
INTRODUCTION
The Wnt-signaling pathway is an important signaling pathway in many types of cancer including colorectal cancer (CRC) [1]. The canonical Wnt-signaling pathway, and the one studied the most with CRC, is mediated via Wnt ligands and their receptors resulting in accumulation of β-catenin [2]. Components of this pathway include the adenomatous polyposis coli (APC) gene which is mutated in roughly 80% of CRC, AXIN 1 and 2, and glycogen synthase kinase 3β (GSK-3β) [3]. Downstream genes in this pathway include c-myc, c-jun, and Cyclin D1 (CCND1). Two non-canonical Wnt-signaling pathways, Wnt/CA2+ and Wnt/planar cell polarity (PCP), also exist [4]. While these Wnt-signaling pathways have been studied less thoroughly than the canonical Wnt/β-catenin pathway, it is felt that the Wnt-signaling pathways do not operate independently of each other. For instance both CaMKII and NFAT in the Wnt/CA2+ pathway influence β-catenin [5]; β-catenin has been linked to JNK in the Wnt/PCP pathway [6]; and drug response may be influenced by both the canonical and non-canonical pathways [7].
Genomic abnormalities in the Wnt-signaling pathway are a component of CRC tumorigenesis. MicroRNAs (miRNA) can directly influence gene expression by binding to the 3′un-translated region of the target mRNA and promoting mRNA degradation and/or inhibit mRNA translation; imperfect base pairing between the miRNA and mRNA can also result in translation repression of the target gene translation [8, 9]. Associations of miRNAs with targeted genes (TG) can be through direct binding to genes or through a pathway effect where down-stream regulation of gene expression from feedback and feedforward loops occur [10]. Several miRNAs, including miR-34, miR-320, miR-200, and Let-7, have been reported as being associated with Wnt-signaling pathway genes [2]. Additionally, miR-34 and miR-25 have been linked to β-catenin in early development [4]. Expression of Wnt-signaling pathway genes, including CTNBB1 (gene that encodes β-catenin), MYC, and CCND1, also have been associated with expression of multiple miRNAs [11]. Peng and colleagues in their review of miRNAs and the Wnt/β-catenin signaling pathway in cancer, provides additional support for the inter-relationship of these factors [12].
In this study we identified genes in the Wnt-signaling pathway using the human Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database. We determined which genes in the Wnt-signaling pathway are differentially expressed in CRC and if those differentially expressed genes are associated with miRNA differential expression. To help determine if the association between the miRNA and mRNA was direct or indirect, i.e. through another TG::miRNA that influences pathway genes, we identified seed-region matches between the TG and miRNA. We evaluate if miRNAs are associated with Wnt-signaling pathway genes and if genes in the canonical and non-canonical pathways are associated with the same miRNAs as other genes in the same canonical or non-canonical Wnt-signaling pathways.
RESULTS
The majority of cases were diagnosed with colon cancer (77.9%) rather than rectal cancer (Table 1). The majority of the study were men (54.4%), non-Hispanic white (74.2%), and had a TP53-mutated tumor (47.5%). MSI samples represented 13.4% of the study and MSS samples comprised 86.6% of the study population.
Table 1: Description of study population
N |
% |
|
|---|---|---|
Site |
||
Colon |
169 |
77.9 |
Rectal |
48 |
22.1 |
Sex |
||
Male |
118 |
54.4 |
Female |
99 |
45.6 |
Age |
||
Mean (SD) |
64.8 |
10.1 |
Race |
||
non-Hispanic White |
161 |
74.2 |
Hispanic |
14 |
6.5 |
non-Hispanic Black |
8 |
3.7 |
Unknown |
34 |
15.7 |
AJCC Stage |
||
1 |
58 |
27.1 |
2 |
61 |
28.5 |
3 |
72 |
33.6 |
4 |
23 |
10.8 |
Tumor Phenotype |
||
TP53 mutated |
103 |
47.5 |
KRAS mutated |
69 |
31.8 |
BRAF-mutated |
21 |
10.1 |
CIMP High |
45 |
20.7 |
MSI |
29 |
13.4 |
Vital Status |
||
Dead |
92 |
42.6 |
Alive |
124 |
57.4 |
Of the 138 Wnt-signaling pathway genes examined (43.5%), 27 were significantly down-regulated (FC<0.67) and 32 genes were significantly up-regulated (FC>1.50) after adjusting for multiple comparisons when evaluating CRC tumors overall (Supplementary Table 1), this compares to 37.5% of all protein-coding genes being dysregulated (6541 dysregulated of 17461 protein-coding genes). Associations were similar when looking at MSS only, although FC were slightly greater for MSS-specific tumors, the difference was minor given that over 85% of all tumor samples were MSS (Supplementary Table 3). The FC of four genes was altered sufficiently to merit further analysis based on our criteria of <0.67 or >1.50 for MSS tumors versus overall (APC2 FCmss 0.65, FCoverall 0.68; FZD6 FCmss 1.54, FCoverall 1.48; CSNK2A1 FCmss 1.54, FCoverall 1.49; DKK1 FCmss 1.87; FCoverall 1.47). Figure 1 visually illustrates the up-regulated genes (red) and the down-regulated pathway genes (yellow) within the Wnt-signaling pathway.
