
INTRODUCTION
T-cell acute lymphoblastic leukemia (T-ALL) is a lymphoid malignancy caused by oncogenic transformation of immature T-cell progenitors [1]. This aggressive hematologic malignancy is typically characterized by diffuse infiltration of the bone marrow by malignant lymphoblasts expressing immature T-cell surface markers. Currently, T-ALL accounts for 20−25% of adult and 10−15% of pediatric cases of ALL [2, 3]. Clinically, patients with T-ALL typically present with high white cell counts in the blood. Patient symptoms include: hematopoiesis inhibition with anaemia, pyrexia, thrombocytopenia, hemorrhage, infection, frequent large thymic masses, as well as pleural, and meningeal infiltration at diagnosis. Abnormal genetic or epigenetic alterations can lead to an arrest of differentiation and uncontrolled expansions of immature thymocytes with distinct gene expression patterns during T-cell development. These alterations end up, resulting in the transformation of normal T-cells into fully transformed T-ALL lymphoblasts [4]. T-ALL can be classified into different clinically relevant biological subgroups according to unique gene expression signatures [5, 6] and distinct immunophenotypes that reflect immature thymocytes arresting at different stages of differentiation [7]. Generally, T-ALL can be classified in three categories. Early T-cell precursor acute lymphoblastic leukemia (ETP-ALL) is blocked at the earliest stages of T-cell differentiation with a distinctive immunophenotype (CD1a−CD8−CD5weak) and presenting stem-cell/myeloid markers [8]. ETP-ALL has a lower frequency of NOTCH1 mutations and homozygous cyclin-dependent kinase inhibitor 2A/2B (CDKN2A/CDKN2B) deletions. Genetically it is characterized by mutations in genes regulating cytokine receptors and signaling that disrupts hematopoietic development, histone-modifying genes, and epigenetic regulators [9, 10]. In contrast, the other two subgroups of T-ALL frequently show NOTCH1 mutations and homozygous CDKN2A/CDKN2B deletions. A second subgroup of T-ALLs with a CD1a+CD4+CD8+ immunophenotype has an excellent prognosis related to the early stages of cortical thymocyte maturation [11]. This subgroup typically shows activation of the T-cell leukemia homeobox 1 (TLX1/HOX11), T-cell leukemia homeobox 3 (TLX3/HOX11L2), NK2 homeobox 1 (NKX2-1) and NK2 homeobox 2 (NKX2-2) genes [5, 12]. Another subgroup of T-ALLs correlated with a late cortical thymocyte immunophenotype (CD4+CD8+CD3high) typically display activation of the T-cell acute lymphocytic leukemia 1/stem cell leukemia (TAL1/SCL) oncogene [5].
T-ALL arises as result of a multistep oncogenic process. Many biologically relevant genetic and epigenetic alterations have been identified as driving events for this process. These events coordinately disrupt key oncogenic, tumor suppressive and developmental pathways responsible for the normal control of cell growth, proliferation, motility, survival and differentiation during thymocyte development. One of the key regulators in early T-cell development and T-ALL oncogenic signals is NOTCH1, which is constitutively active in more than 50% of T-ALLs [13]. NOTCH1-activating mutations frequently co-occur with loss of the CDKN2A locus [9]. Together these result in the aberrant expression of various T-cell fate specific and thymocyte development related transcription factors and epigenetic regulators that can become oncogenes or tumor suppressors. Mutated-NOTCH1 forms regulatory networks with these transcription factors and epigenetic regulators contributing to T-ALL development [1, 14].
Currently, intensified chemotherapy in T-ALL treatment results in a favorable overall survival for patients with 80% in children [15] and 60% in adults [16]. However, the concomitant side effects on the central nervous system and bone development should not be underestimated [17]. The prognosis of T-ALL patients with primary resistance who fail to achieve complete remission or those suffering from relapses after an initial treatment remains poor [18], with a recurrence rate of up to 20% in pediatric and 40% in adult patients [15]. A better understanding of the molecular mechanisms of T-ALL needed to identify more specific targets and develop less toxic anti-leukemic drugs. Over the past few decades, great progress has been made in understanding of the genetics and biology of T-ALL, especially on protein coding genes. More recently, through the study of mutations in the noncoding regions of the genome that lead to aberrant transcription factor expression in T-ALL, a number of microRNAs (miRNAs) were identified. These miRNAs have been linked with these genes and play an important role in pathology of T-ALL [19].
MiRNAs were first discovered in 1993 through the analysis of developmental timing mutants in C. elegans [20, 21]. Since then, over 2,500 miRNAs have been discovered so far in the human genome (http://www.mirbase.org). They are an endogenous, non-coding RNA, composed of 22–24 nucleotides (nt), which function as post-transcriptional gene regulators [22]. MiRNA biogenesis starts with long primary miRNA (pri-miRNA, 60-80nt) transcripts containing one or more hairpin structures produced by RNA polymerase II or III in the nucleus. They are then further processed into a smaller stem-loop (approximately 70 nt) precursor miRNA (pre-miRNA) by RNase III-type endonuclease Drosha. The pre-miRNA is then transported to the cytoplasm by Exportin 5 and is cleaved into a mature double stranded miRNA molecule (approximately 22 nt) by the Dicer ribonuclease. The guide strand is then incorporated into the RNA-induced silencing complex (RISC), guiding RISC to its target messenger RNA (mRNA) through complementary base pairing on the 3′ untranslated region (3′UTR) of the target mRNA. This pairing and binding to the target mRNA leads to mRNA degradation or translation repression [23, 24]. One third of protein encoding genes are regulated by miRNAs, including genes involved in physiological processes [25, 26] as well as with the pathological development and progression of cancer [27, 28]. Recently, miRNAs have been shown to influence various leukemias, including T-ALL. Aberrant expression of miRNAs can function as either oncogenes or tumor suppressors in T-ALL. MiRNAs through the regulation of cell migration, invasion, proliferation, apoptosis, and chemoresistance by targeting key signaling pathways or transcriptional factors play a critical role in T-ALL pathology and progression [19, 29, 30]. Additionally, different miRNAs expression profiles have been linked to specific genetic subtypes of human T-ALL [31, 32]. Furthermore, miRNAs can also act as independent prognostic factors to predict clinical outcomes for T-ALL patients [33]. In the current review, we will focus on the role of miRNAs in the development and progression of T-ALL.
MiRNAs are differentially expressed in T-ALL subpopulation
T-ALLs can be classified into different genetic subtypes according to unique protein coding gene expression signatures and immunophenotypes as described above [5, 8, 11]. Consistently, studies have found that different genetic subgroups of T-ALL also had subtype-specific miRNA expression profiles. Research by Nagel et al. [34] first demonstrated that the miR-17-92 cluster was overexpressed in cell lines and primary cells that were positive for the expression of TLX1, TLX3 and NK2 homeobox 2-5 (NKX2-5). These are members of the NK-like family of homeobox genes which are ectopically activated in the subtype of T-ALL cells with a CD1a+CD4+CD8+ immunophenotype [5, 12]. This miRNA cluster was regulated by these transcription factors leading to the enhanced survival of leukemic T-cells though decreasing E2F transcription factor 1 (E2F1) protein expression [34]. MiR-223 was found overexpressed in a subset of myeloid-like adult T-ALLs which appeared to have an unfavorable clinical outcome [31].
Aberrant expression of homeobox A (HOXA) genes are one of the diverse characteristics of T-cell-specific transcription factors that can function as oncogenes [6]. High expression of miR-196b specifically occurred in MLL-rearranged and T-ALL patients carrying CALM-AF10 or SET-NUP214 fusions and inversion of chromosome 7. These molecular and morphological changes are functionally linked with upregulation of HOXA. Since miR-196b is encoded on the HOXA cluster, miR-196b and HOXA genes might be co-activated in acute lymphoblastic leukemia [35]. In addition, miR-196a and miR-196b expression was associated with an immature (IMM) immunophenotype and expression of CD34 and CD33, with both targeting ETS transcription factor ERG in T-ALL patients [36].
