
INTRODUCTION
Neuroblastoma (NB) is the most common extracranial neoplasm in children and contributes to about 15% of all pediatric cancer-related deaths (1). Despite major advances in therapies over the past decade, the overall outcome for high-risk NB patients is still unacceptable [1]. Current therapies include chemotherapy drugs that are highly toxic to healthy cells and have significant long-term side effects. Therefore, developing novel targeted therapies for high-risk NB is critical to achieve higher efficacy and to alleviate adverse effects. MYCN amplification is a strong characteristic of high-risk NB patients and serves as a genetic marker of disease [2, 3]. Finding therapeutic strategies to directly target MYCN is a difficult task due to its protein structure. Thus, identifying and characterizing druggable targets in MYCN regulators and transcriptional targets in NB may help us to develop efficient therapeutic strategies.
Protein kinases play an essential role in the regulation of cell survival and proliferation [4]. Different kinases such as anaplastic lymphoma kinase (ALK) [5], Aurora kinase [6], RET receptor tyrosine kinase [7] have been shown to be potential therapeutic targets in various cancers, including NB [8]. Maternal embryonic leucine zipper kinase (MELK) is a serine/threonine kinase overexpressed in various organ-specific stem cells and cancers [9, 10]. High MELK expression predicts a poor prognosis of many cancer types, including but not limited to breast cancer [11], astrocytoma [12] and glioblastoma [13]. MELK has been shown to promote cancer cell survival and tumor cell differentiation [14]. Furthermore, although MELK has been suggested to form a heterotrimeric protein complex with the oncogenic transcription factors c-Jun and FoxM1 in cancer stem cells, MELK lacks the binding ability in normal progenitor and stem cells [15, 16]. Therefore, inhibition of the kinase activity of MELK may disrupt MELK-mediated malignancy in cancer cells, while having a minimal effect on normal cells [13].
The molecular mechanism regulating MELK overexpression in cancer cells and the role of MELK in NB tumorigenesis remains ambiguous. In this study, we analyzed a large cohort of NB patients and identified that higher MELK expression is correlated with poor overall survival, prognosis, and overall outcome. Moreover, MELK expression is higher in tumors from high-risk NB patients. Using ChIP-qPCR assays, we showed that MYCN directly binds at the E-box binding motifs present at the MELK gene promoter and promotes transcription. Additionally, the MYCN or MYC knockdown led to decreased MELK mRNA and protein levels in NB cells, whereas MYCN or MYC overexpression led to increased mRNA and protein levels of MELK in NB cells. Furthermore, MELK small molecule inhibitor OTSSP167 inhibited NB growth both in vitro and in vivo by inducing apoptosis. This is the first report that shows the oncogenic role and regulation of MELK in NB. Novel MELK inhibitor OTSSP167 is shown to reduce growth of various cancer types [17]. The orally active compound OTSSP167 is currently in phase-I clinical trials for solid tumors (ClinicalTrails.gov # NCT01910545) and is fast approaching for further clinical development. Our pre-clinical data suggest that MELK is an attractive therapeutic target in MYCN- and MYC-driven cancers, such as NB.
RESULTS
MELK expression is a prognostic marker for high-risk NB
To evaluate how transcription of MELK correlates with NB outcomes, we analyzed large clinical cohorts of NB patients using the R2-data analysis platform. Kaplan-Meier analyses of datasets revealed that low MELK transcript levels strongly correlated with better overall and event-free survival for the Tumor Neuroblastoma SEGC dataset (n = 498) (p < 9.7E-27) and Kocak dataset (n = 649) (p < 1.3e-12) (Figure 1A, 1B). Interestingly, we observed higher MELK expression levels (p < 0.001) in MYCN-amplified NB tumors compared to those in MYCN-nonamplified ones (Figure 1C, 1D). There were significant corrrelations of MELK with MYCN expression levels according to Kocak dataset (Figure 1E). In addition, more aggressive, higher stage tumors had significantly higher MELK expression, suggesting that MELK has a role in de-differentiated invasive malignancy (Figure 1F, 1G). These findings indicate that MELK expression is an important factor in the biology and therapeutic response for high-risk NB.
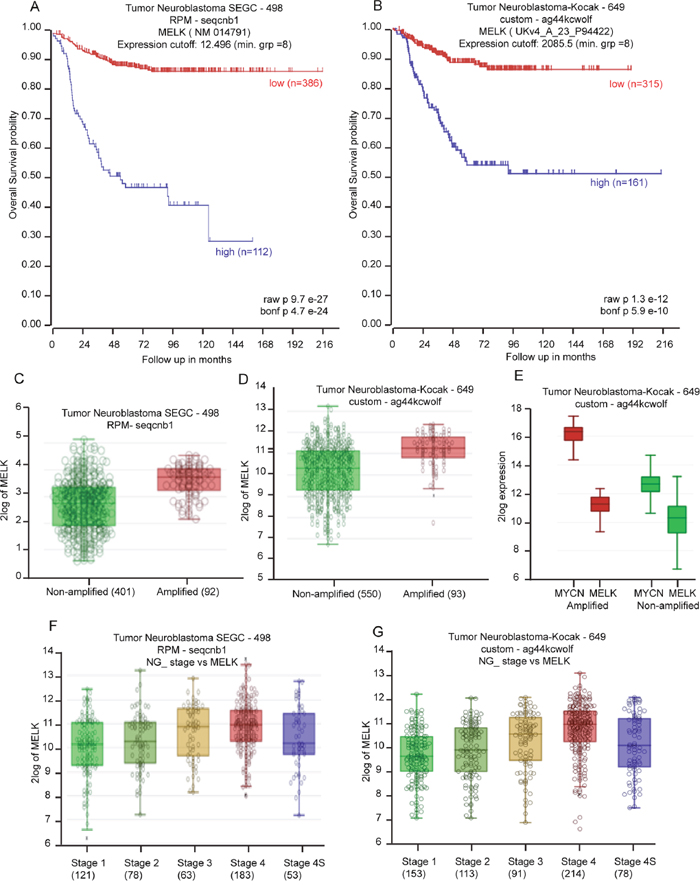
Figure 1: MELK is a prognostic indicator in NB. (A and B) Kaplan-Meier curves show the probability of overall survival based on MELK expression level of 498 patients in the SEGC dataset (A) and 649 patients in the Kocak dataset (B). (C and D) R2-tumor neuroblastoma SEGC dataset (C) and Kocak dataset (D) show higher MELK expression level in MYCN-amplified cases in comparison to MYCN-non-amplified cases. (E) The Kocak dataset shows significant correlations of MELK with MYCN expression levels. (F and G) R2-tumor neuroblastoma SEGC dataset (F) and Kocak dataset (G) show correlation of MELK expression level with NB disease stages.
