
INTRODUCTION
Prostate cancer (PCa) is one of the most common male malignancies and the third leading cause of cancer-related death of men in the United States [1–2]. Small prostatic carcinomas exist in up to 29% of men in their thirties and 64% of men in their sixties, with most of these carcinomas being indolent and/or cured by surgery or radiation [3–5]. However, some men develop an aggressive phenotype that metastasizes and becomes incurable once colonizing the bone [6–7]. These bone metastases produce osteoblastic lesions that are associated with high morbidity and high mortality [8] and attempts at delaying this tumor progression with chemotherapeutic agents have only prolonged survival a few months. [9–10] In order to create more effective treatments where conventional therapeutics have failed, a better understanding of the aggressive phenotype of the disease is of utmost importance and a great unmet medical need.
It is now well-established that reduction-oxidation (redox) regulation of critical transcriptional activators plays an essential role in cell proliferation and survival in a number of different cancers, including prostate cancer [11–13]. Apurinic/apyrimidinic endonuclease 1/redox factor 1 (APE1/Ref-1) is a multifunctional protein that participates in DNA repair and redox transcriptional regulation [14–15]. APE1/Ref-1 has been implicated in the development and progression of numerous cancer types, is conversely correlated to tumor radiation and chemotherapy sensitivity, and is overexpressed in prostate cancer [16–20]. APE1/Ref-1 redox regulation of transcriptional activators occurs through cysteine residues within the DNA binding or transactivation domain of the transcription factor and is essential for full activation of certain transcriptional activators including the oncogenic transcriptional activators AP-1, HIF-1α, NF-κB and STAT3. Treatment with small molecule inhibitors of the redox activity of APE1/Ref-1, such as APX3330 has been shown to diminish the activity of these redox-regulated transcriptional activators [21–23]. Furthermore, the blockade of APE1/Ref-1’s redox activity has been shown to reduce growth-promoting, inflammatory and anti-apoptotic activities in cells [24–25].
The ability of cancer cells to overcome apoptotic signals is crucial for tumor progression. Survivin is an Inhibitor of Apoptosis (IAP) family member, and it is overexpressed in prostate cancer. Survivin has been implicated in resistance to various chemotherapeutic and pro-apoptotic agents [26–28]. Survivin is classically known as an inhibitor of caspases due to its single BIR (Baculovirus IAP Repeat) domain, but recently survivin has been found to be crucial in cell cycle progression as a member of the chromosomal passenger complex (CPC) [29]. As part of the CPC, survivin along with members Borealin, INCENP and Aurora B kinase orient the chromosomes during mitosis. Previously, our lab has demonstrated that survivin is juxtaposed to inflammation in human prostate cancer specimens and may play a role in repair and recovery of prostatic tissue [30]. Attempts at directly targeting survivin have ultimately failed in clinic, therefore new approaches or therapeutics that in some way block the expression or function of survivin are needed.
Accumulating evidence demonstrates that APE1/Ref-1 is a key regulator of cancer cell growth and survival signaling and is upstream of pathways that regulate survivin expression [31]. Here, we report that inhibition of APE1/Ref-1 redox signaling activity decreases prostate cancer cell proliferation, decreases the transcriptional activity of NFκB, and downregulates survivin expression in prostate cancer cells in vitro and in vivo. This is the first report to our knowledge that mechanistically demonstrates that APE1/Ref-1 redox-specific inhibitors are a viable therapeutic option for prostate cancer treatment.