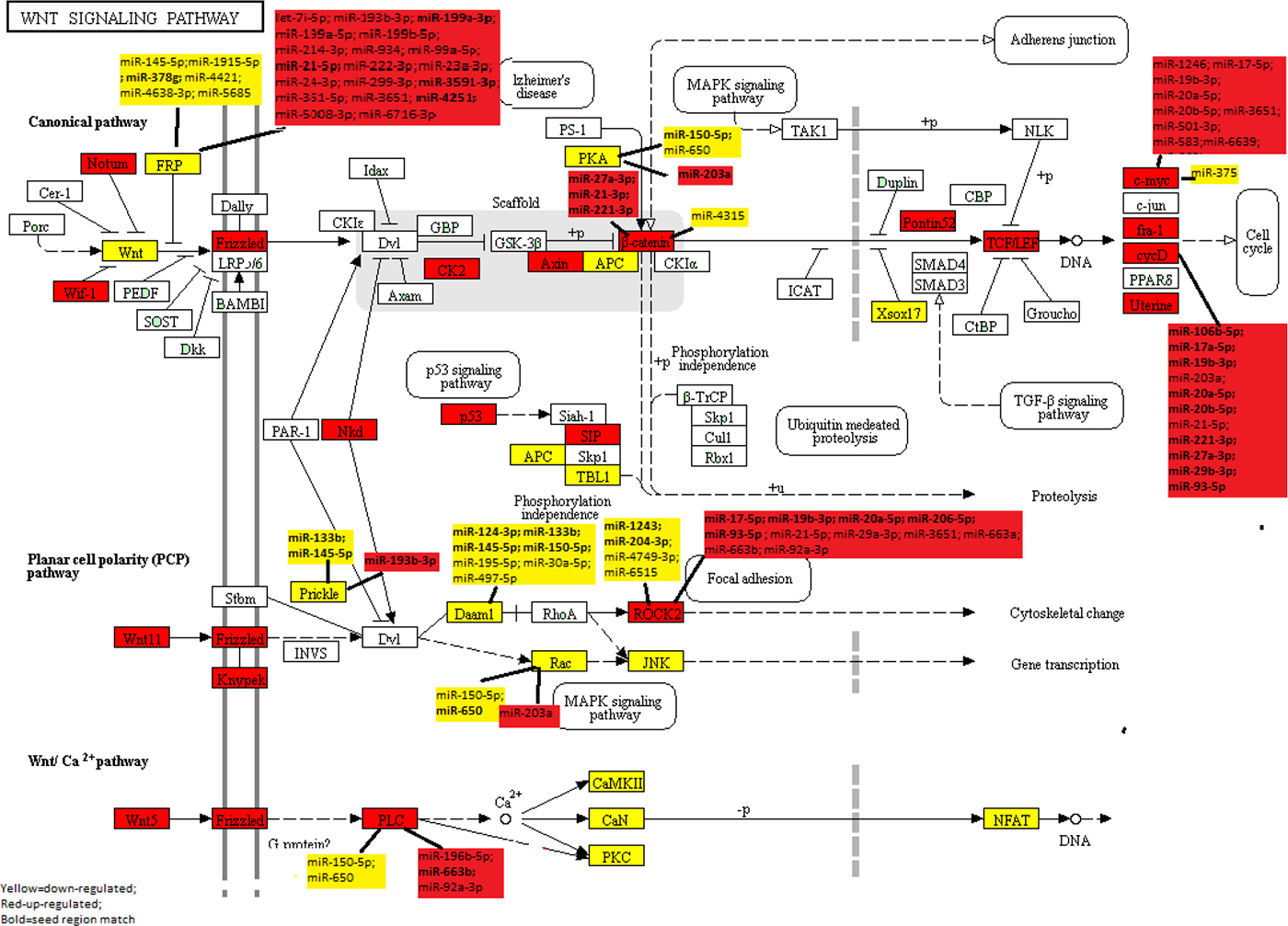
Figure 1: KEGG Wnt-Signaling Pathway: Dysregulated mRNA and miRNA in colorectal cancer.
Further evaluation of significant Wnt-signaling pathway genes with miRNAs showed that 13 of the 66 Wnt-signaling genes that were differentially expressed in CRC tumors overall or specifically with MSI or MSS tumors and were associated with differential expression of miRNAs after adjustment for multiple comparisons (Table 2). Only one gene, LEF1, that was statistically significantly associated with two miRNAs, was excluded because the miRNA’s FCs were outside of the 0.67 and 1.50 cutpoints. Of the 13 genes associated with miRNAs, six were down-regulated in carcinoma tissue and seven were up-regulated in carcinoma tissue relative to normal mucosa. All of the genes were associated with multiple miRNAs, with SFRP5 being associated with 24 miRNAs, ROCK2 with 15 miRNAs, MYC with 13 miRNAs, CCND1 with 11 miRNAs, and SFRP4 with 10 miRNAs. While 33 miRNAs were associated with only one gene, many miRNAs were associated with multiple genes: miR-7i-5p, miR-133b, miR-221-3p, miR-27a-3p, miR-29a-3p, miR-6515-5p, miR-663a, and miR-93-5p were associated with two genes; miR-145-5p, miR-17-5p, miR-193-3p, miR-19b-30, miR-203a, miR-20a-5p, miR-21-5p, miR-3651, miR-650, and miR-663b with three genes, and miR-92a-3p, miR-150-5p, and miR-20b-5p with four genes. Figure 1 further illustrates the pathway relationships between the differentially expressed Wnt-Signaling Pathway genes and miRNAs. As shown in Figure 1, several of the miRNAs associated with these genes are downstream of other genes in the same pathway.