MYCN and MYC promotes MELK expression in NB
To evaluate the correlation between MELK and MYCN/MYC expression in NB cells, we performed an sh-RNA mediated knockdown of MYCN in two MYCN-amplified NB cell lines, NGP and IMR-32, as well as knockdown of MYC in two MYCN-non-amplified NB cell lines, SK-N-AS and SH-SY5Y. Results showed that stable knockdown of MYCN or MYC led to both reduced MELK mRNA and protein in NB cell lines tested (Figure 2A, 2B). These results indicated that MELK is a MYCN/MYC transcriptional target in NB. Furthermore, analysis of published ChIP-sequencing dataset in NB [18] revealed that MYCN directly binds to the E-Box motif present at the MELK 5’UTR (Figure 2C, 2D). To verify these results, we performed individual ChIP-qPCR assays in a MYCN inducible cell line (MYCN3), where MYCN levels can be controlled with doxycycline treatments [18]. With doxyxcline treatment, MYCN binding increased about 17 fold in comparison to the non-treated control (Figure 2E). Similarly, in a ChIP-qPCR assay with IMR-32 cells, anti-MYCN antibody pulled down about a 2 fold more MELK 5’UTR genomic sequence in comparison to the control IgG (Figure 2F). To further confirm the correlation between MELK and MYCN/MYC expressions in NB, we overexpressed MYCN with a 3XFLAG tag in MYCN-amplified IMR-32, NGP cell lines, and MYC with a 3XFLAG tag in MYCN-non-amplified SK-N-AS, SH-SY5Y NB cell lines. Indeed, our results demonstrated that further stable overexpression of MYCN or MYC greatly increased both mRNA and protein levels of MELK in these four NB cell lines tested (Figure 2G, 2H). These data further support that MELK is a MYCN/MYC transcriptional target.
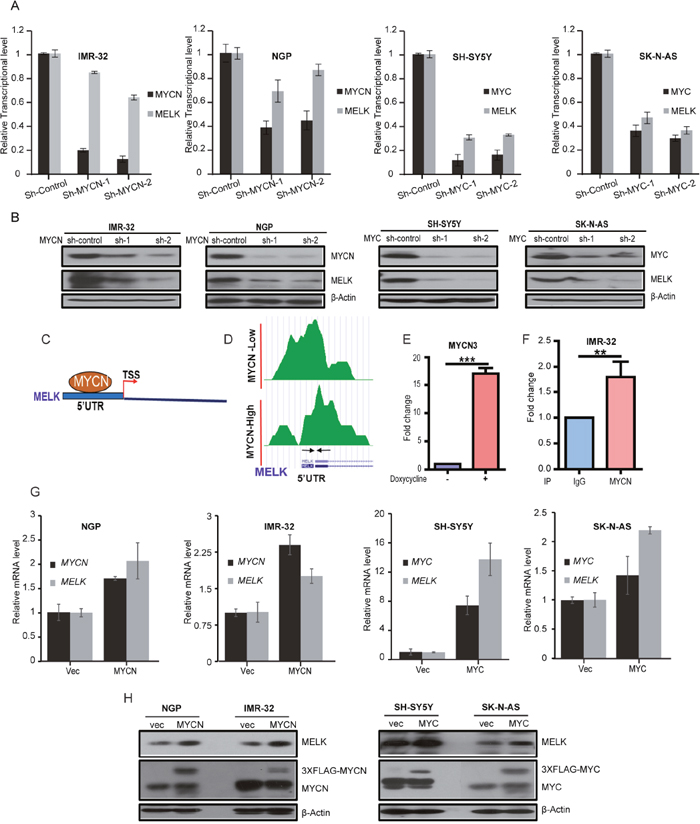
Figure 2: MELK is a transcription target of MYCN/MYC in NB. (A) Stable sh-RNA-mediated knockdown of MYCN/MYC. The qPCR analyses of MYCN/MYC and MELK expression levels were shown. (B) Total proteins of MYCN/MYC and MELK were measured in the indicated cells. (C and D) MYCN ChIP-seq analysis showed binding peaks of MYCN on MELK 5’UTR. TSS- Translation Start Site. Designed primers for ChIP-seq analysis is shown with arrows under the peaks. These primers encompass the E-Box motif. (E) ChIP-qPCR analysis for MYCN binds on MELK 5’UTR site in MYCN3 cell line. Fold change with or without doxycycline treatment on MYCN3 cell line was shown. (F) ChIP-qPCR analysis for MYCN binds on MELK 5’UTR site in IMR-32 NB cell line. IP-IgG and IP-MYCN of MELK-ChIP assay on IMR-32 cells were shown. (G) qPCR analysis of MYCN/MYC and MELK mRNA levels in NB cell lines with stable MYCN/MYC-overexpression were shown. (H) MYCN/MYC and MELK protein levels in NB cell lines with stable MYCN/MYC-overexpression were shown.