RESULTS
APE1/Ref-1 and survivin are nuclear and cytoplasmic localized in human prostate cancer
To confirm APE1/Ref-1 and survivin protein expression in prostate cancer, we performed immunofluorescence using human non-diseased and cancerous prostate specimens (Figure 1A). We found that APE1/Ref-1 and survivin are expressed in non-diseased and cancerous prostate specimens (n = 12). Both proteins are universally expressed in bone metastasis. Localization of both proteins was primarily found to be nuclear and localized to the epithelium but in cancerous prostates cytoplasmic localization was observed (Figure 1A Inlet). To verify if well-characterized prostatic cell lines displayed the same expression pattern, PC-3, C4-2, LNCaP and non-cancerous E7 cells were fractionated into cytoplasmic, nuclear soluble and chromatin bound fractions and immunoblotting was performed evaluating APE1/Ref-1 and survivin protein localization (Figure 1B). E7 cell line is a normal prostatic epithelial cell line that was transformed using the human papillomavirus 16 (HPV16) E7 gene. MEK 1/2, Lamin B1 and Histone H3 were used as the respective controls for each fraction. APE1/Ref-1 protein localization was found to be in all three subcellular fractions in cancerous cell lines but only the nuclear soluble fraction in non-cancerous E7 cells. Survivin protein localization was primarily found in the cytoplasmic and chromatin bound fraction with some variable expression in the nuclear soluble fraction in the cancerous cell lines but localized only to the chromatin bound fraction in the non-cancerous E7 cells. This mirrors the expression pattern found in the human specimens. Additionally, APE1/Ref-1 and survivin protein levels were found to be significantly higher in PC-3, C4-2 and LNCaP cell lines compared to the E7 cell line (Supplementary Figure 1).
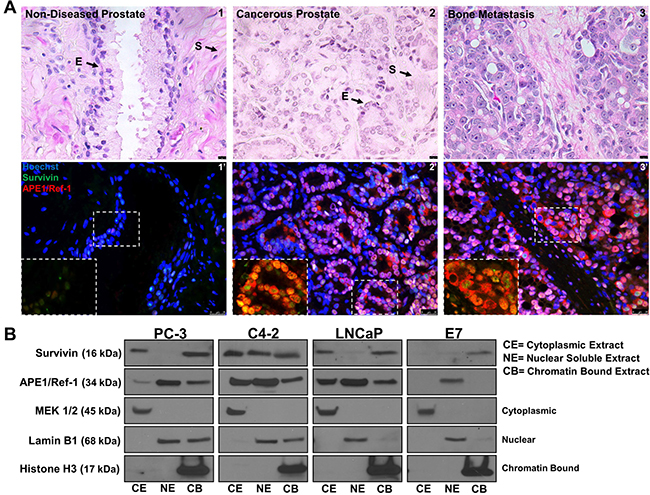
Figure 1: APE1/Ref-1 and survivin are nuclear and cytoplasmic localized in human prostate cancer. (A) Hematoxylin and Eosin staining representing non-diseased (peripheral zone taken from cystoprostatectomy) and cancerous human prostate specimens (1–3). Scale bar = 10 μM. Immunofluorescent images of stained non-diseased and cancerous sections (1′-3′) for APE1/Ref-1 (red) and survivin (green). Scale bar = 25 μm, n = 12. (B) Cellular fractionation representing basal survivin and APE1/Ref-1 protein localization in cancerous (PC-3, C4-2 and LNCaP) and non-cancerous (E7) prostatic cell lines. MEK 1/2 (cytoplasmic), Lamin B1 (nuclear) and Histone H3 (chromatin bound) were used as controls for each subcellular fraction
APE1/Ref-1 redox inhibition decreases prostate cancer cell number
To determine if inhibition of APE1/Ref-1’s redox function affects cell number, prostatic cell lines were treated with increasing concentrations of APE1/Ref-1 redox-specific inhibitors APX3330 and APX2009 for five days and cell number was measured via methylene blue assay (Supplementary Figure 2). RN7-58 is an inactive analogue of the APX3330 and APX2009 chemical families and was used as a negative control. It has been shown to have no effect on APE1/Ref-1 redox function. [32] APX3330 and APX2009 inhibited cell number in a concentration-dependent manner (Figure 2A–2D). Growth IC25’s and IC50’s were determined (Table 1). Student’s t-test was performed to verify statistical IC25 and IC50 differences between APX3330 and APX2009. RN7-58 caused variable cell growth between cell lines but did not contain IC25 or IC50 drug concentrations. APX2009 was found to be 5–10 fold more efficacious than parent compound APX3330 in inhibiting cell proliferation. DMSO control was not significantly different from untreated cells.