Table 2: Associations between dysregulated genes in the Wnt-signaling pathway and miRNAs
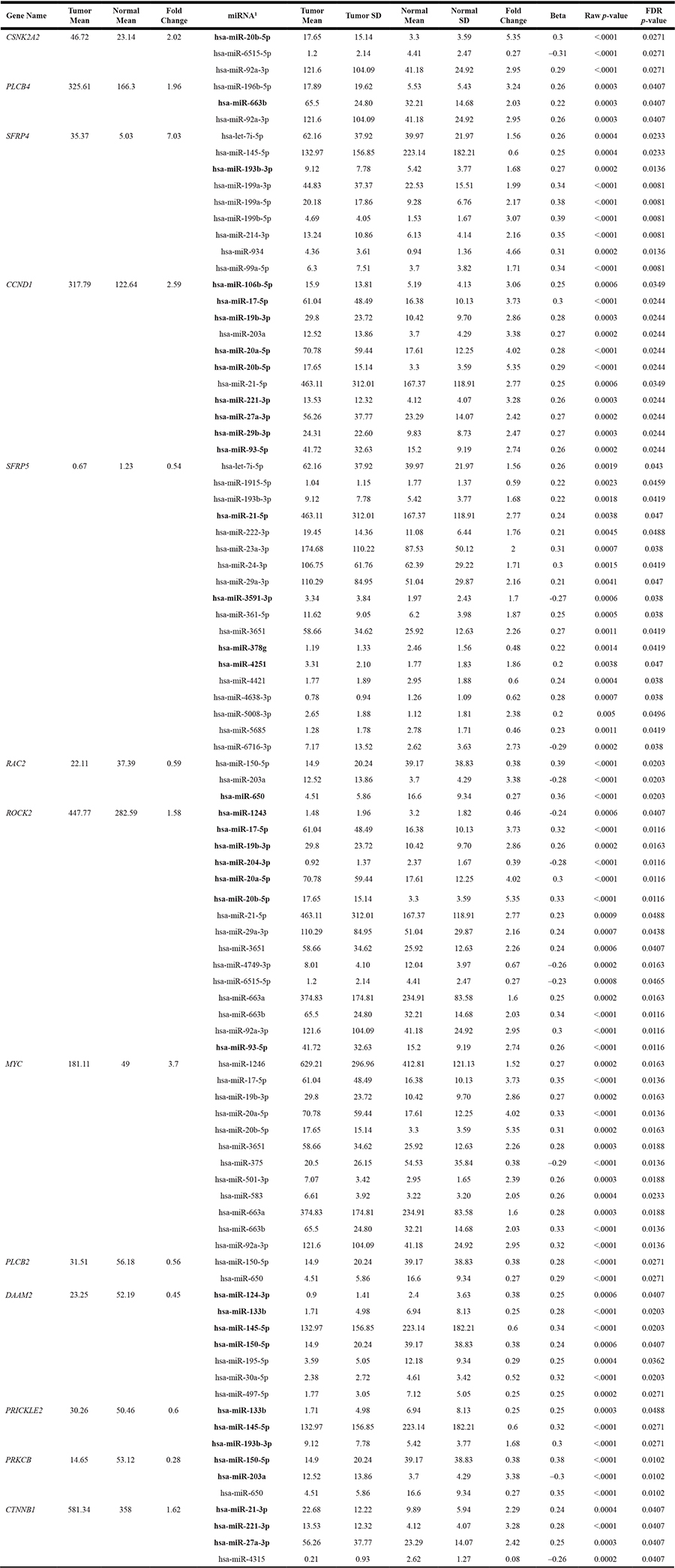
1Bold Text indicates seed match between mRNA and miRNA.
The majority of miRNA::mRNA associations were either both up-regulated or both down-regulated. Looking at seed-region matches between all dysregulated genes and all associated miRNAs there were 36 matches (Table 2 bold text indicates miRNA::mRNA seed matches). Of these 36 seed-region matches, seven had inverse associations between the miRNA and the mRNA. These included: SFRP5 (FC 0.54) with miR-21-5p (FC 2.77), miR-3591-3p (FC 1.70), and miR-4251 (FC 1.86); ROCK2 (FC 1.58) with miR-1243 (FC 0.46) and miR-204-3p (FC 0.39); PRICKLE2 (FC 0.60) with miR-193b-3p (FC 1.68); and PRKCB (FC 0.28) with miR-203a (FC 3.38). However, the majority of seed matches were seen when the miRNA and mRNA were both up-regulated or were both down-regulated, suggesting a greater possibility of an indirect effect.
Evaluation of mRNA and miRNA with colorectal cancer-specific survival did not reveal any significant associations after adjustment for multiple comparisons.
