plasma concentration of trametinib was 62.8 ng/ml (~0.1 μM) [16]. Similar to the target protein evaluation in tumor lysates, protein lysates from AsPC-1 and Panc-1 cells demonstrated a dramatic decrease in ERK phosphorylation and an increase in apoptosis-related proteins by trametinib treatment. These findings further indicate that the antitumor response of trametinib is linked to blocking ERK induced molecular signaling changes that are accountable for tumor cell proliferation and survival.
PDAC progression is multifactorial involving increased tumor cell proliferation, differentiation, migration, angiogenesis, epithelial-to-mesenchymal transition, desmoplasia and immune system evasion, and most of these mechanisms have been shown to be affected by or regulated through the RAS-RAF-MEK-ERK signaling pathway [42, 43]. We have previously demonstrated that nab-paclitaxel-based chemotherapy has antitumor responses based on its antiproliferative, antistromal and proapoptotic effects [44]. Therefore, the possible molecular mechanisms for the improvement in antitumor responses of nab-paclitaxel-based chemotherapy regimen by trametinib may likely be caused by not only augmenting antiproliferative and proapoptotic activities but also by inhibiting other tumorigenic pathways including angiogenesis [45].
Based on the fact that the RAS-RAF-MEK-ERK pathway is one of the most frequently dysregulated pathways in PDAC, MEK inhibitors have shown promising antitumor activity, either alone or in combination with other anticancer drugs [46–48]. Trametinib is a new generation MEK1/2 inhibitor, which is highly selective for these targets. Compared with other MEK inhibitors, trametinib has several advantages including a favorable pharmacokinetic profile, long half-life and manageable toxicity [16, 49]. In the present study, no discernible treatment-associated toxicity was observed during a 2-week therapy period, however, toxicity associated with long-term therapy of trametinib in combination with nab-paclitaxel-based chemotherapy regimens remains to be elucidated. In conclusion, our study indicates that trametinib, a MEK1/2 inhibitor blocking KRAS activating mutation induced RAF-MEK-ERK tumorigenic signaling, is an effective mechanism-specific antitumor agent, and it can improve nab-paclitaxel-based chemotherapy effects in PDAC. These results strongly support the rationale of blocking downstream targets of KRAS-mutation driven signaling and indicate the potential of trametinib as a targeting agent in combination with nab-paclitaxel-based chemotherapy regimens for clinical PDAC therapy.
MATERIALS AND METHODS
Cell culture and reagents
Human PDAC cell lines (AsPC-1, BxPC-3, CFPAC, Mia PaCa-2 and Panc-1) were purchased from the American Type Culture Collection (ATCC, Rockville, MD). Cell lines were tested and authenticated by ATCC. The most common mutations in all the cell lines used in this study are as follows: AsPC-1 (KRAS, p53, p16), Panc-1 (KRAS, p53, p16), Mia PaCa-2 (KRAS, p53, p16) and CFPAC (KRAS, p53, SMAD4) [19]. Cells were initially grown and multiple aliquots were cryopreserved. All the cell lines were used within 6 months after reexpansion in culture. Cells were cultured in DMEM or RPMI 1640 medium (Sigma Chemical Co. St. Louis, MO) containing 10% FBS and maintained at 37°C in a humidified incubator with 5% CO2 and 95% air. Nab-paclitaxel was obtained from Celgene Corporation (Summit, NJ). Gemcitabine and trametinib were purchased from LC labs (Woburn, MA). The cell proliferation reagent WST-1 was purchased from Roche Diagnostic Corporation (Indianapolis, IN).
Cell viability assay
Cell viability was evaluated by the colorimetric WST-1 assay as previously described [50]. Briefly, PDAC cells (4,000 to 5,000 cells per well) were plated in a 96-well plate in regular cell growth medium containing 10% FBS. After 16 hours the medium was replaced with low serum medium containing 2% FBS and the cells were treated with 100 nM to 10 μM concentrations of nab-paclitaxel, gemcitabine and trametinib. After 72 hours, 10 μl WST-1 reagent was added in each well followed by additional incubation for 2 hours. The absorbance was measured at 450 nm using a microplate reader.