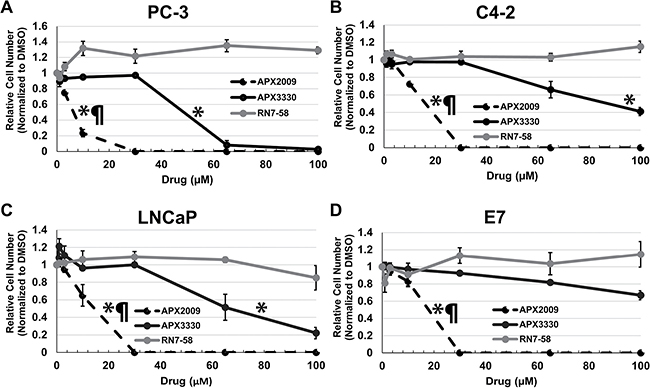
Figure 2: APE1/Ref-1 redox function specific inhibitors decrease cell number in a concentration dependent manner. PC-3 (A), C4-2 (B), LNCaP (C) and E7 (D) cell lines were treated with increasing concentrations of redox-specific inhibitor APX3330, more potent analogue APX2009, and inactive analogue RN7-58 for 5 days. The cells were fixed and stained with methylene blue and measured via spectrophotometry. IC25 and IC50 were determined as the concentrations of drug at which there was a 25% and 50% reduction in absorbance compared to vehicle control (DMSO) and were used for subsequent experiments. n = 3. EC50s were compared between the drugs: * denotes p < 0.05 drug EC50 versus RN7-58, while ¶ denotes p < 0.05, APX3330 versus APX2009.
Table 1: Growth IC25 and IC50’s were determined for each cell line using the 3 growth curves for APX3330 and APX2009
APX3330 (μM) |
APX2009 (μM) |
P value |
||
|---|---|---|---|---|
PC-3 |
IC25 |
36.0 +/− 1.0 |
2.2 +/− .5 |
> 0.0001 |
IC50 |
54.7 +/− 1.6 |
8.9 +/− .7 |
> 0.0001 |
|
C4-2 |
IC25 |
57.4 +/− 3.8 |
7.6 +/− .2 |
0.0002 |
IC50 |
89.5 +/− 7.8 |
14.2 +/− .3 |
0.0006 |
|
LNCaP |
IC25 |
43.8 +/− 4.2 |
6.3 +/− 1.6 |
0.0011 |
IC50 |
71.9 +/− 7.2 |
13 +/− 1.2 |
0.0013 |
|
E7 |
IC25 |
82.7 +/− 8.7 |
9.2 +/− .7 |
0.0011 |
IC50 |
N/A |
16.1+/− .8 |
- |
Data are presented as mean ± s.e.m. P value was determined by comparing IC25 or IC50 values for APX3330 to that of APX2009 averages from the three separate determinations by unpaired Student’s t-test in each cell line.
APE1/Ref-1 redox-specific inhibitors decrease survivin protein levels
Survivin plays an important role in prostate cancer cell proliferation and survival. Since survivin is controlled by APE1/Ref-1-regulated transcription factors in other organ systems such as the pancreas and liver [33–34], we hypothesized that treatment with APE1/Ref-1 redox-specific inhibitors APX3330 and APX2009 would decrease survivin protein levels, at least partially explaining the reduction in proliferative capacity. Prostate cancer cells treated with the respective growth inhibitory IC25 and IC50 drug concentrations of APX3330 and APX2009 (as determined in Table 1) exhibited a significant decrease in survivin protein expression within 48 hours compared to DMSO treated controls (Figure 3A–3D). In contrast, prostate cancer cell total APE1/Ref-1 protein levels were not significantly altered with treatment.