MELK plays oncogenic role in NB
To further understand the role of MELK in NB, we first examined the overall level of MELK protein in a panel of six NB cell lines consisting of three MYCN-amplified, IMR-32, NB-19 and NGP, and three MYCN-non-amplified, CHLA-255, SH-SY5Y and SK-N-AS cell lines (Figure 3A). Results showed abundant levels of MELK in all the cell lines tested, and that MYCN/MYC increases the expression of this kinase protein. Next, we performed an sh-RNA mediated knockdown of MELK in four NB cell lines (IMR-32, NGP, SK-N-AS and SH-SY5Y). As expected, the stable knockdown of MELK reduced MELK protein levels with both of the sh-RNA knockdown constructs used (Figure 3B). MELK knockdown also significantly inhibit the anchorage-independent colony formation and growth in all NB cell lines tested (Figure 3C). These results suggest that MELK contributes to cellular proliferation and is necessary for NB cell survival. MELK kinase regulates cell cycle progression via phosphorylating and activating the tumor suppressor Retinoblastoma (pRb) protein [19]. MYCN is also shown to regulate and inactivate Rb proteins in NB [20, 21]. Consistantly, our results showed that MELK knockdown significantly inhibit the Rb phosphorylation in all the NB cell lines tested (Figure 3B). These results confirm that MELK plays a oncogenic role via regulating the activation of Rb protein in NB.
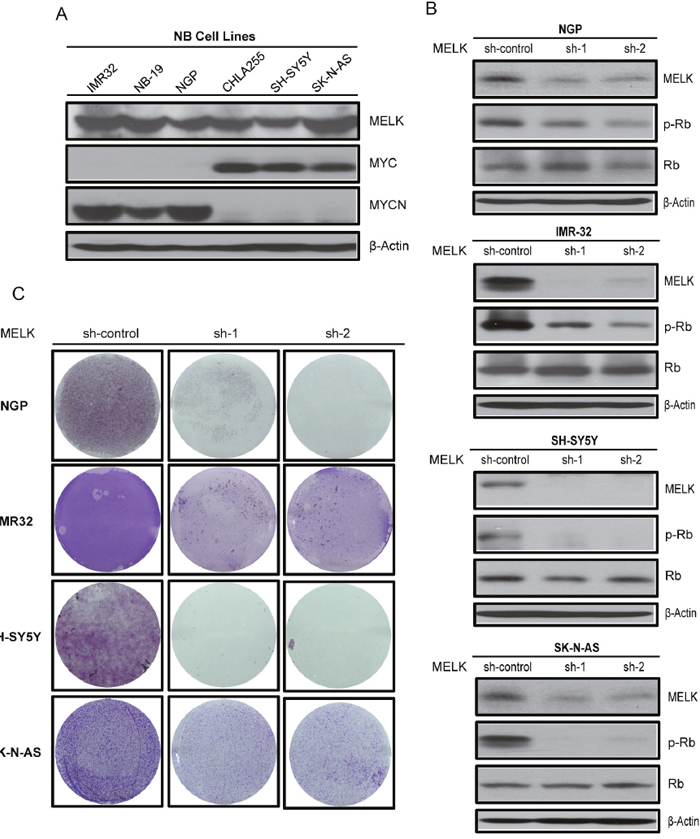
Figure 3: MELK plays an oncogenic role in NB. (A) MELK protein levels were examined by immunoblotting assay in NB cell lines listed. (B) Effects of MELK knockdown on p-Rb levels in NB cell lines were shown by immunoblotting assay. (C) The effect of MELK knockdown on NB colony formation potential were shown.
MELK inhibitor OTSSP167 potently suppresses NB growth in vitro
Our data suggest that MELK inhibition is a potential therapeutic strategy for high-risk NB. We evaluated therapeutic efficacy of a novel small-molecule MELK inhibitor, OTSSP167 [22, 23], in NB cell lines. OTSSP167 significantly inhibit NB proliferation and cellular growth at low nM doses and in a dose-dependent manner, with relatively low IC50 values ranging from most sensitive in IMR-32 (17 nM) to less sensitive in a drug-resiistant NB cell line LA-N-6 (334.8 nM) (Figure 4A, 4B, 4C). Along with reduced growth, cell morphological changes were observed in OTSSP167 treated NB cell lines (Figure 4C). In addition, OTSSP167 induces apoptosis in NB cells, as demonstrated by the PARP and Caspase-3 cleavage assays (Figure 4D). Furthermore, OTSSP167 treatments significantly inhibit anchorage-independent colony formation in a panel of NB cell lines tested (Figure 5A, 5B). This soft-agar colony formnation assay showed that OTSSP167 mediated MELK inhibition significantly inhibit NB growth in vitro. According to prior studies and our data shown above, a depletion of MELK activates the Rb pathway and induces cell cycle arrest [19]. To further validate if OTSSP167 mediated MELK inhibibition can inhibit the Rb phosphorylation and block cell cycle progression, we performed immunoblotting and cell cycle assays in four NB cell lines (Figure 5C, 5D). Indeed, the results showed that OTSSP167 treatment significantly inhibit the MELK levels and consequently inhibit the Rb phosphorylation, and block the cell cycle progression at G1 phase, in a dose-dependent manner and in all the NB cell lines tested (Figure 5C, 5D). These data strongly suggest that pharmacological inhibition of MELK inhibits NB proliferation, colony formation, significantly induce apoptosis by blocking the cell cycle progression.