Western blot analysis
Protein lysates were prepared by treating sub-confluent cells with nab-paclitaxel (10 μM), gemcitabine (10 μM) and trametinib (10 μM), and lysed after 16 hours for Western blotting as previously described [50]. Protein lysates of subcutaneous tumors were prepared by snap freezing tumor tissues in liquid nitrogen and stored at –80°C. These tumor tissues were suspended in lysis buffer and homogenized using the Bullet Blender Homogenizer (Next Generation, Averill Park, NY), and extracts were sonicated on ice. Proteins in supernatants were separated by SDS-PAGE and transferred to PVDF membranes (Bio-Rad, Hercules, CA). The membranes were incubated overnight at 4°C with the following antibodies: total ERK1/2, phospho-ERK1/2 (Thr202/Tyr204), cleaved caspase-3, cleaved PARP-1 and GAPDH (Cell Signaling Technology, Beverly, MA). The membranes were then incubated with the corresponding HRP-conjugated secondary antibodies (Pierce Biotechnologies, Santa Cruz, CA) for 1 to 2 hour. Protein bands were visualized using the enhanced chemiluminescence reagent (SignalFire, Cell Signaling) with an Image360 system and quantitated by densitometry.
Animal experiments
All animals were housed in a pathogen-free facility with access to food and water ad libitum. Animal experiments were performed in accordance with the Institutional Animal Care and Use Committee (IACUC) at the Indiana University School of Medicine (South Bend, IN). Female nonobese diabetic/severe combined immunodeficient (NOD/SCID) mice (4 to 6 weeks old) were subcutaneously injected with AsPC-1 cells (7.5 × 105) or Panc-1 cells (10 × 106) as previously described [44]. Two weeks after tumor cell injection, all mice had a measurable tumor. Mice were then randomized (n = 4 to 6 per group) to receive PBS (control), nab-paclitaxel (5 mg/kg, twice a week), gemcitabine (50 mg/kg, twice a week) and trametinib (1 mg/kg, 5 times a week) via intraperitoneal injection for next 2 weeks. The tumor size was measured twice weekly, and tumor volume (V) was calculated using the formula V = ½ (Length × Width2). Net tumor growth was calculated by subtracting tumor volume on the first therapy day from that on the last day. Mice were euthanized after completion of treatment, tumors were dissected and processed for histological, immunohistochemical and Western blot analysis.
Animal survival studies were performed using female NOD/SCID mice (4–6 weeks of age) as previously described [51]. Briefly, the mice were injected intraperitoneally with AsPC-1 (7.5 × 105) cells and two weeks after tumor cell injection, mice were randomized (n = 6 to 8 per group) to receive PBS (control), nab-paclitaxel (5 mg/kg, twice a week), gemcitabine (50 mg/kg, twice a week) and trametinib (1 mg/kg, 5 times a week) via IP injection for two weeks. Animals were euthanized when moribund according to predefined criteria [52, 53]. Animal survival was evaluated from the first day of treatment until death.
Immunohistochemistry and immunofluorescence
Standard immunohistochemistry protocol was followed to stain the tumor tissue sections, as previously described [54]. Briefly, tumor tissue samples were fixed in 4% paraformaldehyde, embedded in paraffin, tissue sections were cut (5 μm), deparaffinized and rehydrated. The tissue sections were incubated with 1:200 dilution of Ki67 antibody (ab15580, Abcam, Cambridge, MA) followed by incubation with 1:200 dilution of anti-rabbit-Cy3 secondary antibody (Jackson ImmunoResearch Laboratories, West Grove, PA). Slides were mounted using a mounting solution containing 4ʹ,6-diamidino-2-phenylindole (DAPI) (Invitrogen, Carlsbad, CA). The intratumoral proliferative index was determined by calculating the Ki67-positive cells from five different high-power fields (HPF) in a blinded manner in each group. Intratumoral apoptotic activity was evaluated by staining tissue sections with “Apoptag Apoptosis Detection Kit” according to the manufacturer’s (Millipore, Temecula, CA) instructions. The apoptotic index was calculated by dividing the number of TUNEL-positive cells by the total number of cells per HPF in a blinded manner in each group. Fluorescence microscopy was used to detect fluorescent signals using the IX81 Olympus microscope equipped with a Hamamatsu Orca digital camera (Hamamatsu Corporation, Bridgewater, NJ) and a disk-scanning unit (DSU) spinning disk confocal system using Slidebook software (Intelligent Imaging Innovations, Philadelphia, PA).