DISCUSSION
The Wnt-signaling pathway is often dysregulated in CRC [1, 13]. Genes within the pathway, including APC, are among the most commonly mutated genes in CRC [3, 13–15]. In our data, 43.5% of the genes identified in the KEGG Wnt-signaling pathway had significant up or down-regulated expression with a fold change of <0.67 or >1.50, which is slightly greater than when looking at all protein-coding genes in our RNA-Seq data. Thirteen of the 66 dysregulated genes in the Wnt-signaling pathway were associated with miRNA expression. There were 36 seed-region matches between the mRNAs and their associated miRNAs, suggesting that miRNAs had both direct and indirect effects on the Wnt-signaling pathway. Similar miRNAs were associated with genes in the same canonical/non-canonical pathway further suggesting that miRNA::mRNA associations were most likely from feedback and feedforward loops.
The Wnt-signaling pathway has three arms, the canonical Wnt/β-catenin pathway and two non-canonical Wnt-signaling pathways exist, Wnt/CA2+ and Wnt/planar cell polarity [4]. The Wnt/β-catenin pathway is most often studied with CRC and includes CTNBB1 (that codes β-catenin), APC, SMAD3 and SMAD4, c-myc, c-jun, CCND1, TCF7L2, TP53, MMP7, and MAP3K7, all genes that have been associated with CRC [1, 16–22]. β-catenin is a central component of this pathway in terms of regulation of cell growth and metastasis. Wnt-signaling triggers destabilization of β-catenin by the Axin complex. The tumor suppressor, APC, down-regulates β-catenin, as do Axins I and II (also tumor suppressors). Truncating mutations of APC alter the Axin binding sites for β-catenin and thus influences the stability of the β-catenin complex [23, 24]. Frizzled protein and Secreted frizzled-related protein (SFRP) modulate Wnt signaling through direct interaction with Wnt genes and have a role in regulating cell growth and differentiation. SFRP5 has been shown to inhibit PAR1-induced β-catenin stabilization. Hypermethylation of SFRP1 during chronic inflammation has been shown to lead to the occurrence of CRC [25]; SFRP2 also is known to modulate Wnt signaling and can increase B-catenin expression [26].
In our study, expression of several Wnt genes, including WNT5B, WNT1, WNT10B, WNT2B, WNT9A, WNT4, WNT10A, WNT16, and WNT8B were down-regulated with FC of <0.67, while others such as WNT2, WNT11, and WNT5A were up-regulated. Also down-regulated at that level were SFRP1 and SFRP5. APC2 and APC were both statistically significantly down-regulated but with higher FCs (FCs 0.68 and 0.76 respectively). While APC was down-regulated in tumor tissue, this does not necessarily equate mutation, although roughly 80% of the tumors in this study had an APC mutation. APC mutations are usually stop mutations and frame shifts, which would lead to loss of functional protein and possibly less stable mRNA through nonsense-mediated RNA decay [27, 28]. AXIN2, was highly up-regulated in our data. Additionally, expression of CTNNB1 was increased in tumor samples in our data, suggesting that several elements of Wnt-signaling were dysregulated and that the stabilization of β-catenin needed to control cell growth was destroyed. Down-stream of CTNNB1 in this arm of the Wnt-signaling pathway, LEF1, MYC, CCND1, and MMP7 were up-regulated, and TCF7L1 down-regulated.
Several miRNAs were associated with differential gene expression in the canonical Wnt/β-catenin pathway in our data. Most notably, SFRP4 and SFRP5 had nine and 18 associations with miRNAs, CCND1 was associated with 11 miRNAs, MYC was associated with 12 miRNA, and CTNNB1 was associated with four miRNAs. SFRP4 and SFRP5 were both associated with let-7i-5p and with 193b-3p. MiRNA-201a had a seed match with both PRKCB and CCND1. MiR-21-3p had a seed-region match with CTNNB1 while miR-21-5p had a seed-region match with SFRP5 and also was associated with CCND1. There were several miRNAs that had seed region matches with CCND1 that also were associated with MYC, including miR-17-5p, miR-19b-3p, miR-20a-5p and miR-20b-5p. Seed region matches suggest a greater likelihood of a direct association between the mRNA and the miRNA. In most instances of seed region matches in our data both the miRNA and mRNA were either up-regulated or down-regulated, suggesting feedback or feedforward loops influenced the expression profiles though an indirect mechanism. Our data suggest that within the Wnt-signaling pathway miRNAs are associated both directly and indirectly with TG to alter gene expression.
The non-canonical arms of the Wnt-signaling pathways are thought to have indirect associations to the Wnt/β-catenin pathway [1, 2, 7]. Both the Wnt/CA2+ and Wnt/PCP pathways have genes that were either up or down-regulated in our data and some of these genes were associated with miRNA differential expression. The Wnt/PCP pathway is associated with cell orientation during development but is thought also to have a role in metastasis [29]. The Wnt/CA2+-signaling pathway controls intracellular calcium influence and can activate several downstream kinases including CAMKII which has been shown to inhibit β-catenin-dependent transcription [2]. WNT11 is central to the WNT/PCP pathway while WNT5A is involved in the Wnt/CA2+ pathway. Both WNT11 and WNT5A were significantly up-regulated in our data.
Several genes in the Wnt/PCP pathway were differentially expressed in our CRC samples. Several of these genes, including PRICKLE2, DAAM2, ROCK2, and RAC2, also were associated with miRNA expression. RAC2 had a seed-region match with miR-650, ROCK2 had a seed-region match with seven miRNAs, with miR-1243 and miR-204-3p expression being inversely related to ROCK2 expression; DAAM2 had seed region matches with four miRNAs and PRICKLE2 has a seed region match with three miRNAs. There was overlap between miRNAs differentially expressed and genes within the pathways, such as miR-133b being associated with both DAAM2 and PRICKLE2. Again, these findings would support that miRNAs have both direct and indirect associations with these pathways.