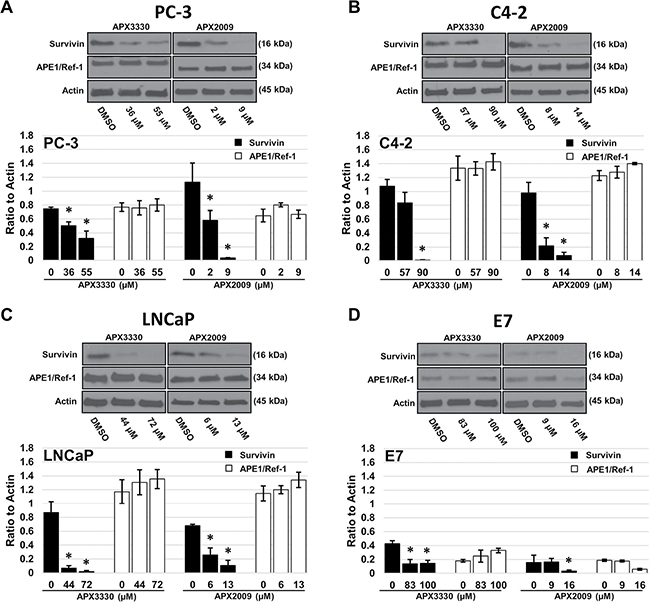
Figure 3: Treatment with APX3330 and APX2009 decreases survivin protein levels. PC-3 (A), C4-2 (B), LNCaP (C) and E7 (D) cell lines were treated with DMSO, or the growth inhibitory IC25 and IC50 drug concentrations of APX3330 or APX2009 for 48 hours. Immunoblotting for survivin, APE1/Ref-1 and Actin as labeled. Data presented are representative of three determinations with densitometry quantification, n = 3, *-denoting p < 0.05 (DMSO vs. IC25 and IC50 Drug Concentrations) as assessed by ANOVA.
APE1/Ref-1 siRNA reduces proliferation and survivin protein levels
Using siRNA specific to APE1/Ref-1, we investigated if APE1/Ref-1 knockdown reduces cell growth and survivin protein levels. PC-3 and C4-2 cell lines were transfected with two distinct sequences of 50 nM APE1/Ref-1 siRNA (verified > 70% knockdown by immunoblotting) and growth was compared to scrambled siRNA-transfected cells (Figure 4A). Those cells transfected with APE1/Ref-1 siRNA grew at a significantly slower rate compared to those cells transfected with the scrambled siRNA. Representative pictures of fixed and methylene blue stained C4-2 and PC-3 scrambled siRNA (Scr), survivin siRNA #1 (siAPE1 #1) and #2 (siAPE1 #2) were taken (Figure 4B). Immunoblotting was performed 72 hours post transfection and survivin protein levels were found to be decreased compared to scrambled control (Figure 4C).
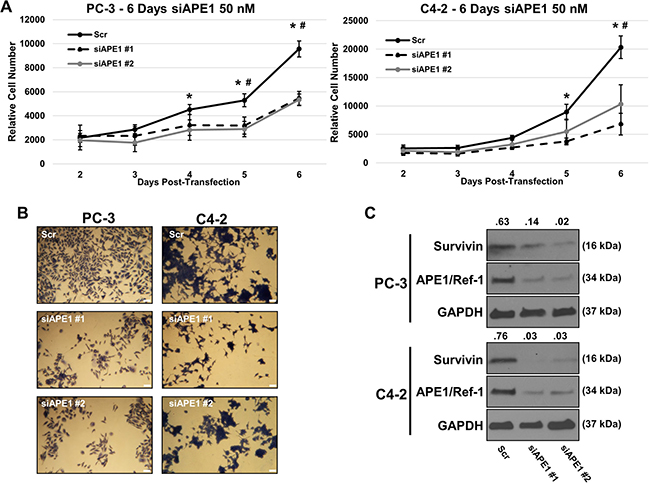
Figure 4: APE1/Ref-1 siRNA knockdown decreases cell proliferation and survivin protein levels. (A) Separate aliquots of PC-3 and C4-2 cell lines were transfected with two distinct sequences of 50 nM APE1/Ref-1 siRNA (verified >70% knockdown by immunoblotting) and growth was compared to scrambled siRNA-transfected cells. n = 3, *-denoting p < 0.05 within ANOVA (Scr vs siAPE#1), #- denoting p < 0.05 within ANOVA (Scr vs siAPE#2). (B) Representative pictures of fixed and methylene blue stained C4-2/PC-3 scrambled siRNA (Scr), survivin siRNA #1 (siAPE1 #1) and #2 (siAPE1 #2). (C) Immunoblotting was performed using antibodies for APE1/Ref-1, survivin and GAPDH as labeled after 72 hours post-transfection.