Furthermore, Coskun et al. [32] identified a set of miRNAs that were differently expressed in adult ETP-ALL by comparing miRNA profiles between ETP-ALL and non-ETP T-ALL patients. Their results included two of the most upregulated (miR-221 and miR-222) and six downregulated miRNAs (miR-151-3p, miR-19a, miR-20b, miR-342-3p, miR-363, and miR-576-3p). It was also discovered that miR-222 inhibited proliferation, caused cell cycle arrest and apoptosis in leukemic cells by directly inhibiting the expression of the proto-oncogene ETS1 through in vitro studies, which has been shown to be concomitant with the poor prognosis of ETP-ALL.
These data suggest that miRNAs are differentially expressed in subpopulations of T-ALL as genetic alterations and the illumination of subtype-specific miRNA expression profiling could contribute to better diagnosis and treatment for T-ALL patients. However, more comprehensive studies remain to be reported.
MiRNAs expression and signaling pathways in T-ALL
Oncogenic NOTCH1 signal in T-ALL
The NOTCH1 signaling pathway is prominent for early T-cell fate specification, which is the process by which multipotent hematopoietic progenitors commit to T-cell lineage, as well as further thymocyte development [37, 38]. Aberrant, constitutively active NOTCH1 signaling is the predominant oncogenic event involved in the pathogenesis of T-ALL [13]. NOTCH1 is a class I transmembrane glycoprotein composed of functional extracellular (NECD), transmembrane (TM), and intracellular (NICD) domains. NOTCH1 functions as a receptor for membrane-bound ligands Jagged1, Jagged2 and Delta1, in order to regulate cell-fate determination. Upon ligand activation, the NICD is released through cleavage by a member of a disintegrin and metalloproteinase (ADAM) family of proteases and the gamma secretase complex, and then translocates to the nucleus. In the nucleus, NICD binds to the CBF-1/suppressor of hairless/Lag-1 (CSL) transcription factor complex, resulting in the subsequent activation of the canonical Notch target genes [39]. Many canonical downstream targets that mediate oncogenic activity of NOTCH1 in T-ALL have been described [1, 40]. Recently, it has become increasingly evident that alterations in the expression of miRNAs also play an important role in the NOTCH1 regulatory network during T-ALL progression.
NOTCH1-driven T-ALL mouse model
The oncogenic role of NOTCH1 in T-ALL can be illustrated by the rapid development of acute leukemia in murine models. Currently a mouse model is being used that transplants hematopoietic progenitors with a constitutively active, intracellular form of NOTCH1 [41]. Much progress has been made in elucidation of the oncogenic or tumor suppressor role of miRNAs in NOTCH1-driven T-ALL with this model. To establish this model, fetal liver cells with hematopoietic progenitor cells (HPCs) isolated from pregnant mice were retrovirally transduced with an intracellular NOTCH1 (ICN1) construct as well as a construct encoding antagomiR or premiR. These HPCs were subsequently injected into the tail vein of lethally irradiated recipient mice [41, 42]. This model has also been modified where, the transduced HPCs can also be directly injected into irradiated syngeneic mice via the tail vein [43]. The T-ALL onset was then monitored and the miRNA profiles could be used to identify potential oncogenic or tumor suppressive miRNAs as compared with control mice.
Konstantinos et al. [44] demonstrated that a novel translocation targeting the miR-17-92 cluster coincided with a second rearrangement that activates NOTCH1 in T-ALL. Among these are miR-19, which showed the highest expression of all members of the miR-17-92 cluster in human T-ALL. This expression of miR-19 is sufficient enough to promote leukemogenesis in NOTCH1-induced T-ALL in vivo through coordination a phosphatidylinositol-3-OH kinase (PI3K) pathway related program of cell survival. The PI3K pathway related program directly targets its components including BIM (BCL2L11), AMP-activated kinase (PRKAA1), and tumor suppressor phosphatases PTEN and PP2A (PPP2R5E). These observations indicated a collaborative role of NOTCH1 and the miR-17-92 cluster in T-ALL development. Moreover, four miRNAs in the miR-17-92 cluster and four important genes (CYLD, HOXA9, BCL2L11 and RUNX1) were found in a “miRNAs and genes co-regulatory network” by a target prediction algorithms assay of genes and miRNAs known to be involved in T-ALL via bioinformatics [45]. MiR-19 was confirmed to regulate NF-κB signaling through direct targeting of CYLD with further in vitro experiments [45]. Mavrakis et al. [46] further identified five miRNAs (miR-19b, miR-20a, miR-26a, miR-92 and miR-223) that contributed to leukemogenesis and acted as multi-targeted regulators of several tumor suppressor genes (IKAROS/IKZF1, PTEN, BIM, PHF6, NF1 and FBXW7). The Mavrakis’s research discovered these miRNAs through cross-comparison of miRNA expression profiles in human T-ALL with an unbiased genetic screen and computational analyses. Three of these miRNAs (miR-19b, miR-20a, and miR-92) belong to the oncogenic miR-17-92 cluster. All five miRNAs could promote T-ALL development in a NOTCH1-driven mouse model. These miRNAs had overlapping and cooperative effects on tumor suppressor genes with miR-19b directly targeting PTEN and BIM; miR-20a directly targeting PTEN, BIM and PHF6; miR-92 directly targeting IKAROS/IKZF1, PTEN, BIM, NF1 and FBXW7, and miR-223 directly targeting FBXW7, respectively. MiR-223 was subsequently shown to be activated by TAL1 [47] and NOTCH1 [48], two important T-ALL oncogenes. Mets et al. further investigated miRNAs that directly target the tumor suppressor PHF6 by performing an unbiased PHF6 3′UTR-microRNA library screen and combined the results with microRNA profiling data of samples from patients with T-ALL. MiR-128-3p was selected as a candidate for PHF6-targeting, which could significantly accelerate leukemia onset in a NOTCH1-induced T-ALL mouse model upon over-expression [49]. In addition, another study demonstrated that Dicer1-mediated miRNA biogenesis was essential for development, progression, and maintenance of NOTCH1-driven T-ALL in leukemic mice, which is a key component of the miRNA processing machinery [50]. With conditional knockout, the biallelic loss of Dicer1 resulted in the abrogation of established NOTCH-induced T-ALLs, indicating that the NOTCH-on transformed state is dependent on one or more miRNAs. They further delineated that miR-21 promoted survival of T-ALL cells, in part through the repression of the tumor suppressor gene programmed cell death 4 (PDCD4) in NOTCH-addicted murine and human T-ALL cell lines based on miRNA expression profile data and in vitro experiments.
Many tumor-suppressive miRNAs have also been identified with NOTCH1-sensitized T-ALL mouse model. Sanghvi et al. characterized a set of functionally interconnected tumor-suppressive miRNAs with this model [43]. First, they screened miRNAs that exhibited decreased expression by at least 10-fold in primary T-ALL samples as compared to normal tissues. They then performed functional assessments by in vitro proliferation assays and in vivo miRNA loss-of-function studies evaluating their leukemogenesis effects. They accomplished this in a murine, NOTCH1-driven, T-ALL model with miRNAs knockout, miRNA sponges or lentiviral miRZips approaches. After selection, five miRNAs were characterized (miR-29, miR-31, miR-150, miR-155, and miR-200), all with tumor-suppressive effects in NOTCH1-driven T-ALL model. Using a computational strategy to eliminate genes that were exclusively targeted by the tumor-suppressive miRNAs, they identified the known T-ALL oncogene MYB (target of miR-150, miR-155, and miR-200). They also discovered a potential new oncogene in T-ALL, HBP1 (target of miR-29, miR-31, miR-155, and miR-200), which encodes a transcription factor. Their research then focused on these genes as key targets of this tumor-suppressive miRNA network. The expression of both genes was increased in T-ALL patient samples, and each gene promoted the progression of T-ALL in mice. Interestingly, it was also noted that the oncogenic NOTCH1/MYC pathway inhibited miR-31, miR-150, and miR-155 expression. This suggests that repression of the expression of a subset of tumor suppressor miRNAs in T-ALL cells might be a result of NOTCH and MYC activation. Mets et al. [42] focused on the oncogene MYB with post-transcriptional regulation by miRNAs in T-ALL through an unbiased, high-throughput dual luciferase-based MYB 3′UTR-miRNA library screen. They employed subsequent analyses of miRNA-mRNA correlation data during normal T-cell differentiation and in primary T-ALL patient samples. Followed by in vitro luciferase reporter assay, miR-193b-3p was detected as a direct negative regulator of MYB. Remarkably, downregulation of miR-193b-3p by antagomiR in the NOTCH1-induced mouse model significantly accelerated leukemia onset in vivo, suggesting the inhibition of tumor-suppressor miR-193b-3p can cooperate with NOTCH1 in T-ALL pathogenesis.