The Wnt-signaling pathway has been associated with miRNAs in other studies. Several of these studies have been reviewed by Peng and colleagues [12]. While the review by Peng included non-CRC associations, there were several genes in the Wnt-signaling pathway that were associated with miRNAs and CRC. They reported CRC-specific previous associations between miR-101 and miR-320 and β-catenin, miR-224 and GSK3β, and miR-490-3p and FRAT1. We did not see any of these associations, however we only examined miRNA associations with genes that had a more meaningful FC and since we were examining multiple genes and miRNAs, our correction for multiple comparisons was greater. MiR-221 has been shown to be associated with β-catenin pathways and MYC [30]. In our data miR-221-3p was associated with CTNNB1 and CCND1 (both with seed matches). MiR-29 and miR-30e have been shown to influence Wnt signaling; miR-23b has been shown to inhibit FZD7 translation; miR93 has been shown to down-regulate expression of genes encoding β-catenin, Axin, Myc and Cyclin D; and miR-135a/5 have been shown to suppress APC expression [4]. In our data miR-29b-3p and miR-93-5p had a seed region match with CCND1, and miR-29a-3p was also associated with SFRP5 and ROCK2; while miR-30a-5p was associated with DAAM2, suggesting miRNA regulation of both the canonical and non-canonical Wnt-signaling pathways.
There are strengths and limitations of this study to consider. Although our sample size is small, it is one of the largest available samples with paired tumor/normal data. We focused only those genes that were statistically significant and also had a FC of 1.5 or greater or 0.67 or less. Using these criteria, we did not examine all statistically significant genes and miRNAs that were differentially expressed. Thus, genes like APC, which were statistically significantly down-regulated but the FC was greater than 0.67, were not examined further with miRNAs. A biologically important FC is not well defined, and by using set values for further follow-up we could have missed Wnt-signaling genes associated with miRNAs. Additionally, in our current analysis, we utilized a negative binomial model with a random subject effect. Previously we reported results from DESEQ2 for some of these genes; DESEQ2 uses a fixed effect model and additional normalization and variance reduction methods. Our results vary slightly between the two analytic methods in terms of FC and adjusted p values, although the interpretation of findings is consistent. We exclusively used the KEGG pathway database to identify Wnt-signaling pathway genes. Some genes, such as WTX and AMER1, that have been shown to influence Wnt-signaling [31, 32] were not considered by KEGG to be part of the Wnt-signaling pathway, and therefore we did not include them in our analysis. Thus, other genes may importantly alter Wnt-signaling as well as influence miRNA expression and subsequently mRNA expression within those components of Wnt-signaling that we examined. When evaluating miRNA with mRNAs, we could miss important gene associations since miRNAs have their impact post-transcriptionally. However, much of the current information on miRNA target genes comes from gene expression data and association observed may have important biological meaning, but must be acknowledged as being incomplete [33, 34].
Given the number of genes in the Wnt-signaling pathway that were dysregulated in our study, our data support the importance of this pathway in CRC. Our data also support the hypothesis that miRNAs are involved in this signaling pathway, either through direct binding to the mRNA or through indirect mechanisms. We encourage others to both replicate these findings and to conduct targeted research on the identified associations to further our understanding of this important signaling pathway in the carcinogenic process.
METHODS
Study participants
Study participants come from two population-based case-control studies that included all incident colon and rectal cancer patients diagnosed between 30 to 79 years of age in Utah or who were members of the Kaiser Permanente of Northern California (KPNC). Participants were non-Hispanic white, Hispanic, or black for the colon cancer study; the rectal cancer study also included people of Asian race [35, 36]. Case diagnosis was verified by tumor registry data as a first primary adenocarcinoma of the colon or rectum and occurred between October 1991 and September 1994 (colon study) and between May 1997 and May 2001 (rectal study) [37]. The Institutional Review Boards at the University of Utah and at KPNC approved the study.
RNA processing
Formalin-fixed paraffin embedded tissue from the initial biopsy or surgery was used to extract RNA. RNA was extracted, isolated and purified from carcinoma tissue and adjacent normal mucosa as previously described [38]. We observed no differences in RNA quality based on age of the tissue.
mRNA: RNA-seq sequencing library preparation and data processing
Total RNA from 245 colorectal carcinoma and normal mucosa pairs was chosen for sequencing based on availability of RNA and high quality miRNA data in order to have both mRNA and miRNA from the same individuals; the 217 pairs that passed quality control (QC) were used in these analyses [39]. RNA library construction was performed with the Illumina TruSeq Stranded Total RNA Sample Preparation Kit with Ribo-Zero. The samples were then fragmented and primed for cDNA synthesis, adapters were then ligated onto the cDNA, and the resulting samples were then amplified using PCR; the amplified library was then purified using Agencount AMPure XP beads. A more detailed description of the methods can be found in our previous work [40]. Illumina TruSeq v3 single read flow cell and a 50 cycle single-read sequence run were performed on an Illumina HiSeq instrument. Reads were aligned to a sequence database containing the human genome (build GRCh37/hg19, February 2009 from genome.ucsc.edu) and alignment was performed using novoalign v2.08.01. Total gene counts were calculated for each exon and UTR of the genes using gene coordinates obtained from http://genome.ucsc.edu. We disregarded genes that were not expressed in our RNA-Seq data or for which the expression was missing for the majority of samples [40].