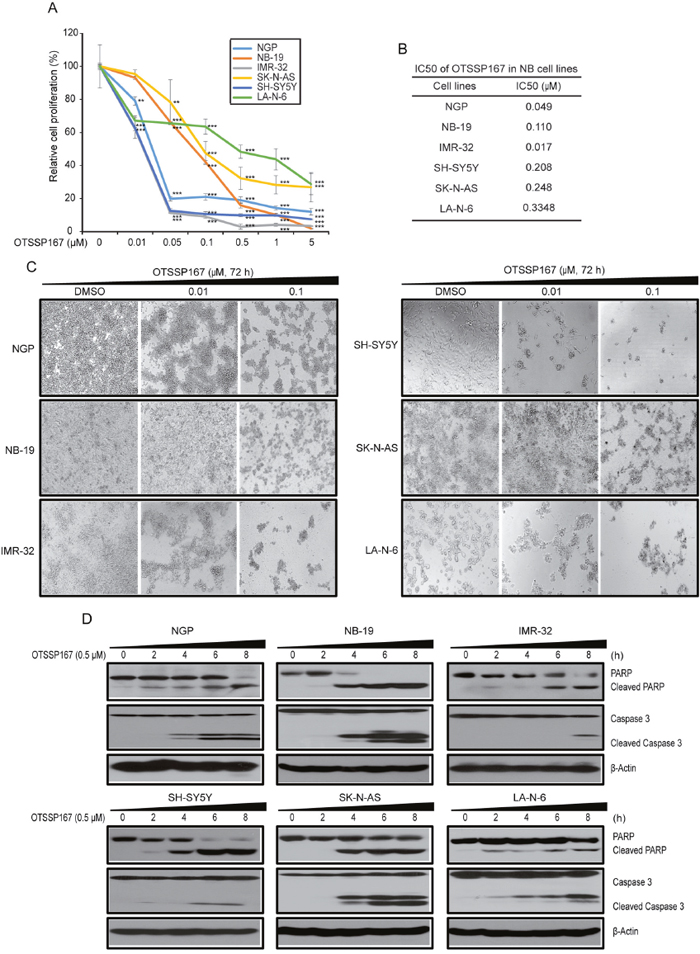
Figure 4: MELK inhibitor OTSSP167 inhibits cell proliferation and induces apoptosis in NB. (A) The effects of OTSSP167 on the proliferation of a panel of six NB cell lines using cck-8 assay were shown. (B) IC50 calculation of cell lines showen in (A) by using PRISM 5.0. (C) Effects of OTSSP167 on NB cell morphology were shown. (D) Effects of OTSSP167 on PARP and Caspase-3 cleavages in NB cells were shown by immunoblotting assay.
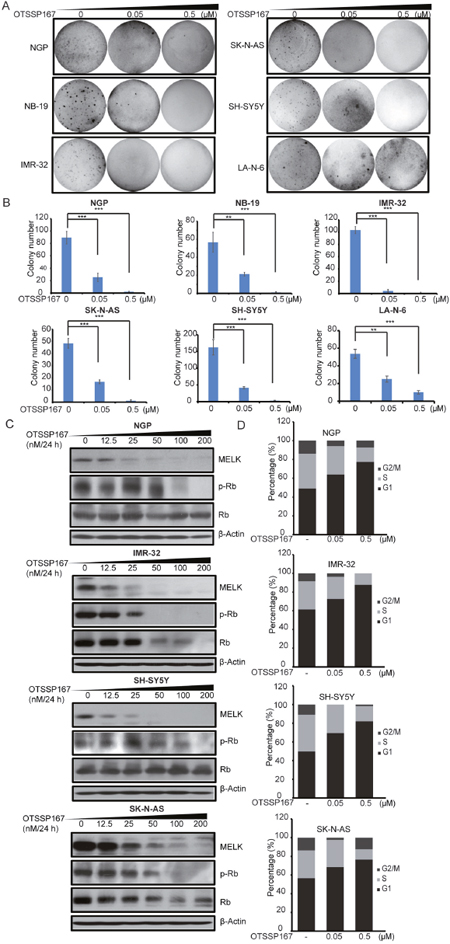
Figure 5: MELK inhibitor OTSSP167 exhibits inhibitory effects in NB. (A) The effects of OTSSP167 on anchorage-independent colony formation and growth in soft agar assay were shown. (B) Number counts of (A). (C) The effects of OTSSP167 on p-Rb level in NB cells were shown by immunoblotting assay. The anti-β-actin antibody was used as a loading control for whole cell extracts. (D) The effects of OTSSP167 on NB cell cycle by flow cytometry analysis.
MELK inhibitor OTSSP167 potently inhibits NB tumor growth in vivo
To determine the in vivo effects of OTSSP167, we used an orthotopic NB xenograft mouse model. This mouse model recapitulate the highly aggressive and invasive growth pattern of human NB [24]. NGP-luciferase cell xenografts were generated, imaged, randomized into two groups, and treated with either OTSSP167 (10 mg/kg/d) or DMSO (vehicle control) daily for 14 days. Subsequently, tumors were harvested, photographed, and weighted. Results showed that OTSSP167 significantly inhibit in vivo NB tumor growth in comparison to controls (Figure 6A, 6B). We then tested the effect of OTSSP167 on Rb phosphorylation in NB in vivo. In this experiment, we treated mice bearing the established tumors with either OTSSP167 (10 mg/kg/d) or DMSO (vehicle control) daily for 48 hours. Subsequently, tumors were harvested and analyzed. We found that MELK and p-Rb levels were significantly inhibited and PARP cleavage levels were increased in tumors from the OTPSS167 treated mice in comparison to tumors from vehicle treated mice (Figure 6C). These in vivo results further confirmed our in vitro observations that inhibion of MELK inhibit the Rb protein function to block cell cycle progression and induce apoptosis in NB. Based on these data, we suggest a working model for MELK function in NB cells (Figure 6D). Our results demonstrate the potential of MELK inhibition by OTSSP167 as a novel therapeutic approach for high-risk NB.