MiRNAs directly involved in NOTCH1 oncogenic signaling in T-ALL
In NOTCH1-dependent T-ALL models, sustained NOTCH1-receptor signaling activates a broad array of genes involved in anabolic pathways, among which NOTCH1/MYC regulatory axis plays a major role. This results in NOTCH1-induced transformation through a feed-forward loop transcriptional circuitry [51]. This critical feed-forward loop regulatory network can be directly regulated by miRNAs [52, 53]. MiR-451 and miR-709 have been shown to modulate MYC expression and also act as downstream targets of NOTCH1 in murine T-ALL [52]. In this study, activation of NOTCH1 signaling by induction of ICN1 led to repression of miR-451 and miR-709 in an established mouse model of T-ALL. These repression effects were mediated by degradation of the E2A tumor suppressor followed by ICN1 induction, which transcriptionally activates the genes encoding miR-451 and miR-709. Importantly, both miR-451 and miR-709 directly repressed MYC expression and miR-709 also directly targeted the AKT and Ras-GRF1 oncogenes. It was also demonstrated that decreased expression of miR-451 and miR-709 was necessary for initiation and maintenance of mouse T-ALL. Thus, miR-451 and miR-709 functioned as suppressors of oncogenesis within the NOTCH1/MYC regulatory axis of murine T-ALL. The NOTCH1/miR-451/MYC axis also played a part in human T-ALL (only miR-451 has human homologue). Moreover, MYC could also modulate NOTCH1 expression through a feed-forward loop via a miRNA intermediary [53]. The expression level of miR-30a, a member of a family of miRNAs that are transcriptionally suppressed by MYC [54], was negatively correlated with NOTCH1 mutational status in T-ALL patient samples. Sustained NOTCH1 signaling upregulated MYC expression which resulted in decreased expression of miR-30a. MiR-30a could directly bind to 3′UTR of NOTCH1 and inhibit NOTCH1 expression. These discoveries depict a MYC/miR-30a/NOTCH1 feedback regulatory loop that by MYC-mediated inhibition of miR-30a de-repressed NOTCH1, eventually increasing its own expression [53]. Another miRNA that directly targeted NOTCH1 was miR-101, which was downregulated in T-ALL patient specimens and T-ALL cell lines [55]. MiR-101 acts as a tumor suppressor through the repression of proliferation and invasion, induction of apoptosis, and enhancement of chemotherapeutic sensitivity in T-ALL cells in vitro mediated by inhibiting NOTCH1 [19]. As discussed above, it was also shown that the oncogenic NOTCH1/MYC pathway inhibited the expression of tumor suppressor miRNAs, including miR-31, miR-150, and miR-155 [43]. Thus, NOTCH, MYC, and miRNAs formed a complicated and very important feedback regulatory network with each other which contributed to pathogenesis of T-ALL.
MiR-223 has been found to be overexpressed in a subset of T-ALLs and directly targeted FBXW7, a known tumor suppressor gene in T-ALL [31, 46]. Further studies by putative promoter analysis, luciferase and chromatin immunoprecipitation (ChIP) assays revealed that both NOTCH and NF-κB signal could directly and cooperatively activate the transcriptional activity of miR-223 promoter. Moreover, it also confirmed the negative regulation of FBXW7 by miR-223 [48]. Similarly, another group [56] identified that deletion of miR-181ab1 expression inhibited the development of NOTCH1-induced T-ALL in mouse models and human cells through direct targeting of NOTCH regulated ankyrin repeat protein (NRARP), a negative regulator of NOTCH1 downstream signaling. Intriguingly, miR-181ab1 could control the strength and threshold of NOTCH activity in tumorigenesis where the miR-181ab1 deletion appeared to have stronger inhibitory effects on T-ALL cells with lower levels of ICN1 expression. It was also shown that miR-181ab1 could be specifically targeted to inhibit tumor development with little toxicity to normal development, in that the effects of miR-181ab1 deletion were compensated for during normal thymic progenitor development but not during T-ALL development [56].
An overview of the described miRNAs involved in NOTCH-driven T-ALL and in NOTCH1/MYC axis can be found in Figures 1, 2 and in Table 1.
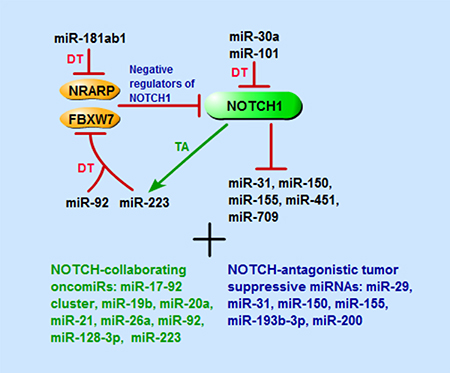
Figure 1: Schematic representation of miRNAs that are involved in NOTCH1-driven T-ALL, including miRNAs directly target or be targeted by NOTCH1 signaling pathway components, and that have collaborating or antagonistic effects with NOTCH pathway. TA, transcriptional activation; DT, direct targeting.
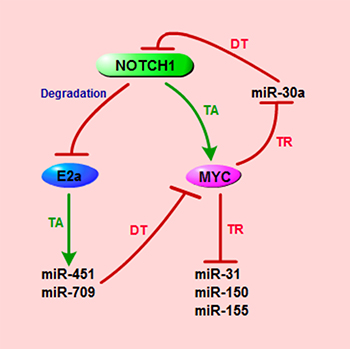
Figure 2: Schematic representation of miRNAs that are implicated in NOTCH1/MYC axis in T-ALL. TA, transcriptional activation; TR, transcriptional repression; DT, direct targeting.