Statistical analysis
Statistical analysis for in vivo tumor growth studies was performed by one-way ANOVA for multiple group comparisons and Student’s t-test for the individual group comparisons. Survival study statistics were performed using logrank group comparison (GraphPad Prism 6.0). P values less than 0.05 were considered statistically significant. In vitro cell proliferation data are expressed as the mean ± standard deviation. Statistical significance was analyzed by the two-tailed Student’s t-test using GraphPad Prism 6.0 Software (GraphPad Software, San Diego, CA) for individual group comparisons.
ACKNOWLEDGMENTS AND FUNDING
This work was financially supported in part by the Walther Cancer Foundation, Seeding Research in Cancer Grant (N Awasthi) and Indiana University School of Medicine funds.
CONFLICTS OF INTEREST
The authors indicate no conflicts of interest.
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016; 66:7–30.
2. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014; 74:2913–2921.
3. Gillen S, Schuster T, Meyer Zum Buschenfelde C, Friess H, Kleeff J. Preoperative/neoadjuvant therapy in pancreatic cancer: a systematic review and meta-analysis of response and resection percentages. PLoS Med. 2010; 7:e1000267.
4. Burris HA 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, Nelson R, Dorr FA, Stephens CD, Von Hoff DD. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997; 15:2403–2413.
5. Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, Adenis A, Raoul JL, Gourgou-Bourgade S, de la Fouchardiere C, Bennouna J, Bachet JB, Khemissa-Akouz F, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011; 364:1817–1825.
6. Kundranda MN, Niu J. Albumin-bound paclitaxel in solid tumors: clinical development and future directions. Drug Des Devel Ther. 2015; 9:3767–3777.
7. Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, Harris M, Reni M, Dowden S, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013; 369:1691–1703.
8. Shapiro P. Ras-MAP kinase signaling pathways and control of cell proliferation: relevance to cancer therapy. Crit Rev Clin Lab Sci. 2002; 39:285–330.
9. Thomas RK, Baker AC, Debiasi RM, Winckler W, Laframboise T, Lin WM, Wang M, Feng W, Zander T, MacConaill L, Lee JC, Nicoletti R, Hatton C, et al. High-throughput oncogene mutation profiling in human cancer. Nat Genet. 2007; 39:347–351.
10. Surade S, Blundell TL. Structural biology and drug discovery of difficult targets: the limits of ligandability. Chem Biol. 2012; 19:42–50.
11. Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007; 26:3291–3310.
12. Gilmartin AG, Bleam MR, Groy A, Moss KG, Minthorn EA, Kulkarni SG, Rominger CM, Erskine S, Fisher KE, Yang J, Zappacosta F, Annan R, Sutton D, et al. GSK1120212 (JTP-74057) is an inhibitor of MEK activity and activation with favorable pharmacokinetic properties for sustained in vivo pathway inhibition. Clin Cancer Res. 2011; 17:989–1000.
13. Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N, Kudchadkar R, Burris HA 3rd, Falchook G, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012; 367:1694–1703.
14. Rissmann R, Hessel MH, Cohen AF. Vemurafenib/dabrafenib and trametinib. Br J Clin Pharmacol. 2015; 80:765–767.
15. Walters DM, Lindberg JM, Adair SJ, Newhook TE, Cowan CR, Stokes JB, Borgman CA, Stelow EB, Lowrey BT, Chopivsky ME, Gilmer TM, Parsons JT, Bauer TW. Inhibition of the growth of patient-derived pancreatic cancer xenografts with the MEK inhibitor trametinib is augmented by combined treatment with the epidermal growth factor receptor/HER2 inhibitor lapatinib. Neoplasia. 2013; 15:143–155.
16. Infante JR, Fecher LA, Falchook GS, Nallapareddy S, Gordon MS, Becerra C, DeMarini DJ, Cox DS, Xu Y, Morris SR, Peddareddigari VG, Le NT, Hart L, et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. Lancet Oncol. 2012; 13:773–781.
17. Infante JR, Somer BG, Park JO, Li CP, Scheulen ME, Kasubhai SM, Oh DY, Liu Y, Redhu S, Steplewski K, Le N. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur J Cancer. 2014; 50:2072–2081.
18. Janne PA, Shaw AT, Pereira JR, Jeannin G, Vansteenkiste J, Barrios C, Franke FA, Grinsted L, Zazulina V, Smith P, Smith I, Crino L. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol. 2013; 14:38–47.
19. Deer EL, Gonzalez-Hernandez J, Coursen JD, Shea JE, Ngatia J, Scaife CL, Firpo MA, Mulvihill SJ. Phenotype and genotype of pancreatic cancer cell lines. Pancreas. 2010; 39:425–435.