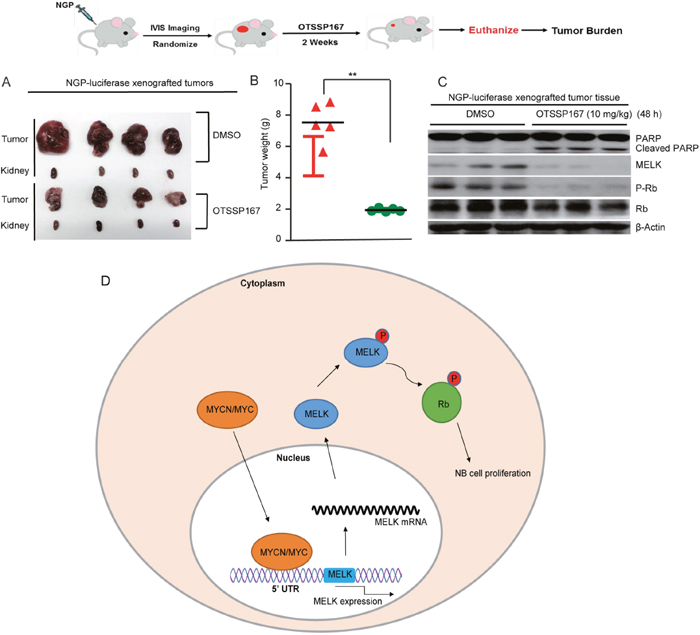
Figure 6: MELK inhibitor OTSSP167 inhibits Nb tumor growth. (A and B) NGP-luciferase xenografts were treated with DMSO or 10 mg/kg of OTSSP167 daily for 14 days. After treatment cycles, tumors and corresponding kidneys were photographed (A) and weighted (B). (C) Mice with NB tumors were treated with DMSO or 10 mg/kg of OTSSP167 daily for 48 hours. Effects of OTSSP167 on Rb phosphorylation and apoptotic marker PARP cleavage in the established NB tumor in vivo were shown. (D) A working model of MELK function in NB cells was shown.
DISCUSSION
Several transcription factors, such as oncoprotein FoxM1 and c-JUN, have been reported to promote MELK in cancer cells [25, 26]. However, in NB, the mechanism of MELK regulation still remains unclear. The gene expression analysis of NB tumors from patients revealed that MELK strongly correlates with MYCN expression and amplification. We therefore hypothesize that MYCN might promote MELK expression in NB. Our results demonstrate that MELK is a direct transcription target of MYCN/MYC in NB cells. Genetic and small molecule-mediated MELK inhibition was able to significantly suppress cell proliferation and induce apoptosis. MELK inhibition is a promising therapeutic strategy for high-risk NB patients.
Some studies have reported that MELK mutagenization mediated by CRISPR/Cas9 had no effects on basal breast cancer cell lines or cell lines from six other cancer types [27]. In contrast, our results showed that MELK knockdown using sh-RNA in four different NB cell lines dramatically inhibited cell viability and colony formation, a key indicator of the crucial role of MELK on cell survival in NB cells. It is likely that MELK is required for cell proliferation in NB but is not essential in some of other cancer types. Further study is needed to clarify the role of MELK in different cancer types.
MELK inhibitor OTSSP167 with a potential off-target effect should be considered. Studies have reported that MELK-knockout cells remain sensitive to OTSSP167 since OTSSP167 blocks cell proliferation through an off-target mechanism [27]. In addition, OTSSP167 is reported to show off-target activity against Aurora B, BUB1, and Haspin kinases in HeLa and MCF7 cells [28]. According to Kinome scan data, OTSSP167 is in fact a non-selective kinase inhibitor (http://lincs.hms.harvard.edu/db/sm/10337-102). Although related off-target effects are yet to be elucidated, OTSSP167 is still capable in directly reducing MELK levels and inducing apoptosis in NB cell lines tested.
Another confounding issue is to determine which signaling pathways regulate MELK kinase activity. MELK kinase mediates a large variety of cell characteristics including but not limited to cell cycle control, cell proliferation, apoptosis, cell migration, oncogenesis, cancer resistance, and recurrence [10]. Further, MELK functions through multifaceted signaling involving proteins such as p53 [29], Mcl-1 [30], Rb [31], CDK1, CDK25B, MAPK [32, 33]. In our study, we found that MELK kinase inhibition in NB showed inhibitory effect on Rb phosphorylation and cell proliferation. Signaling pathway that modulates MELK activity in NB cells should be further investigated.
In summary, our data showed that MELK is a transcriptional target of MYCN/MYC and plays an oncogenic role in NB. Inhibition of MELK, inhibit NB growth by blocking the cell cycle progression due to inhibition of Rb functions. The small molecule MELK inhibitor, OTSSP167 potently inhibit NB tumor growth, and reprents a promising therapeutic strategy for high-risk NB.
MATERIALS AND METHODS
Clinical patient cohorts
The Kocak neuroblastoma patient dataset (N = 649) and tumor neuroblastoma dataset SEGC (N = 498) that includes microarray profiles of unique primary tumors are publically available in the R2: Genomic Analysis and Visualization Platform database (http://hgserver1.amc.nl/cgibin/r2/main.cgi). This platform also support the multi-parametric analysis of NB patient outcome with gene expression.
Cell culture and reagents
NB cell lines used in this study were routinely cultured and maintained as described previously [18, 19]. Cell lines were verified via genotyping within the past 6 months and were tested for Mycoplasma contaminations on a monthly basis. Anti-MELK (A303-136A) was purchased from Bethyl Laboratories. Anti-PARP (9532), anti-Caspase 3 (9662), anti-p-Rb (9308), anti-Rb (9309), anti-Mouse (7076), and anti-Rabbit (7074) antibodies were purchased from Cell Signaling Technology. Anti-MYCN (sc-56729) and anti-MYC (sc-40) were from Santa Cruz Biotechnology. Anti-β-actin (A2228) primary antibody was obtained from Sigma-Aldrich Corp (St. Louis, MO, USA). OTSSP167 (HY-15512) was purchased from Medchem Express (Monmouth Junction, NJ, USA).