miRNA
The Agilent Human miRNA Microarray V19.0 was used. Data were required to pass stringent QC parameters established by Agilent that included tests for excessive background fluorescence, excessive variation among probe sequence replicates on the array, and measures of the total gene signal on the array to assess low signal. Samples failing to meet quality standards were re-labeled, hybridized to arrays, and re-scanned. If a sample failed QC assessment a second time, the sample was excluded from analysis. The repeatability associated with this microarray was extremely high (r = 0.98) [37]; comparison of miRNA expression levels obtained from the Agilent microarray to those obtained from qPCR had an agreement of 100% in terms of directionality of findings and the FCs were almost identical [41]. To normalize differences in miRNA expression that could be attributed to the array, amount of RNA, location on array, or factors that could erroneously influence miRNA expression levels, total gene signal was normalized by multiplying each sample by a scaling factor which was the median of the 75th percentiles of all the samples divided by the individual 75th percentile of each sample [42].
WNT-signaling genes
The Kyoto Encyclopedia of Genes and Genomes (KEGG) (www.genome.jp/kegg-gin/show_pathway?hsa04310) Pathway map for Wnt-signaling was used to identify genes associated with the canonical and non-canonical Wnt-signaling pathway. Using this map, we identified 138 genes (Supplementary Table 2) in this signaling pathway.
Statistical methods
We utilized negative binomial mixed effects model in SAS (accounting for carcinoma/normal status as well as subject effect) to determine which genes in the Wnt-signaling pathway had a significant difference in expression between individually paired colorectal carcinoma and normal mucosa and their related fold changes (FC). In the negative binomial model we offset the overall exposure as the log of the expression of all identified protein-coding genes (n = 17461). The Benjamini and Hochberg [43] procedure was used to control the false discovery rate (FDR) using a value of 0.05 or less. A FC greater than one indicates a positive differential expression (i.e. up-regulated in carcinoma tissue) while a FC between zero and one indicates a negative differential expression (i.e. down-regulated in carcinoma tissue). We generated the level of expression of each gene by dividing the total expression for that gene in an individual by the total expression of all protein-coding genes per million transcripts (RPMPCG or reads per million protein-coding genes). We considered overall CRC differential expression as well as differential expression for microsatellite unstable (MSI) and stable (MSS) tumors separately since the Wnt-signaling pathway is thought to have a larger role in MSS tumor development.
We arbitrarily focused on those genes and miRNAs with FCs of >1.50 or <0.67 in order to have more meaningful differences between tumor and normal samples. There were 814 miRNAs expressed in greater than 20% of normal colorectal mucosa samples that were analyzed; differential expression was calculated using subject-level paired data as the expression in the carcinoma tissue minus the expression in the normal mucosa. In these analyses, we fit a least squares linear regression model to the RPMPCG differential expression levels and miRNA differential expression levels. P-values were generated using the bootstrap method by creating a distribution of 10,000 F statistics derived by resampling the residuals from the null hypothesis model of no association between gene expression and miRNA expression using the boot package in R. Linear models were adjusted for age and sex. Multiplicity adjustments for gene/miRNA associations were made at the gene level using the FDR by Benjamini and Hochberg [43].
Bioinformatics analysis
We analyzed miRNAs and targeted mRNAs for seed region matches. The mRNA 3’ UTR FASTA as well as the seed region sequence of the associated miRNA were analyzed to determine seed region pairings between miRNA and mRNA. MiRNA seed regions were calculated as described in our previous work [44]; we calculated and included seeds of six, seven, and eight nucleotides in length. Our hypothesis is that a seed match would increase the likelihood that identified genes associated with a specific miRNA were more likely to have a direct association given a higher propensity for binding. As miRTarBase [33] uses findings from many different investigations spanning across years and alignments, we used FASTA sequences generated from both GRCh37 and GRCh38 Homo sapiens alignments, using UCSC Table Browser (https://genome.ucsc.edu/cgi-bin/hgTables) [45]. We downloaded FASTA sequences that matched our Ensembl IDs and had a consensus coding sequences (CCDS) available. Analysis was done using scripts in R 3.2.3 and in perl 5.018002.
Author contributions
MS obtained funding, planned study, oversaw study data collection and analysis, and wrote the manuscript. JS provided input into the statistical analysis. LM conducted bioinformatics analysis and helped write manuscript. LS provided data and input into the manuscript. RW oversaw laboratory analysis and gave input into data interpretation. WS reviewed and edited the manuscript and did pathology overview for the study. JH conducted statistical analysis and managed data. All authors approved final manuscript.
ACKNOWLEDGMENTS AND FUNDING
The contents of this manuscript are solely the responsibility of the authors and do not necessarily represent the official view of the National Cancer Institute. We acknowledge Sandra Edwards for data oversight and study management, and Michael Hoffman and Erica Wolff for miRNA analysis. We acknowledge Dr. Bette Caan and the staff at the Kaiser Permanente Medical Research Program for sample and data collection. This study was supported by NCI grants CA163683 and CA48998.
CONFLICTS OF INTEREST
None.