Table 1: miRNAs involved in T-ALL biology
miRNAs in/with |
Deregulation |
Direct targets |
Function |
References |
|---|---|---|---|---|
Pathways |
||||
NOTCH |
||||
miR-30a |
downregulation |
NOTCH1 |
MYC repressed; forms a MYC/miR-30a/NOTCH1 feed-forward regulatory loop in NOTCH-driven T-ALL; represses growth; promotes apoptosis |
[53] |
miR-31 |
downregulation |
HBP1 |
tumor suppressor in NOTCH1-driven T-ALL model; NOTCH1/MYC pathway repressed |
[43] |
miR-92 |
upregulation |
FBXW7 |
oncomiR; induces NOTCH1 expression |
[46] |
miR-101 |
downregulation |
NOTCH1 |
represses proliferation and invasion; induces apoptosis; enhances chemotherapeutic sensitivity |
[19] |
miR-150 |
downregulation |
MYB |
tumor suppressor in NOTCH1-driven T-ALL model; NOTCH1/MYC pathway repressed |
[43] |
miR-155 |
downregulation |
MYB, HBP1 |
tumor suppressor in NOTCH1-driven T-ALL model; NOTCH1/MYC pathway repressed |
[43] |
miR-181ab1 |
upregulation |
NRARP |
controls the activity of NOTCH in tumorigenesis of NOTCH1-driven T-ALL |
[56] |
miR-223 |
upregulation |
FBXW7 |
myeloid-like high; NOTCH1 activated; induces NOTCH1 expression; promotes growth |
|
miR-451 |
downregulation |
MYC |
NOTCH1 repressed; tumor suppressor among NOTCH1/MYC regulatory axis of mouse and human T-ALL |
[52] |
miR-709 |
downregulation |
MYC, AKT, Ras-GRF1 |
NOTCH1 repressed; tumor suppressor among NOTCH1/MYC regulatory axis of mouse T-ALL |
[52] |
PI3K/AKT |
||||
miR-19 |
upregulation |
PP2A, PRKAA1, BIM, PTEN |
increases phosphorylation of AKT and the ribosomal S6 protein; promotes survival |
[44] |
NF-κB |
||||
miR-19 |
upregulation |
CYLD |
induces downstream of NF-κB; |
[45] |
miR-181a |
upregulation |
− |
induces chemoresistance through activating AKT |
[59] |
miR-223 |
upregulation |
FBXW7 |
NF-κB activated |
|
cAMP/PKA |
||||
miR-142-3p |
upregulation |
GRa, cAMP/ PKA |
promotes growth; induces glucocorticoid resistance; correlates with poor prognosis |
[57] |
JAK/STAT |
||||
miR-21 |
upregulation |
− |
inhibits STAT3 protein expression, promotes proliferation and invasion; decreases apoptosis |
[58] |
Transcription factors |
||||
TAL1 |
||||
miR-101 |
downregulation |
TAL1 |
targets TAL1 |
[55] |
miR-140-5p |
downregulation |
TAL1 |
targets TAL1 |
[55] |
miR-146b-5p |
downregulation |
− |
TAL1 repressed; inhibits migration and delays T-ALL progression |
[63] |
miR-223 |
upregulation |
FBXW7 |
TAL1 repressed; mediates TAL1-induced growth |
[47] |
miR-448 |
downregulation |
TAL1 |
targets TAL1 |
[55] |
miR-485-5p |
downregulation |
TAL1 |
targets TAL1 |
[55] |
miR-520d-5p |
− |
TAL1 |
targets TAL1 |
[55] |
HOXA |
||||
miR-196a |
HOXA high |
ERG |
associated with an IMM and expression of CD34 and CD33 |
[36] |
miR-196b |
HOXA and IMM high |
ERG |
co-activated with HOXA; associated with an IMM and expression of CD34 and CD33 |
[36] |
SOX4 |
||||
miR-204 |
downregulation |
SOX4 |
inhibits proliferation and metastasis |
[64] |
JunB |
||||
miR-149* |
upregulation |
JunB |
promotes proliferation and suppresses apoptosis |
[30] |
EGR1 |
||||
miR-181a |
upregulation |
EGR1 |
enhances proliferation |
[66] |
Other oncogenes or tumor suppressors |
||||
miR-16 |
− |
− |
negatively correlates with DFS and overall survival of childhood T-ALL patients |
[82] |
miR-19a |
ETP-ALL low |
− |
ETP-ALL low |
[32] |
miR-19b |
upregulation |
PTEN, BIM |
oncomiR; inhibits apoptosis |
[46] |
miR-20a |
upregulation |
PTEN, BIM, PHF6 |
oncomiR; inhibits apoptosis |
[46] |
miR-20b |
ETP-ALL low |
− |
ETP-ALL low |
[32] |
miR-21 |
upregulation |
PDCD4 |
promotes survival |
[50] |
miR-29a |
downregulation |
− |
negatively correlates with DFS |
[84] |
miR-29 |
downregulation |
HBP1 |
tumor suppressor in NOTCH1-driven T-ALL model |
[43] |
miR-92 |
upregulation |
IKAROS/IKZF1, PTEN, FBXW7, BIM, NF1 |
oncomiR; induces NOTCH1 expression; inhibits apoptosis |
[46] |
miR-128-3p |
− |
PHF6 |
oncomiR in a NOTCH1- driven T-ALL model |
[49] |
miR-151-3p |
ETP-ALL low |
− |
ETP-ALL low |
[32] |
miR-193b-3p |
downregulation |
MYB |
tumor suppressor in NOTCH1-driven T-ALL model |
[42] |
miR-200 |
downregulation |
MYB, HBP1 |
tumor suppressor in NOTCH1-driven T-ALL model |
[43] |
miR-221 |
ETP-ALL high |
− |
ETP-ALL high; an independent predictive factor for shorter overall survival |
|
miR-222 |
ETP-ALL high |
ETS1 |
ETP-ALL high; inhibits proliferation and causes apoptosis |
[32] |
miR-342-3p |
ETP-ALL low |
− |
ETP-ALL low |
[32] |
miR-363 |
ETP-ALL low |
− |
ETP-ALL low |
[32] |
miR-576-3p |
ETP-ALL low |
− |
ETP-ALL low |
[32] |
miR-590 |
upregulation |
RB1 |
promotes proliferation, migration, and invasion |
[29] |
miR-664 |
upregulation |
PLP2 |
promotes proliferation, migration, and invasion |
[75] |
MiRNAs with other oncogenic signals in T-ALL
In addition to the well-known oncogenic NOTCH signaling pathway, there are many other biologically relevant pathways controlling cell growth, proliferation and survival, such as PI3K/AKT [44], NF-κB [45, 48], cAMP/PKA [57], and JAK/STAT pathway [58]; all of whose interactions with miRNAs also play important roles in T-ALL pathology.
As mentioned above, miR-19, which belongs to the miR-17-92 cluster, controlled multiple regulators (PP2A, PRKAA1, BIM, and PTEN) of PI3K signaling which resulted in increased phosphorylation of AKT and the ribosomal S6 protein, which subsequently promoted survival of T-ALL cells [44]. Additionally, miR-181a could induce chemoresistance in Jurkat T-ALL cells through activating AKT, which will be discussed in detail later [59]. Moreover, luciferase assay experiments showed that miR-19 directly repressed the expression of CYLD, which plays a predominant role in the negative regulation of NF-κB, inducing activation of the NF-κB downstream program [45]. Furthermore, miR-223 was transcriptionally activated by NF-κB signaling pathway [48]. Together, this research demonstrates that, the miRNAs and the NF-κB signaling pathway form a feedback regulatory loop.
MiR-142-3p has been shown to be highly expressed in human T-leukemic cells and has been correlated with poor prognosis of T-cell leukemia patients. MiR-142-3p acted in an oncogenic role in T-ALL by promoting leukemic cell growth and inducing glucocorticoid (GC) resistance through targeting both glucocorticoid receptor alpha (GRa) and cyclic adenosine monophosphate (cAMP)/protein kinase A (PKA) pathways [57]. MiR-21 has been identified as an oncogenic miRNA involved in NOTCH-mediated induction of T-ALL [50]. Recently it was also found upregulated in Jurkat cells, a T-ALL cell line, promoting cell proliferation, invasion, and decreased the apoptosis rate through the inhibition of signal transducer and activator of transcription 3 (STAT3) expression at the protein level [58].
MiRNAs expression and transcription factors in T-ALL
In T-ALL development, the transcriptional program is strictly regulated. Pathological development of T-ALL requires aberrant expression of intact master developmental regulatory transcription factors that can function as oncogenes. One of these oncogenic transcription factors is TAL1/SCL, a member of basic helix-loop-helix (bHLH) family, which was aberrantly expressed in 60% of human T-ALL cases [60]. TAL1/SCL has been shown to initiate T-ALL in murine models [61, 62]. MiRNAs directly controlled by TAL1 were investigated via global miRNA expression profiling after depletion of TAL1 and genome-wide analysis of TAL1 occupancy by ChIP coupled to parallel DNA sequencing [47]. In this study, miR-223 was identified as the most upregulated miRNA by TAL1, and ChIP-sequence revealed that miR-223 was a direct target of TAL1. It was also shown that the expression of TAL1 and miR-223 was closely correlated during normal T-cell development as well as T-ALL, with high expression in early thymocytes and marked downregulation after the double-negative-2 stage of maturation. Moreover, it was confirmed that miR-223 mediated overexpression of TAL1-induced growth of T-ALL cells through the direct inhibition of the expression of tumor suppressor, FBXW7. FBXW7 has been shown to be able to repress MYC, MYB, NOTCH1, and CYCLIN E expression. Furthermore, another study of TAL1-regulated miRNAs in T-ALLs also showed the direct activation of miR-223, and inversely, the direct repression of miR-146b-5p by TAL1 [63]. Although transcription factor TAL1 is characterized as an oncogene during T-ALL progression, its aberrant activation is attributed to chromosomal rearrangement. However, there are some T-ALL patients who lack the TAL1 locus rearrangement but have TAL1 activation. Unfortunately, to date the underlying mechanisms around this anomaly are still largely unknown. The same group further investigated whether epigenetic regulation of TAL1 by specific miRNAs that may contribute to the ectopic expression of TAL1 in some T-ALL cases [55]. By performing computational target prediction, luciferase reporter system, and mutagenesis assays; five candidate miRNAs (miR-101, miR-520d-5p, miR-140-5p, miR-448 and miR-485-5p) were found to directly target TAL1. Of these five targets, four miRNAs (miR-101, miR-140-5p, miR-448 and miR-485-5p) were downregulated in T-ALL patient specimens and T-ALL cell lines [55].