Immunoblotting assay
Immunoblotting assays were performed as described previously [34]. Briefly, cells were lysed using RIPA buffer and cell lysates were collected after centrifuging for 15 minutes at 13,000 rpm. Protein concentrations were determined using Bradford assay following the manufacturer’s instructions (Bio-Rad). Protein samples were separated by SDS-PAGE, transferred to PVDF membranes (Bio-Rad), blocked with 5% milk, and probed with indicated primary antibodies overnight at 4°C. The membranes were then incubated with anti-mouse or anti-rabbit IgG conjugated with horseradish peroxidase for 1 hour and blots were developed using ECL-Plus Western detection system (GE HealthCare) for visualization.
Cell cycle analysis
Cells were treated with indicated concentrations of OTSSP167 and washed with ice-cold PBS. Cells were then harvested and centrifuged for 5 min at 500 X g and 4°C. Solutions were then fixed with 3 mL 70% ice cold methanol. The fixed cells were stained with 50 μg/mL Propidium iodide (PI) solution and were analyzed by flow cytometry (FCS Express 4 Software). The data showed the distribution of the three cell cycle phases (G1, S, G2/M).
Cell proliferation and soft agar colony formation assay
Cell proliferation was measured using Cell Counting Kit-8 (Dojindo Laboratories) in accordance to the manufacturer’s instructions. NB cells were seeded at 5000 cells/well in a 96-well microtiter plate and after 24 hours of incubation, were treated with increasing concentrations of OTSSP167. Cellular proliferation was measured 72 hours post-incubation by adding CCK-8 reagent and was monitored for optical density at 450 nm using a microplate reader. Each experiment was performed in replicates of six and background reading of the media was subtracted from each well for result standardization. Soft agar colony formation assays were performed using standard conditions as described previously [24]. All assays were performed in triplicates and repeated three times with proper controls.
Generation of gene knockdown and overexpression NB cell lines
Trc2 lentiviral shRNA vectors (Sigma-Aldrich Inc.) were used to generate MYC, MYCN and MELK knockdown cell lines. The following were shRNA sequences used: MYCN-sh-1: 5’- AATTCTTACACTGCCTGTATA-3’, MYCN-sh-2: 5’-AATCTCTGTTATGTACTGTAC-3’. MYC-sh-1: AATGTCCTGAGCAATCACCTA, MYC-sh-2: AATGTCCTGAGCAATCACCTA. MELK-sh-1: 5’-AAGTTCATTGGAACTACCAAC-3’, MELK-sh-2: 5’-AATTGATGGATTCTTCCATCC-3’. The lenti-viral expression vectors encoding human MYCN and MYC open reading frame (ORF) were used to generate MYC and MYCN overexpression NB cell lines. These vectors were used to generate virus and transduce NB cell lines as described previously [34].
Quantitative reverse transcription-PCR
Gene transcription levels were measured using the qRT-PCR method as described previously [24]. Total RNA was extracted using TRIzol LS Reagent (Invitrogen) and the concentration was measured. Quantitative PCR was performed in triplicate using SensiFAST SYBR Hi-ROX One-Step Kit according to the manufacturer’s instructions (Bio-73005, Bioline). The mRNA level for each gene was detected by Applied Biosystems™ Real-Time PCR Instruments. Primers used in this study were MYCN: Forward 5’-AGAGGACACCCTGAGCGATTC-3’, Reverse 5’-CATAGTTGTGCTGCTGGTGGA-3’; MYC: Forward 5’-CTCCATGAGGAGACACCGCCCA-3’, Reverse 5’-AAGGTGATCCAGACTCTGACCT-3’; MELK: Forward 5’-ATAGCTACCATCTCTCCAGTA-3’ and Reverse 5’-CTTGCAAGAGGACTATGAAAG-3’, respectively.
ChIP-qPCR assay
ChIP-qPCR assays were performed using the ChIP-IT Express Kit (Active Motif) in accordance to the manufacturer’s instructions as described previously [19]. The antibodies used for chromatin immune-precipitation were anti-MYCN (OP13, Calbiochem Inc.) and control mouse IgG (12-371, EMD Millipore). The ChIP-purified DNA was analyzed by qPCR. ChIP-MELK primers were designed using our previously published MYCN ChIP-seq dataset which used an MYCN3 cell line under both MYCN amplified and non-amplified conditions [18]. ChIP-MELK primers were designed using the UCSC genome browser and Primer3 software (www.SimGene.com). ChIP-MELK primers were Forward- 5’-GAGAACTGTGACTGCCAGAGG-3’ and Reverse- 5’-TGTGGAGCCGTGAAAGGGAT-3’. ChIP-negative control primers were Forward- 5’- ATGGTTGCCACTGGGGATCT -3’ and Reverse- 5’- TGCCAAAGCCTAGGGGAAGA -3’.
In vivo xenograft assays
Four to six week-old female inbred athymic immunodeficient Nude mice (Nu/Nu) were purchased from Taconic Biosciences and used for all xenograft studies. Mice were implanted using a previously described orthotopic xenograft model [24, 34]. All mice were handled according to protocols approved by the Institutional Animal Care and Use Committee of Baylor College of Medicine. In the mice, 1 x 106 NGP NB cells were surgically implanted in the sub-renal capsule through a transverse incision over the left flank. Tumor growth was monitored twice a week with bioluminescent imaging (IVIS Lumina XR System, Caliper Life Sciences). Mice bearing similar sized tumors were randomly divided into groups and were treated with either DMSO or OTSSP167 (10 mg/kg, intraperitoneal injection once daily) for 14 days. After treatment, tumors were harvested. Tumors were either weighted and photographed for analysis or lysed for immunoblotting.