REFERENCES
1. Polakis P. Wnt signaling in cancer. Cold Spring Harb Perspect Biol. 2012; 4.
2. Onyido EK, Sweeney E, Nateri AS. Wnt-signalling pathways and microRNAs network in carcinogenesis: experimental and bioinformatics approaches. Mol Cancer. 2016; 15:56.
3. Samowitz WS, Slattery ML, Sweeney C, Herrick J, Wolff RK, Albertsen H. APC mutations and other genetic and epigenetic changes in colon cancer. Mol Cancer Res. 2007; 5:165–70.
4. Song JL, Nigam P, Tektas SS, Selva E. microRNA regulation of Wnt signaling pathways in development and disease. Cell Signal. 2015; 27:1380–91.
5. Ishitani T, Kishida S, Hyodo-Miura J, Ueno N, Yasuda J, Waterman M, Shibuya H, Moon RT, Ninomiya-Tsuji J, Matsumoto K. The TAK1-NLK mitogen-activated protein kinase cascade functions in the Wnt-5a/Ca(2+) pathway to antagonize Wnt/beta-catenin signaling. Mol Cell Biol. 2003; 23:131–9.
6. Saadeddin A, Babaei-Jadidi R, Spencer-Dene B, Nateri AS. The links between transcription, beta-catenin/JNK signaling, and carcinogenesis. Mol Cancer Res. 2009; 7:1189–96.
7. Bordonaro M, Tewari S, Cicco CE, Atamna W, Lazarova DL. A switch from canonical to noncanonical Wnt signaling mediates drug resistance in colon cancer cells. PLoS One. 2011; 6:e27308.
8. Cai X, Hagedorn CH, Cullen BR. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA. 2004; 10:1957–66.
9. Forstemann K, Tomari Y, Du T, Vagin VV, Denli AM, Bratu DP, Klattenhoff C, Theurkauf WE, Zamore PD. Normal microRNA maturation and germ-line stem cell maintenance requires Loquacious, a double-stranded RNA-binding domain protein. PLoS Biol. 2005; 3:e236.
10. Wu Q, Qin H, Zhao Q, He XX. Emerging role of transcription factor-microRNA-target gene feed-forward loops in cancer. Biomed Rep. 2015; 3:611–6.
11. Slattery ML, Herrick JS, Mullany LE, Samowitz WS, Sevens JR, Sakoda L, Wolff RK. The co-regulatory networks of tumor suppressor genes, oncogenes, and miRNAs in colorectal cancer. Genes Chromosomes Cancer. 2017; 56:769–87.
12. Peng Y, Zhang X, Feng X, Fan X, Jin Z. The crosstalk between microRNAs and the Wnt/β-catenin signaling pathway in cancer. Oncotarget. 2017; 8:14089–14106. https://doi.org/10.18632/oncotarget.12923.
13. Schneikert J, Behrens J. The canonical Wnt signalling pathway and its APC partner in colon cancer development. Gut. 2007; 56:417–25.
14. Curtin K, Wolff RK, Herrick JS, Abo R, Slattery ML. Exploring multilocus associations of inflammation genes and colorectal cancer risk using hapConstructor. BMC Med Genet. 2010; 11: 170.
15. Jass JR, Barker M, Fraser L, Walsh MD, Whitehall VL, Gabrielli B, Young J, Leggett BA. APC mutation and tumour budding in colorectal cancer. J Clin Pathol. 2003; 56:69–73.
16. Brudvik KW, Paulsen JE, Aandahl EM, Roald B, Tasken K. Protein kinase A antagonist inhibits beta-catenin nuclear translocation, c-Myc and COX-2 expression and tumor promotion in Apc(Min/+) mice. Mol Cancer. 2011; 10:149.
17. Ciznadija D, Tothill R, Waterman ML, Zhao L, Huynh D, Yu RM, Ernst M, Ishii S, Mantamadiotis T, Gonda TJ, Ramsay RG, Malaterre J. Intestinal adenoma formation and MYC activation are regulated by cooperation between MYB and Wnt signaling. Cell Death Differ. 2009; 16:1530–8.
18. Al-Kuraya K, Novotny H, Bavi P, Siraj AK, Uddin S, Ezzat A, Sanea NA, Al-Dayel F, Al-Mana H, Sheikh SS, Mirlacher M, Tapia C, Simon R, et al. HER2, TOP2A, CCND1, EGFR and C-MYC oncogene amplification in colorectal cancer. J Clin Pathol. 2007; 60:768–72.
19. Lan F, Yue X, Han L, Shi Z, Yang Y, Pu P, Yao Z, Kang C. Genome-wide identification of TCF7L2/TCF4 target miRNAs reveals a role for miR-21 in Wnt-driven epithelial cancer. Int J Oncol. 2012; 40:519–26.
20. Slattery ML, Folsom AR, Wolff R, Herrick J, Caan BJ, Potter JD. Transcription Factor 7-like 2 Polymorphism and Colon Cancer. Cancer Epidemiol Biomarkers Prev. 2008; 17:978–82.
21. Broussard EK, Kim R, Wiley JC, Marquez JP, Annis JE, Pritchard D, Disis ML. Identification of putative immunologic targets for colon cancer prevention based on conserved gene upregulation from preinvasive to malignant lesions. Cancer Prev Res (Phila). 2013; 6:666–74.