MiR-204 expression was decreased in T-leukemic cells compared to normal T-cell samples. Ectopic expression of miR-204 could inhibit proliferation, migration and invasion of T-ALL cells, indicating that miR-204 may act as a tumor suppressor in the regulation of cell growth and metastasis in T-ALL cells [64]. Moreover, the sex determining region Y-box 4 (SOX4), a transcription factor in development which belongs to the C subgroup of SRY-related HMG box (SOX) transcription factor family [65], was a direct target of miR-204. Its inactivation could partially mediate the tumor suppressor role of miR-204 in T-ALL [64]. On the contrary, miR-149*, acting as an oncomiR, was highly expressed in T-ALL cell lines and T-ALL patients’ bone marrow samples. MiR-149* could promote proliferation and reduce apoptosis in T-ALL cells by directly targeting JunB, a transcription factor involved in regulating gene activity following the primary growth factor response [30]. Another example is the negative correlation between miR-181a and the early growth response 1 (EGR1) [66]. EGR1 is a member of the EGR gene family encoding for C2H2-type zinc-finger transcription factors [67], acting as a tumor suppressor to promote G0/G1 cell-cycle arrest [68]. Over-expression of miR-181a in Jurkat T-ALL cells could decrease EGR1 expression, increase cell proliferation and enhance the cell-cycle progression from G0/G1 to S phase [66]. Dual luciferase assays revealed that miR-181a directly targeted EGR1 followed by downregulation of TGF1, BCL2, p53 and p73, whose transcription were promoted by EGR1 [69–74]. Strikingly, the rescue of EGR1 protein could revert the phenotype originally observed in Jurkat-miR-181a-overexpressing cells, suggesting that miR-181a behaved as an oncomiR in T-ALL by downregulating EGR1.
MiRNAs expression and cell biological behavior deregulation in T-ALL
Aberrant expression of miRNAs has been implicated in the deregulation of various important cellular functions such as cell growth, apoptosis, migration, and invasion. All of these factors might contribute to or block the progression of leukemogenesis in T-ALL. As mentioned above, many miRNAs are involved in cell survival regulation during T-ALL progression via modulating key survival molecules or signaling pathways. The miR-17-92 cluster could suppress apoptosis and enhance survival of leukemic T-cells through decreasing E2F1 protein expression [34]. MiR-19 activated the PI3K/AKT pro-survival pathway via downregulation of several negative regulators [44]. MiR-142-3p targets the cAMP/PKA pathway to promote leukemic T-cell proliferation [57]. The oncomiR miR-21 was shown to promote cell proliferation, invasion, as well as decrease the apoptosis rate in Jurkat cells [58]. MiR-149* was also classified as an oncomiR that promoted cell proliferation and suppressed apoptosis by targeting JunB in T-ALL cells [30]. Overexpression of miR-181a in Jurkat T-ALL cells promoted proliferation and enhanced the cell-cycle transition from G0/G1 to S phase through direct targeting of the transcription factor EGR1 [66]. Moreover, miR-664 could promote proliferation, migration and invasion of T-ALL cells; and negatively regulate PLP2 expression through direct binding of its 3′UTR [75]. MiR-590 was shown to inhibit RB1 expression and promote proliferation and invasion of T-ALL cells [29]. In this study, miR-590 was found to be highly expressed in T-ALL samples as compared with normal healthy T-cell controls. It was shown to be negatively correlated with RB1 [76], a negative regulator of cell cycle, which was confirmed in 395 T-ALL patients samples. Overexpression of miR-590 promoted T-ALL cell proliferation by increasing G1/S transition, enhancing migration and invasion. Further studies revealed that miR-590 directly targeted and negatively regulated RB1 in T-ALL cells, indicating that repressed RB1 might mediate the biological effects of miR-590 on T-ALL cells.
On the contrary, there are many tumor-suppressive miRNAs that function as T-ALL progression inhibitors by suppressing cell proliferation, inducing cell apoptosis, and impeding cell migration and invasion capabilities. Overexpression of miR-222 inhibited proliferation and caused cell cycle arrest and apoptosis in leukemic cells by directly suppressing ETS1 expression [32]. Enforced expression of a set of tumor suppressor miRNAs (miR-29, miR-31, miR-150, miR-155, and miR-200) had anti-proliferative effects in four human T-ALL cell lines [43]. MiR-101 repressed proliferation and invasion, and induced apoptosis in Jurkat cells by direct downregulation of NOTCH1 expression [19]. MiR-204 was decreased in T-ALL, and its ectopic expression suppressed cell proliferation, migration and invasion [64]. Overexpression of the tumor suppressive miRNA miR-146b-5p was confirmed to repress the in vitro migration and invasion of T-ALL cells, and extend mouse survival in a xenotransplantation model of human T-ALL in vivo [77].
MiRNAs expression and drug resistance and prognosis in T-ALL
Leukemia drug resistance is a particularly troubling challenge in T-ALL, and is associated with relapse and dismal prognosis despite intensified chemotherapy [78, 79]. This leads to a considerable proportion of patients with T-ALL who fail to obtain complete remission and eventually die as a result of disease progression [80, 81]. Epigenetic studies have started to reveal the relevance between chemoresistance, prognosis and deregulation of miRNAs in T-ALL; however, much efforts remain to be devoted to research in these areas.
MiR-142-3p was upregulated in human T-leukemic cell lines and primary T-leukemic cells, and could induce glucocorticoid resistance by decreasing GRa protein expression through directly targeting the 3′UTR of GRa mRNA. The suppression of the GRa was concomitant with the higher expression of miR-142-3p in T-ALL patients with poor prognosis as compared to those with good prognosis [57]. The high expression of miR-181a in T-ALLs was associated with multi-drug resistance through the activation of the AKT signaling pathway [59]. In this study, overexpression of miR-181a in the Jurkat cell line could increase AKT phosphorylation. MiR-181a and phosphorylated AKT were significantly increased when Jurkat cells exposed to chemotherapeutic agents, including doxorubicin (DOX), cyclophosphamide (CTX), cytarabine (Ara-C), and cisplatin for 48 h. Specific inhibition of miR-181a in Jurkat cells and its resistant sublines increased cell sensitivity to DOX, in accordance with decreased AKT phosphorylation; and decreased miR-181a expression was also related to enhanced cell sensitivity to cisplatin and CTX in doxorubicin-resistant Jurkat cells [59]. In contrast, the tumor suppressive miRNA, miR-101 was reported to enhance drug sensitivity of Jurkat cells [19]. In this study, miR-101 expression was significantly decreased in Jurkat cells following treatment with Adriamycin.
The association of distinct miRNAs expression in relation to risk categories in T-ALL has been investigated in a number of publications. High miR-16 expression was found to be associated with hyperleukocytosis and poor cytogenetic groups in 93 childhood ALL cases [82]. In T-ALL cases, a significant trend was found between the shortening of a patient’s survival rate associated with the lowest to the highest miR-16 levels for both disease-free survival (DFS) and overall survival [82]. MiR-221 was highly expressed in T-ALL samples from 48 T-ALL patients and was an independent predictive factor for shorter overall survival [83]. Oliveira et al. found that miR-29a expression level was extremely reduced in T-ALL cells compared to normal T-cells, and lower level of miR-29a is associated with higher blast counts in the bone marrow and increased DFS in T-ALL patients [84].
CONCLUSIONS
The identification and molecular characterization of new oncogenic and tumor suppressive miRNAs in T-ALL have provided new insights into the pathogenesis of this disease. Since miRNA regulates or can be regulated by multiple genes and signaling pathways that affect various cell functions, it can be used as markers for disease diagnosis, progression, treatment response and clinical outcome [32, 51, 57, 84]. A number of key signaling pathways that play critical role in T-ALL pathology and progression are involved in the miRNA regulatory network including the NOTCH pathway, whose aberrant constitutive activation is believed to be the predominant oncogenic event in the pathogenesis of T-ALL [13]. With NOTCH1-driven T-ALL mouse model, many miRNAs with oncogenic or tumor suppressive roles in T-ALL onset and progression have been elucidated (Figure 1). Moreover, miRNA expression profiles, target prediction algorithms of miRNAs, and key protein-coding genes known to be involved in T-ALL, as well as functional studies have created a complicated co-regulatory network that eventually influence leukemia pathogenesis, progression, treatment, and prognosis (Table 1).