20. Hidalgo M. Pancreatic cancer. N Engl J Med. 2010; 362:1605–1617.
21. Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Kamiyama H, Jimeno A, Hong SM, Fu B, Lin MT, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008; 321:1801–1806.
22. Jaffee EM, Hruban RH, Canto M, Kern SE. Focus on pancreas cancer. Cancer Cell. 2002; 2:25–28.
23. Maitra A, Hruban RH. Pancreatic cancer. Annu Rev Pathol. 2008; 3:157–188.
24. Bryant KL, Mancias JD, Kimmelman AC, Der CJ. KRAS: feeding pancreatic cancer proliferation. Trends Biochem Sci. 2014; 39:91–100.
25. Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, Rustgi AK, Chang S, Tuveson DA. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005; 7:469–483.
26. Bardeesy N, Aguirre AJ, Chu GC, Cheng KH, Lopez LV, Hezel AF, Feng B, Brennan C, Weissleder R, Mahmood U, Hanahan D, Redston MS, Chin L, et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc Natl Acad Sci U S A. 2006; 103:5947–5952.
27. Philip PA, Mooney M, Jaffe D, Eckhardt G, Moore M, Meropol N, Emens L, O’Reilly E, Korc M, Ellis L, Benedetti J, Rothenberg M, Willett C, et al. Consensus report of the national cancer institute clinical trials planning meeting on pancreas cancer treatment. J Clin Oncol. 2009; 27:5660–5669.
28. Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT, Salton M, Dahlman KB, Tadi M, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature. 2011; 480:387–390.
29. Poulikakos PI, Rosen N. Mutant BRAF melanomas—dependence and resistance. Cancer Cell. 2011; 19:11–15.
30. Sebolt-Leopold JS. Advances in the development of cancer therapeutics directed against the RAS-mitogen-activated protein kinase pathway. Clin Cancer Res. 2008; 14:3651–3656.
31. Lindberg JM, Newhook TE, Adair SJ, Walters DM, Kim AJ, Stelow EB, Parsons JT, Bauer TW. Co-treatment with panitumumab and trastuzumab augments response to the MEK inhibitor trametinib in a patient-derived xenograft model of pancreatic cancer. Neoplasia. 2014; 16:562–571.
32. Tolcher AW, Bendell JC, Patnaik A, Papadopoulos K, Bellew KM, Cox DS. A phase Ib study of the MEK inhibitor GSK1120212 combined with gemcitabine in patients with solid tumors: interim results. J Clin Oncol 2011; 29:278.
33. Hatzivassiliou G, Haling JR, Chen H, Song K, Price S, Heald R, Hewitt JF, Zak M, Peck A, Orr C, Merchant M, Hoeflich KP, Chan J, et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS- versus BRAF-driven cancers. Nature. 2013; 501:232–236.
34. Lito P, Saborowski A, Yue J, Solomon M, Joseph E, Gadal S, Saborowski M, Kastenhuber E, Fellmann C, Ohara K, Morikami K, Miura T, Lukacs C, et al. Disruption of CRAF-mediated MEK activation is required for effective MEK inhibition in KRAS mutant tumors. Cancer Cell. 2014; 25:697–710.
35. Awasthi N, Zhang C, Schwarz AM, Hinz S, Wang C, Williams NS, Schwarz MA, Schwarz RE. Comparative benefits of Nab-paclitaxel over gemcitabine or polysorbate-based docetaxel in experimental pancreatic cancer. Carcinogenesis. 2013; 34:2361–2369.
36. Schwarz RE, Konduri S, Awasthi N, Cafasso D, Schwarz MA. An antiendothelial combination therapy strategy to increase survival in experimental pancreatic cancer. Surgery. 2009; 146:241–249.
37. Newhook TE, Lindberg JM, Adair SJ, Kim AJ, Stelow EB, Rahma OE, Parsons JT, Bauer TW. Adjuvant Trametinib Delays the Outgrowth of Occult Pancreatic Cancer in a Mouse Model of Patient-Derived Liver Metastasis. Ann Surg Oncol. 2016; 23:1993–2000.
38. Xu J, Knox JJ, Ibrahimov E, Chen E, Serra S, Tsao M, Cao P, Vines D, Green DE, Metran-Nascente C, McNamara MG, Hedley DW. Sequence dependence of MEK inhibitor AZD6244 combined with gemcitabine for the treatment of biliary cancer. Clin Cancer Res. 2013; 19:118–127.