22. Slattery ML, Lundgreen A, Wolff RK. MAP kinase genes and colon and rectal cancer. Carcinogenesis. 2012; 33:2398–408.
23. Polakis P. Drugging Wnt signalling in cancer. EMBO J. 2012; 31:2737–46.
24. Bienz M, Clevers H. Linking colorectal cancer to Wnt signaling. Cell. 2000; 103:311–20.
25. Katoh Y, Katoh M. Hedgehog signaling pathway and gastrointestinal stem cell signaling network (review). Int J Mol Med. 2006; 18:1019–23.
26. Mirotsou M, Zhang Z, Deb A, Zhang L, Gnecchi M, Noiseux N, Mu H, Pachori A, Dzau V. Secreted frizzled related protein 2 (Sfrp2) is the key Akt-mesenchymal stem cell-released paracrine factor mediating myocardial survival and repair. Proc Natl Acad Sci USA. 2007; 104:1643–8.
27. Popp MW, Maquat LE. Leveraging Rules of Nonsense-Mediated mRNA Decay for Genome Engineering and Personalized Medicine. Cell. 2016; 165:1319–22.
28. Martin L, Grigoryan A, Wang D, Wang J, Breda L, Rivella S, Cardozo T, Gardner LB. Identification and characterization of small molecules that inhibit nonsense-mediated RNA decay and suppress nonsense p53 mutations. Cancer Res. 2014; 74:3104–13.
29. Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene. 2017; 36:1461–73.
30. Lupini L, Bassi C, Ferracin M, Bartonicek N, D’Abundo L, Zagatti B, Callegari E, Musa G, Moshiri F, Gramantieri L, Corrales FJ, Enright AJ, Sabbioni S, et al. miR-221 affects multiple cancer pathways by modulating the level of hundreds messenger RNAs. Front Genet. 2013; 4:64.
31. Tanneberger K, Pfister AS, Kriz V, Bryja V, Schambony A, Behrens J. Structural and functional characterization of the Wnt inhibitor APC membrane recruitment 1 (Amer1). J Biol Chem. 2011; 286:19204–14.
32. Major MB, Camp ND, Berndt JD, Yi X, Goldenberg SJ, Hubbert C, Biechele TL, Gingras AC, Zheng N, Maccoss MJ, Angers S, Moon RT. Wilms tumor suppressor WTX negatively regulates WNT/beta-catenin signaling. Science. 2007; 316:1043–6.
33. Chou CH, Chang NW, Shrestha S, Hsu SD, Lin YL, Lee WH, Yang CD, Hong HC, Wei TY, Tu SJ, Tsai TF, Ho SY, Jian TY, et al. miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 2016; 44:D239–47.
34. Slattery ML, Herrick JS, Stevens JR, Wolff RK, Mullany LE. An Assessment of Database-Validated microRNA Target Genes in Normal Colonic Mucosa: Implications for Pathway Analysis. Cancer Inform. 2017; 16:1176935117716405.
35. Slattery ML, Potter J, Caan B, Edwards S, Coates A, Ma KN, Berry TD. Energy balance and colon cancer—beyond physical activity. Cancer research. 1997; 57:75–80.
36. Slattery ML, Caan BJ, Benson J, Murtaugh M. Energy balance and rectal cancer: an evaluation of energy intake, energy expenditure, and body mass index. Nutr Cancer. 2003; 46:166–71.
37. Slattery ML, Herrick JS, Pellatt DF, Stevens JR, Mullany LE, Wolff E, Hoffman MD, Samowitz WS, Wolff RK. MicroRNA profiles in colorectal carcinomas, adenomas and normal colonic mucosa: variations in miRNA expression and disease progression. Carcinogenesis. 2016; 37:245–61.
38. Slattery ML, Herrick JS, Mullany LE, Valeri N, Stevens J, Caan BJ, Samowitz W, Wolff RK. An evaluation and replication of miRNAs with disease stage and colorectal cancer-specific mortality. Int J Cancer. 2015; 137:428–38.
39. Pellatt AJ, Slattery ML, Mullany LE, Wolff RK, Pellatt DF. Dietary intake alters gene expression in colon tissue: possible underlying mechanism for the influence of diet on disease. Pharmacogenet Genomics. 2016; 26:294–306.
40. Slattery ML, Pellatt DF, Mullany LE, Wolff RK, Herrick JS. Gene expression in colon cancer: A focus on tumor site and molecular phenotype. Genes Chromosomes Cancer. 2015; 54:527–41.
41. Pellatt DF, Stevens JR, Wolff RK, Mullany LE, Herrick JS, Samowitz W, Slattery ML. Expression Profiles of miRNA Subsets Distinguish Human Colorectal Carcinoma and Normal Colonic Mucosa. Clin Transl Gastroenterol. 2016; 7:e152.
42. Agilent Technologies I. Agilent GeneSpring User Manual Santa Clara, CA: Aglient Technologies Inc; 2013.
43. Benjamini YH. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser A Stat Soc. 1995; 57:289–300.
44. Mullany LE, Herrick JS, Wolff RK, Slattery ML. MicroRNA Seed Region Length Impact on Target Messenger RNA Expression and Survival in Colorectal Cancer. PLoS One. 2016; 11:e0154177.
45. Karolchik D, Hinrichs AS, Furey TS, Roskin KM, Sugnet CW, Haussler D, Kent WJ. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004; 32:D493–96.