MiRNAs might be better therapeutic targets because their expression are often tissue specific. Additionally research has shown that there is one specific subtype of miRNA that plays different roles during normal thymic T-cell development and leukemogenesis. For example, in a study of examination of the roles of three mir-181 genes (mir-181ab1, mir-181ab2 and mir-181cd) in normal thymocyte development and in T-ALL development [56], it was found that deletion of mir-181ab1, but not mir-181ab2 and mir-181cd, effectively inhibited NOTCH1-induced T-ALL [56]. More important, the effects of mir-181ab1 deletion were compensated for during normal thymic progenitor development but not during T-ALL development. As a result, deletion of mir-181ab1 gene could specifically inhibit the activity of the NOTCH oncogene in tumorigenesis without causing significant defects in normal development [56]. Thus, drugs targeting these tissue specific miRNAs could be less off-target and cause less toxicity to normal tissues. Better understanding of the co-regulatory molecular mechanism between miRNAs and protein coding genes involved in T-ALL will contribute to more targeted-therapies for patients. Although the delivery efficiency remains challenging, therapies with small interfering RNAs and antisense oligonucleotides or a miRNA mimic targeting specific miRNA could show promising prospects with technical improvements in the future.
ACKNOWLEDGMENTS AND FUNDING
The author gratefully acknowledges the financial support from the Science and Technology Commission Foundation of Chaoyang district, Beijing (CYSF151510).
CONFLICTS OF INTEREST
The author has declared no conflicts of interest.
REFERENCES
1. Belver L, Ferrando A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat Rev Cancer. 2016; 16:494–507.
2. Hunger SP, Mullighan CG. Acute Lymphoblastic Leukemia in Children. N Engl J Med. 2015; 373:1541–52.
3. Litzow MR, Ferrando AA. How I treat T-cell acute lymphoblastic leukemia in adults. Blood. 2015; 126:833–41.
4. Koch U, Radtke F. Mechanisms of T cell development and transformation. Annu Rev Cell Dev Biol. 2011; 27:539–62.
5. Ferrando AA, Neuberg DS, Staunton J, Loh ML, Huard C, Raimondi SC, Behm FG, Pui CH, Downing JR, Gilliland DG, Lander ES, Golub TR, Look AT. Gene expression signatures define novel oncogenic pathways in T cell acute lymphoblastic leukemia. Cancer Cell. 2002; 1:75–87.
6. Soulier J, Clappier E, Cayuela JM, Regnault A, Garcia-Peydro M, Dombret H, Baruchel A, Toribio ML, Sigaux F. HOXA genes are included in genetic and biologic networks defining human acute T-cell leukemia (T-ALL). Blood. 2005; 106:274–86.
7. Meijerink JP. Genetic rearrangements in relation to immunophenotype and outcome in T-cell acute lymphoblastic leukaemia. Best Pract Res Clin Haematol. 2010; 23: 307–18.
8. Coustan-Smith E, Mullighan CG, Onciu M, Behm FG, Raimondi SC, Pei D, Cheng C, Su X, Rubnitz JE, Basso G, Biondi A, Pui CH, Downing JR, et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 2009; 10:147–56.
9. Van Vlierberghe P, Ambesi-Impiombato A, De Keersmaecker K, Hadler M, Paietta E, Tallman MS, Rowe JM, Forne C, Rue M, Ferrando AA. Prognostic relevance of integrated genetic profiling in adult T-cell acute lymphoblastic leukemia. Blood. 2013; 122:74–82.
10. Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M, Lu C, Chen SC, Wei L, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012; 481:157–63.
11. Niehues T, Kapaun P, Harms DO, Burdach S, Kramm C, Korholz D, Janka-Schaub G, Gobel U. A classification based on T cell selection-related phenotypes identifies a subgroup of childhood T-ALL with favorable outcome in the COALL studies. Leukemia. 1999; 13:614–7.
12. Homminga I, Pieters R, Langerak AW, de Rooi JJ, Stubbs A, Verstegen M, Vuerhard M, Buijs-Gladdines J, Kooi C, Klous P, van Vlierberghe P, Ferrando AA, Cayuela JM, et al. Integrated transcript and genome analyses reveal NKX2-1 and MEF2C as potential oncogenes in T cell acute lymphoblastic leukemia. Cancer Cell. 2011; 19:484–97.
13. Weng AP, Ferrando AA, Lee W, Morris JP 4th, Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT, Aster JC. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004; 306:269–71.
14. Wallaert A, Durinck K, Taghon T, Van Vlierberghe P, Speleman F. T-ALL and thymocytes: a message of noncoding RNAs. J Hematol Oncol. 2017; 10:66.
15. Pui CH, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet. 2008; 371:1030–43.
16. Stock W, Johnson JL, Stone RM, Kolitz JE, Powell BL, Wetzler M, Westervelt P, Marcucci G, DeAngelo DJ, Vardiman JW, McDonnell D, Mrozek K, Bloomfield CD, et al. Dose intensification of daunorubicin and cytarabine during treatment of adult acute lymphoblastic leukemia: results of Cancer and Leukemia Group B Study 19802. Cancer. 2013; 119:90–8.
17. den Hoed MA, Pluijm SM, te Winkel ML, de Groot-Kruseman HA, Fiocco M, Hoogerbrugge P, Leeuw JA, Bruin MC, van der Sluis IM, Bresters D, Lequin MH, Roos JC, Veerman AJ, et al. Aggravated bone density decline following symptomatic osteonecrosis in children with acute lymphoblastic leukemia. Haematologica. 2015; 100:1564–70.
18. Sutton R, Shaw PJ, Venn NC, Law T, Dissanayake A, Kilo T, Haber M, Norris MD, Fraser C, Alvaro F, Revesz T, Trahair TN, Dalla-Pozza L, et al. Persistent MRD before and after allogeneic BMT predicts relapse in children with acute lymphoblastic leukaemia. Br J Haematol. 2015; 168:395–404.
19. Qian L, Zhang W, Lei B, He A, Ye L, Li X, Dong X. MicroRNA-101 regulates T-cell acute lymphoblastic leukemia progression and chemotherapeutic sensitivity by targeting Notch1. Oncol Rep. 2016; 36:2511–6.
20. Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993; 75:843–54.
21. Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993; 75:855–62.
22. de Oliveira JC, Brassesco MS, Scrideli CA, Tone LG, Narendran A. MicroRNA expression and activity in pediatric acute lymphoblastic leukemia (ALL). Pediatr Blood Cancer. 2012; 59:599–604.
23. Beezhold KJ, Castranova V, Chen F. Microprocessor of microRNAs: regulation and potential for therapeutic intervention. Mol Cancer. 2010; 9:134.
24. Nana-Sinkam SP, Croce CM. MicroRNAs as therapeutic targets in cancer. Transl Res. 2011; 157:216–25.
25. Feng S, Cong S, Zhang X, Bao X, Wang W, Li H, Wang Z, Wang G, Xu J, Du B, Qu D, Xiong W, Yin M, et al. MicroRNA-192 targeting retinoblastoma 1 inhibits cell proliferation and induces cell apoptosis in lung cancer cells. Nucleic Acids Res. 2011; 39:6669–78.
26. Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005; 120:15–20.
27. Tufekci KU, Oner MG, Meuwissen RL, Genc S. The role of microRNAs in human diseases. Methods Mol Biol. 2014; 1107:33–50.
28. Croce CM, Calin GA. miRNAs, cancer, and stem cell division. Cell. 2005; 122:6–7.
29. Miao MH, Ji XQ, Zhang H, Xu J, Zhu H, Shao XJ. miR-590 promotes cell proliferation and invasion in T-cell acute lymphoblastic leukaemia by inhibiting RB1. Oncotarget. 2016; 7:39527–34. http://doi.org/10.18632/oncotarget.8414.
30. Fan SJ, Li HB, Cui G, Kong XL, Sun LL, Zhao YQ, Li YH, Zhou J. miRNA-149* promotes cell proliferation and suppresses apoptosis by mediating JunB in T-cell acute lymphoblastic leukemia. Leuk Res. 2016; 41:62–70.