Statistical analysis
All values were presented as the mean ± standard deviation (SD). A two-tailed Student’s t-test and ANVOA were used to determine the statistical significance among drug treatment groups. Each assay was repeated at least twice, and representative results were presented. P < 0.05 was considered to be statistically significant. The IC50 value was calculated with Graphpad Prism 5 software (La Jolla, CA). Survival analyses were performed using Kaplan-Meier method and two-sided log-rank tests.
Author contributions
S.G. and J.L. designed and performed experiments, developed methodology, acquired and analyzed data, wrote the paper. Y.Z., Y.Y., H.L., Z.C., H.L., M.P., S.B., X.X., Z.S. and K.G. performed experiments, provided materials. X.C. and W.S. analyzed data. S.A. and J.Y. supervised the study, designed and performed experiments, developed methodology, acquired and analyzed data, and wrote the paper.
ACKNOWLEDGMENTS
The authors would like to thank Dr. Jason Shohet for providing valuable suggestion. This work is dedicated to all the children suffering from neuroblastoma who inspire us to work hard to find a cure.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
GRANT SUPPORT
This work is supported by grants from NIH/NINDS grants 1R01NS072420, 1R21NS085467 and 5R21NS094654 to JY. SA is supported by the Wipe Out Kids’ Cancer Foundation grant, Baylor College of Medicine Pediatric Pilot Award, and the David’s Warriors St. Baldrick’s Foundation Scholar Career Development Award.
REFERENCES
1. Maris JM. Recent advances in neuroblastoma. N Engl J Med. 2010; 362:2202-2211.
2. Esposito MR, Aveic S, Seydel A, Tonini GP. Neuroblastoma treatment in the post-genomic era. J Biomed Sci. 2017; 24:14.
3. Newman EA, Nuchtern JG. Recent biologic and genetic advances in neuroblastoma: implications for diagnostic, risk stratification, and treatment strategies. Semin Pediatr Surg. 2016; 25:257-264.
4. Makita N, Iiri T. Tyrosine kinase inhibitor-induced thyroid disorders: a review and hypothesis. Thyroid. 2013; 23:151-159.
5. Bresler SC, Weiser DA, Huwe PJ, Park JH, Krytska K, Ryles H, Laudenslager M, Rappaport EF, Wood AC, McGrady PW, Hogarty MD, London WB, Radhakrishnan R, et al. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell. 2014; 26:682-694.
6. Bogen D, Wei JS, Azorsa DO, Ormanoglu P, Buehler E, Guha R, Keller JM, Mathews Griner LA, Ferrer M, Song YK, Liao H, Mendoza A, Gryder BE, et al. Aurora B kinase is a potent and selective target in MYCN-driven neuroblastoma. Oncotarget. 2015; 6:35247-35262. https://doi.org/10.18632/oncotarget.6208.
7. Lambertz I, Kumps C, Claeys S, Lindner S, Beckers A, Janssens E, Carter DR, Cazes A, Cheung BB, De Mariano M, De Bondt A, De Brouwer S, Delattre O, et al. Upregulation of MAPK negative feedback regulators and RET in mutant ALK neuroblastoma: implications for targeted treatment. Clin Cancer Res. 2015; 21:3327-3339.
8. Pillai P, Surenya RS, Nair SV, Lakshmanan VK. Cancer kinases and its novel inhibitors: past, present and future challenges. Curr Drug Targets. 2015; 16:1233-1245.
9. Alachkar H, Mutonga MB, Metzeler KH, Fulton N, Malnassy G, Herold T, Spiekermann K, Bohlander SK, Hiddemann W, Matsuo Y, Stock W, Nakamura Y. Preclinical efficacy of maternal embryonic leucine-zipper kinase (MELK) inhibition in acute myeloid leukemia. Oncotarget. 2014; 5:12371-12382. https://doi.org/10.18632/oncotarget.2642.
10. Ganguly R, Mohyeldin A, Thiel J, Kornblum HI, Beullens M, Nakano I. MELK-a conserved kinase: functions, signaling, cancer, and controversy. Clin Transl Med. 2015; 4:11.
11. Speers C, Zhao SG, Kothari V, Santola A, Liu M, Wilder- Romans K, Evans J, Batra N, Bartelink H, Hayes DF, Lawrence TS, Brown PH, Pierce LJ, Feng FY. Maternal embryonic leucine zipper kinase (MELK) as a novel mediator and biomarker of radioresistance in human breast cancer. Clin Cancer Res. 2016; 22:5864-5875.
12. Marie SK, Okamoto OK, Uno M, Hasegawa AP, Oba-Shinjo SM, Cohen T, Camargo AA, Kosoy A, Carlotti CG Jr, Toledo S, Moreira-Filho CA, Zago MA, Simpson AJ, Caballero OL. Maternal embryonic leucine zipper kinase transcript abundance correlates with malignancy grade in human astrocytomas. Int J Cancer. 2008; 122:807-815.
13. Ganguly R, Hong CS, Smith LG, Kornblum HI, Nakano I. Maternal embryonic leucine zipper kinase: key kinase for stem cell phenotype in glioma and other cancers. Mol Cancer Ther. 2014; 13:1393-1398.
14. Xia H, Kong SN, Chen J, Shi M, Sekar K, Seshachalam VP, Rajasekaran M, Goh BK, Ooi LL, Hui KM. MELK is an oncogenic kinase essential for early hepatocellular carcinoma recurrence. Cancer Lett. 2016; 383:85-93.
15. Gu C, Banasavadi-Siddegowda YK, Joshi K, Nakamura Y, Kurt H, Gupta S, Nakano I. Tumor-specific activation of the C-JUN/MELK pathway regulates glioma stem cell growth in a p53-dependent manner. Stem Cells. 2013; 31:870-881.