39. Chen N, Brachmann C, Liu X, Pierce DW, Dey J, Kerwin WS, Li Y, Zhou S, Hou S, Carleton M, Klinghoffer RA, Palmisano M, Chopra R. Albumin-bound nanoparticle (nab) paclitaxel exhibits enhanced paclitaxel tissue distribution and tumor penetration. Cancer Chemother Pharmacol. 2015; 76:699–712.
40. Hamidi H, Lu M, Chau K, Anderson L, Fejzo M, Ginther C, Linnartz R, Zubel A, Slamon DJ, Finn RS. KRAS mutational subtype and copy number predict in vitro response of human pancreatic cancer cell lines to MEK inhibition. Br J Cancer. 2014; 111:1788–1801.
41. Hanrahan AJ, Solit DB. RAF/MEK dependence of KRAS-mutant pancreatic ductal adenocarcinomas. Cancer Discov. 2012; 2:666–669.
42. Maurer G, Tarkowski B, Baccarini M. Raf kinases in cancer-roles and therapeutic opportunities. Oncogene. 2011; 30:3477–3488.
43. McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M, Tafuri A, Stivala F, Libra M, Basecke J, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007; 1773:1263–1284.
44. Awasthi N, Zhang C, Schwarz AM, Hinz S, Schwarz MA, Schwarz RE. Enhancement of nab-paclitaxel antitumor activity through addition of multitargeting antiangiogenic agents in experimental pancreatic cancer. Mol Cancer Ther. 2014; 13:1032–1043.
45. Bridgeman VL, Wan E, Foo S, Nathan MR, Welti JC, Frentzas S, Vermeulen PB, Preece N, Springer CJ, Powles T, Nathan PD, Larkin J, Gore M, et al. Preclinical Evidence That Trametinib Enhances the Response to Antiangiogenic Tyrosine Kinase Inhibitors in Renal Cell Carcinoma. Mol Cancer Ther. 2016; 15:172–183.
46. Hofmann I, Weiss A, Elain G, Schwaederle M, Sterker D, Romanet V, Schmelzle T, Lai A, Brachmann SM, Bentires-Alj M, Roberts TM, Sellers WR, Hofmann F, et al. K-RAS mutant pancreatic tumors show higher sensitivity to MEK than to PI3K inhibition in vivo. PLoS One. 2012; 7:e44146.
47. Zhao C, Xiao H, Wu X, Li C, Liang G, Yang S, Lin J. Rational combination of MEK inhibitor and the STAT3 pathway modulator for the therapy in K-Ras mutated pancreatic and colon cancer cells. Oncotarget. 2015; 6:14472–14487. http://doi.org/10.18632/oncotarget.3991.
48. Franco J, Witkiewicz AK, Knudsen ES. CDK4/6 inhibitors have potent activity in combination with pathway selective therapeutic agents in models of pancreatic cancer. Oncotarget. 2014; 5:6512–6525. http://doi.org/10.18632/oncotarget.2270.
49. Salama AK, Kim KB. MEK inhibition in the treatment of advanced melanoma. Curr Oncol Rep. 2013; 15:473–482.
50. Awasthi N, Schwarz MA, Verma V, Cappiello C, Schwarz RE. Endothelial monocyte activating polypeptide II interferes with VEGF-induced proangiogenic signaling. Lab Invest. 2009; 89:38–46.
51. Schwarz RE, Awasthi N, Konduri S, Cafasso D, Schwarz MA. EMAP II-based antiangiogenic-antiendothelial in vivo combination therapy of pancreatic cancer. Ann Surg Oncol. 2010; 17:1442–1452.
52. Awasthi N, Yen PL, Schwarz MA, Schwarz RE. The efficacy of a novel, dual PI3K/mTOR inhibitor NVP-BEZ235 to enhance chemotherapy and antiangiogenic response in pancreatic cancer. J Cell Biochem. 2012; 113:784–791.
53. Awasthi N, Kirane A, Schwarz MA, Toombs JE, Brekken RA, Schwarz RE. Smac mimetic-derived augmentation of chemotherapeutic response in experimental pancreatic cancer. BMC Cancer. 2011; 11:15.
54. Awasthi N, Zhang C, Ruan W, Schwarz MA, Schwarz RE. Evaluation of poly-mechanistic antiangiogenic combinations to enhance cytotoxic therapy response in pancreatic cancer. PLoS One. 2012; 7:e38477.