31. Chiaretti S, Messina M, Tavolaro S, Zardo G, Elia L, Vitale A, Fatica A, Gorello P, Piciocchi A, Scappucci G, Bozzoni I, Fozza C, Candoni A, et al. Gene expression profiling identifies a subset of adult T-cell acute lymphoblastic leukemia with myeloid-like gene features and over-expression of miR-223. Haematologica. 2010; 95:1114–21.
32. Coskun E, Neumann M, Schlee C, Liebertz F, Heesch S, Goekbuget N, Hoelzer D, Baldus CD. MicroRNA profiling reveals aberrant microRNA expression in adult ETP-ALL and functional studies implicate a role for miR-222 in acute leukemia. Leuk Res. 2013; 37:647–56.
33. Xia H, Yamada S, Aoyama M, Sato F, Masaki A, Ge Y, Ri M, Ishida T, Ueda R, Utsunomiya A, Asai K, Inagaki H. Prognostic impact of microRNA-145 downregulation in adult T-cell leukemia/lymphoma. Hum Pathol. 2014; 45:1192–8.
34. Nagel S, Venturini L, Przybylski GK, Grabarczyk P, Schmidt CA, Meyer C, Drexler HG, Macleod RA, Scherr M. Activation of miR-17-92 by NK-like homeodomain proteins suppresses apoptosis via reduction of E2F1 in T-cell acute lymphoblastic leukemia. Leuk Lymphoma. 2009; 50:101–8.
35. Schotte D, Lange-Turenhout EA, Stumpel DJ, Stam RW, Buijs-Gladdines JG, Meijerink JP, Pieters R, Den Boer ML. Expression of miR-196b is not exclusively MLL-driven but is especially linked to activation of HOXA genes in pediatric acute lymphoblastic leukemia. Haematologica. 2010; 95:1675–82.
36. Coskun E, von der Heide EK, Schlee C, Kuhnl A, Gokbuget N, Hoelzer D, Hofmann WK, Thiel E, Baldus CD. The role of microRNA-196a and microRNA-196b as ERG regulators in acute myeloid leukemia and acute T-lymphoblastic leukemia. Leuk Res. 2011; 35:208–13.
37. Radtke F, Wilson A, Stark G, Bauer M, van Meerwijk J, MacDonald HR, Aguet M. Deficient T cell fate specification in mice with an induced inactivation of Notch1. Immunity. 1999; 10:547–58.
38. Radtke F, MacDonald HR, Tacchini-Cottier F. Regulation of innate and adaptive immunity by Notch. Nat Rev Immunol. 2013; 13:427–37.
39. Dumortier A, Wilson A, MacDonald HR, Radtke F. Paradigms of notch signaling in mammals. Int J Hematol. 2005; 82:277–84.
40. Paganin M, Ferrando A. Molecular pathogenesis and targeted therapies for NOTCH1-induced T-cell acute lymphoblastic leukemia. Blood Rev. 2011; 25:83–90.
41. Pear WS, Aster JC, Scott ML, Hasserjian RP, Soffer B, Sklar J, Baltimore D. Exclusive development of T cell neoplasms in mice transplanted with bone marrow expressing activated Notch alleles. J Exp Med. 1996; 183:2283–91.
42. Mets E, Van der Meulen J, Van Peer G, Boice M, Mestdagh P, Van de Walle I, Lammens T, Goossens S, De Moerloose B, Benoit Y, Van Roy N, Clappier E, Poppe B, et al. MicroRNA-193b-3p acts as a tumor suppressor by targeting the MYB oncogene in T-cell acute lymphoblastic leukemia. Leukemia. 2015; 29:798–806.
43. Sanghvi VR, Mavrakis KJ, Van der Meulen J, Boice M, Wolfe AL, Carty M, Mohan P, Rondou P, Socci ND, Benoit Y, Taghon T, Van Vlierberghe P, Leslie CS, et al. Characterization of a set of tumor suppressor microRNAs in T cell acute lymphoblastic leukemia. Sci Signal. 2014; 7:ra111.
44. Mavrakis KJ, Wolfe AL, Oricchio E, Palomero T, de Keersmaecker K, McJunkin K, Zuber J, James T, Khan AA, Leslie CS, Parker JS, Paddison PJ, Tam W, et al. Genome-wide RNA-mediated interference screen identifies miR-19 targets in Notch-induced T-cell acute lymphoblastic leukaemia. Nat Cell Biol. 2010; 12:372–9.
45. Ye H, Liu X, Lv M, Wu Y, Kuang S, Gong J, Yuan P, Zhong Z, Li Q, Jia H, Sun J, Chen Z, Guo AY. MicroRNA and transcription factor co-regulatory network analysis reveals miR-19 inhibits CYLD in T-cell acute lymphoblastic leukemia. Nucleic Acids Res. 2012; 40:5201–14.
46. Mavrakis KJ, Van Der Meulen J, Wolfe AL, Liu X, Mets E, Taghon T, Khan AA, Setty M, Rondou P, Vandenberghe P, Delabesse E, Benoit Y, Socci NB, et al. A cooperative microRNA-tumor suppressor gene network in acute T-cell lymphoblastic leukemia (T-ALL). Nat Genet. 2011; 43:673–8.
47. Mansour MR, Sanda T, Lawton LN, Li X, Kreslavsky T, Novina CD, Brand M, Gutierrez A, Kelliher MA, Jamieson CH, von Boehmer H, Young RA, Look AT. The TAL1 complex targets the FBXW7 tumor suppressor by activating miR-223 in human T cell acute lymphoblastic leukemia. J Exp Med. 2013; 210:1545–57.
48. Kumar V, Palermo R, Talora C, Campese AF, Checquolo S, Bellavia D, Tottone L, Testa G, Miele E, Indraccolo S, Amadori A, Ferretti E, Gulino A, et al. Notch and NF-kB signaling pathways regulate miR-223/FBXW7 axis in T-cell acute lymphoblastic leukemia. Leukemia. 2014; 28:2324–35.
49. Mets E, Van Peer G, Van der Meulen J, Boice M, Taghon T, Goossens S, Mestdagh P, Benoit Y, De Moerloose B, Van Roy N, Poppe B, Vandesompele J, Wendel HG, et al. MicroRNA-128-3p is a novel oncomiR targeting PHF6 in T-cell acute lymphoblastic leukemia. Haematologica. 2014; 99:1326–33.
50. Junker F, Chabloz A, Koch U, Radtke F. Dicer1 imparts essential survival cues in Notch-driven T-ALL via miR-21-mediated tumor suppressor Pdcd4 repression. Blood. 2015; 126:993–1004.
51. Sanchez-Martin M, Ferrando A. The NOTCH1-MYC highway toward T-cell acute lymphoblastic leukemia. Blood. 2017; 129:1124–33.
52. Li X, Sanda T, Look AT, Novina CD, von Boehmer H. Repression of tumor suppressor miR-451 is essential for NOTCH1-induced oncogenesis in T-ALL. J Exp Med. 2011; 208:663–75.
53. Ortega M, Bhatnagar H, Lin AP, Wang L, Aster JC, Sill H, Aguiar RC. A microRNA-mediated regulatory loop modulates NOTCH and MYC oncogenic signals in B- and T-cell malignancies. Leukemia. 2015; 29:968–76.
54. Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM, Dang CV, Thomas-Tikhonenko A, Mendell JT. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet. 2008; 40:43–50.
55. Correia NC, Melao A, Povoa V, Sarmento L, Gomez de Cedron M, Malumbres M, Enguita FJ, Barata JT. microRNAs regulate TAL1 expression in T-cell acute lymphoblastic leukemia. Oncotarget. 2016; 7:8268–81. http://doi.org/10.18632/oncotarget.6987.
56. Fragoso R, Mao T, Wang S, Schaffert S, Gong X, Yue S, Luong R, Min H, Yashiro-Ohtani Y, Davis M, Pear W, Chen CZ. Modulating the strength and threshold of NOTCH oncogenic signals by mir-181a-1/b-1. PLoS Genet. 2012; 8:e1002855.
57. Lv M, Zhang X, Jia H, Li D, Zhang B, Zhang H, Hong M, Jiang T, Jiang Q, Lu J, Huang X, Huang B. An oncogenic role of miR-142-3p in human T-cell acute lymphoblastic leukemia (T-ALL) by targeting glucocorticoid receptor-alpha and cAMP/PKA pathways. Leukemia. 2012; 26:769–77.