16. Joshi K, Banasavadi-Siddegowda Y, Mo X, Kim SH, Mao P, Kig C, Nardini D, Sobol RW, Chow LM, Kornblum HI, Waclaw R, Beullens M, Nakano I. MELK-dependent FOXM1 phosphorylation is essential for proliferation of glioma stem cells. Stem Cells. 2013; 31:1051-1063.
17. Chung S, Suzuki H, Miyamoto T, Takamatsu N, Tatsuguchi A, Ueda K, Kijima K, Nakamura Y, Matsuo Y. Development of an orally-administrative MELK-targeting inhibitor that suppresses the growth of various types of human cancer. Oncotarget. 2012; 3:1629-1640. https://doi.org/10.18632/oncotarget.790.
18. Shohet JM, Ghosh R, Coarfa C, Ludwig A, Benham AL, Chen Z, Patterson DM, Barbieri E, Mestdagh P, Sikorski DN, Milosavljevic A, Kim ES, Gunaratne PH. A genome-wide search for promoters that respond to increased MYCN reveals both new oncogenic and tumor suppressor microRNAs associated with aggressive neuroblastoma. Cancer Res. 2011; 71:3841-3851.
19. Kig C, Beullens M, Beke L, Van Eynde A, Linders JT, Brehmer D, Bollen M. Maternal embryonic leucine zipper kinase (MELK) reduces replication stress in glioblastoma cells. J Biol Chem. 2013; 288:24200-24212.
20. MYCN drives RB-null retinoblastoma initiation. Cancer Discov. 2017; 7:OF13.
21. Huang M, Weiss WA. Neuroblastoma and MYCN. Cold Spring Harb Perspect Med. 2013; 3:a014415.
22. Cho YS, Kang Y, Kim K, Cha YJ, Cho HS. The crystal structure of MPK38 in complex with OTSSP167, an orally administrative MELK selective inhibitor. Biochem Biophys Res Commun. 2014; 447:7-11.
23. Kohler RS, Kettelhack H, Knipprath-Meszaros AM, Fedier A, Schoetzau A, Jacob F, Heinzelmann-Schwarz V. MELK expression in ovarian cancer correlates with poor outcome and its inhibition by OTSSP167 abrogates proliferation and viability of ovarian cancer cells. Gynecol Oncol. 2017; 145:159-166.
24. Agarwal S, Lakoma A, Chen Z, Hicks J, Metelitsa LS, Kim ES, Shohet JM. G-CSF promotes neuroblastoma tumorigenicity and metastasis via STAT3-dependent cancer stem cell activation. Cancer Res. 2015; 75:2566-2579.
25. Kato T, Inoue H, Imoto S, Tamada Y, Miyamoto T, Matsuo Y, Nakamura Y, Park JH. Oncogenic roles of TOPK and MELK, and effective growth suppression by small molecular inhibitors in kidney cancer cells. Oncotarget. 2016; 7:17652-17664. https://doi.org/10.18632/oncotarget.7755.
26. Inoue H, Kato T, Olugbile S, Tamura K, Chung S, Miyamoto T, Matsuo Y, Salgia R, Nakamura Y, Park JH. Effective growth-suppressive activity of maternal embryonic leucine-zipper kinase (MELK) inhibitor against small cell lung cancer. Oncotarget. 2016; 7:13621-13633. https://doi.org/10.18632/oncotarget.7297.
27. Lin A, Giuliano CJ, Sayles NM, Sheltzer JM. CRISPR/ Cas9 mutagenesis invalidates a putative cancer dependency targeted in on-going clinical trials. Elife. 2017.
28. Ji W, Arnst C, Tipton AR, Bekier ME 2nd, Taylor WR, Yen TJ, Liu ST. OTSSP167 abrogates mitotic checkpoint through inhibiting multiple mitotic kinases. PLoS One. 2016; 11:e0153518.
29. Chung S, Kijima K, Kudo A, Fujisawa Y, Harada Y, Taira A, Takamatsu N, Miyamoto T, Matsuo Y, Nakamura Y. Preclinical evaluation of biomarkers associated with antitumor activity of MELK inhibitor. Oncotarget. 2016; 7:18171-18182. https://doi.org/10.18632/oncotarget.7685.
30. Edupuganti R, Taliaferro JM, Wang Q, Xie X, Cho EJ, Vidhu F, Ren P, Anslyn EV, Bartholomeusz C, Dalby KN. Discovery of a potent inhibitor of MELK that inhibits expression of the anti-apoptotic protein Mcl-1 and TNBC cell growth. Bioorg Med Chem. 2017; 25:2609-2616.
31. Badouel C, Korner R, Frank-Vaillant M, Couturier A, Nigg EA, Tassan JP. M-phase MELK activity is regulated by MPF and MAPK. Cell Cycle. 2006; 5:883-889.
32. Li S, Li Z, Guo T, Xing XF, Cheng X, Du H, Wen XZ, Ji JF. Maternal embryonic leucine zipper kinase serves as a poor prognosis marker and therapeutic target in gastric cancer. Oncotarget. 2016; 7:6266-6280. https://doi.org/10.18632/oncotarget.6673.
33. Jiang P, Zhang D. Maternal embryonic leucine zipper kinase (MELK): a novel regulator in cell cycle control, embryonic development, and cancer. Int J Mol Sci. 2013; 14:21551-21560.
34. Agarwal S, Ghosh R, Chen Z, Lakoma A, Gunaratne PH, Kim ES, Shohet JM. Transmembrane adaptor protein PAG1 is a novel tumor suppressor in neuroblastoma. Oncotarget. 2016; 7:24018-24026. https://doi.org/10.18632/oncotarget.8116.