58. Shi C, Zhang X, Li X, Zhang L, Li L, Sun Z, Fu X, Wu J, Chang Y, Li W, Chen Q, Zhang M. Effects of microRNA-21 on the biological functions of T-cell acute lymphoblastic lymphoma/leukemia. Oncol Lett. 2016; 12:4173–80.
59. Yan ZX, Zheng Z, Xue W, Zhao MZ, Fei XC, Wu LL, Huang LM, Leboeuf C, Janin A, Wang L, Zhao WL. MicroRNA181a Is Overexpressed in T-Cell Leukemia/Lymphoma and Related to Chemoresistance. Biomed Res Int. 2015; 2015:197241.
60. Begley CG, Aplan PD, Davey MP, Nakahara K, Tchorz K, Kurtzberg J, Hershfield MS, Haynes BF, Cohen DI, Waldmann TA, Kirsch IR. Chromosomal translocation in a human leukemic stem-cell line disrupts the T-cell antigen receptor delta-chain diversity region and results in a previously unreported fusion transcript. Proc Natl Acad Sci U S A. 1989; 86:2031–5.
61. Condorelli GL, Facchiano F, Valtieri M, Proietti E, Vitelli L, Lulli V, Huebner K, Peschle C, Croce CM. T-cell-directed TAL-1 expression induces T-cell malignancies in transgenic mice. Cancer Res. 1996; 56:5113–9.
62. Kelliher MA, Seldin DC, Leder P. Tal-1 induces T cell acute lymphoblastic leukemia accelerated by casein kinase IIalpha. EMBO J. 1996; 15:5160–6.
63. Correia NC, Durinck K, Leite AP, Ongenaert M, Rondou P, Speleman F, Enguita FJ, Barata JT. Novel TAL1 targets beyond protein-coding genes: identification of TAL1-regulated microRNAs in T-cell acute lymphoblastic leukemia. Leukemia. 2013; 27:1603–6.
64. Yin JJ, Liang B, Zhan XR. MicroRNA-204 inhibits cell proliferation in T-cell acute lymphoblastic leukemia by downregulating SOX4. Int J Clin Exp Pathol. 2015; 8:9189–95.
65. van de Wetering M, Oosterwegel M, van Norren K, Clevers H. Sox-4, an Sry-like HMG box protein, is a transcriptional activator in lymphocytes. EMBO J. 1993; 12:3847–54.
66. Verduci L, Azzalin G, Gioiosa S, Carissimi C, Laudadio I, Fulci V, Macino G. microRNA-181a enhances cell proliferation in acute lymphoblastic leukemia by targeting EGR1. Leuk Res. 2015; 39:479–85.
67. Sukhatme VP, Kartha S, Toback FG, Taub R, Hoover RG, Tsai-Morris CH. A novel early growth response gene rapidly induced by fibroblast, epithelial cell and lymphocyte mitogens. Oncogene Res. 1987; 1:343–55.
68. Gibbs JD, Liebermann DA, Hoffman B. Egr-1 abrogates the E2F-1 block in terminal myeloid differentiation and suppresses leukemia. Oncogene. 2008; 27:98–106.
69. Liu C, Adamson E, Mercola D. Transcription factor EGR-1 suppresses the growth and transformation of human HT-1080 fibrosarcoma cells by induction of transforming growth factor beta 1. Proc Natl Acad Sci U S A. 1996; 93:11831–6.
70. Huang RP, Fan Y, deBelle I, Ni Z, Matheny W, Adamson ED. Egr-1 inhibits apoptosis during the UV response: correlation of cell survival with Egr-1 phosphorylation. Cell Death Differ. 1998; 5:96–106.
71. de Belle I, Huang RP, Fan Y, Liu C, Mercola D, Adamson ED. p53 and Egr-1 additively suppress transformed growth in HT1080 cells but Egr-1 counteracts p53-dependent apoptosis. Oncogene. 1999; 18:3633–42.
72. Krones-Herzig A, Adamson E, Mercola D. Early growth response 1 protein, an upstream gatekeeper of the p53 tumor suppressor, controls replicative senescence. Proc Natl Acad Sci U S A. 2003; 100:3233–8.
73. Krones-Herzig A, Mittal S, Yule K, Liang H, English C, Urcis R, Soni T, Adamson ED, Mercola D. Early growth response 1 acts as a tumor suppressor in vivo and in vitro via regulation of p53. Cancer Res. 2005; 65:5133–43.
74. Pignatelli M, Luna-Medina R, Perez-Rendon A, Santos A, Perez-Castillo A. The transcription factor early growth response factor-1 (EGR-1) promotes apoptosis of neuroblastoma cells. Biochem J. 2003; 373:739–46.
75. Zhu H, Miao MH, Ji XQ, Xue J, Shao XJ. miR-664 negatively regulates PLP2 and promotes cell proliferation and invasion in T-cell acute lymphoblastic leukaemia. Biochem Biophys Res Commun. 2015; 459:340–5.
76. Carreira S, Goodall J, Aksan I, La Rocca SA, Galibert MD, Denat L, Larue L, Goding CR. Mitf cooperates with Rb1 and activates p21Cip1 expression to regulate cell cycle progression. Nature. 2005; 433:764–9.
77. Correia NC, Fragoso R, Carvalho T, Enguita FJ, Barata JT. MiR-146b negatively regulates migration and delays progression of T-cell acute lymphoblastic leukemia. Sci Rep. 2016; 6:31894.
78. Bhojwani D, Pui CH. Relapsed childhood acute lymphoblastic leukaemia. Lancet Oncol. 2013; 14:e205-17.
79. Burke MJ, Verneris MR, Le Rademacher J, He W, Abdel-Azim H, Abraham AA, Auletta JJ, Ayas M, Brown VI, Cairo MS, Chan KW, Diaz Perez MA, Dvorak CC, et al. Transplant Outcomes for Children with T Cell Acute Lymphoblastic Leukemia in Second Remission: A Report from the Center for International Blood and Marrow Transplant Research. Biol Blood Marrow Transplant. 2015; 21:2154–9.
80. Goldstone AH, Richards SM, Lazarus HM, Tallman MS, Buck G, Fielding AK, Burnett AK, Chopra R, Wiernik PH, Foroni L, Paietta E, Litzow MR, Marks DI, et al. In adults with standard-risk acute lymphoblastic leukemia, the greatest benefit is achieved from a matched sibling allogeneic transplantation in first complete remission, and an autologous transplantation is less effective than conventional consolidation/maintenance chemotherapy in all patients: final results of the International ALL Trial (MRC UKALL XII/ECOG E2993). Blood. 2008; 111:1827–33.
81. Kozlowski P, Astrom M, Ahlberg L, Bernell P, Hulegardh E, Hagglund H, Karlsson K, Markuszewska-Kuczymska A, Tomaszewska-Toporska B, Smedmyr B, Amini RM, Hallbook H, Swedish Adult ALL Group. High relapse rate of T cell acute lymphoblastic leukemia in adults treated with Hyper-CVAD chemotherapy in Sweden. Eur J Haematol. 2014; 92:377–81.
82. Kaddar T, Chien WW, Bertrand Y, Pages MP, Rouault JP, Salles G, Ffrench M, Magaud JP. Prognostic value of miR-16 expression in childhood acute lymphoblastic leukemia relationships to normal and malignant lymphocyte proliferation. Leuk Res. 2009; 33:1217–23.
83. Gimenes-Teixeira HL, Lucena-Araujo AR, Dos Santos GA, Zanette DL, Scheucher PS, Oliveira LC, Dalmazzo LF, Silva-Junior WA, Falcao RP, Rego EM. Increased expression of miR-221 is associated with shorter overall survival in T-cell acute lymphoid leukemia. Exp Hematol Oncol. 2013; 2:10.
84. Oliveira LH, Schiavinato JL, Fraguas MS, Lucena-Araujo AR, Haddad R, Araujo AG, Dalmazzo LF, Rego EM, Covas DT, Zago MA, Panepucci RA. Potential roles of microRNA-29a in the molecular pathophysiology of T-cell acute lymphoblastic leukemia. Cancer Sci. 2015; 106:1264–77.