Treatment with APX2009 induces G1 cell arrest but not cell death
Based on the increased potency of APX2009 over APX3330 in PCa cells, we focused on the second generation compound, APX2009 for the remainder of our molecular studies. To determine if inhibition of APE1/Ref-1 via APX2009 results in cell death due to loss of survival signaling, PC-3 and C4-2 cells were treated with either vehicle (DMSO) or previously-determined IC50 concentrations of APX2009 (9 μM in PC3 and 14 μM in C4-2) for 48 hours (Figure 5A) and cell lysates were collected for immunoblotting (Figure 5B). After APX2009 treatment, both PC-3 and C4-2 cells displayed an altered, flattened cellular morphology. However, treatment with these compounds did not induce cell death as determined by both a lack of increased caspase 3 cleavage (Figure 5B) and TUNEL labeling (data not shown). Because no increase in apoptosis was detected and cell cycle proteins Cdc2 and Cyclin B1 were dramatically decreased by APE1/Ref-1 inhibition (undetectable in PC-3 cells and a ~97% decrease in C4-2 cells), cell cycle analysis was performed using Propidium Iodide (PI) staining. PC-3 and C4-2 cells were treated with APX2009 (9 μM and 14 μM, respectively) for 48 hours, stained with PI, and analyzed by flow cytometry (Figure 5C). We found that the percentage of cells in G1 significantly increased, p ˂ 0.05 via Student’s t-test, from 58 to 68% and 63 to 74% in PC3 and C4-2 cells, respectively, indicating G1 arrest of prostate cancer cells in response to APE1/Ref-1 inhibition. These effects on the cell cycle progression are similar to other recent reports of APE1/Ref-1 redox inhibition in cancer [35–36].
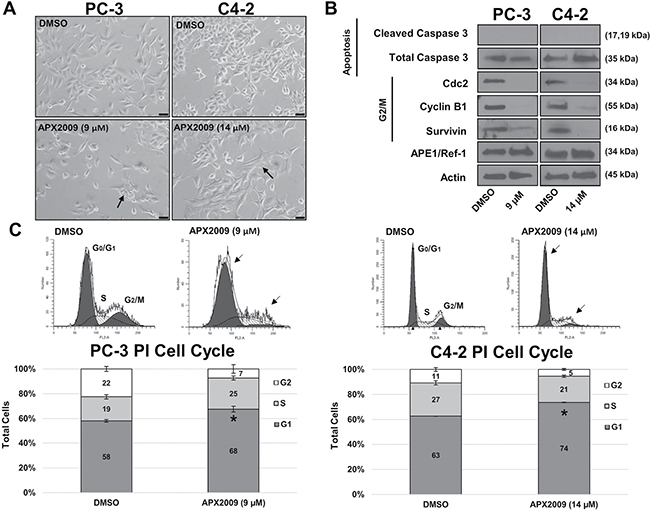
Figure 5: APE1/Ref-1 redox inhibition induces G1 cell arrest. (A) PC-3 and C4-2 cell lines were treated with DMSO or APX2009 (9 and 14 μM, respectively) for 48 hours. Representative images were taken at 20× Magnification. Scale bar = 50 μm. (B) Immunoblotting was performed and membranes were probed with antibodies for Cleaved Caspase 3, Total Caspase, Cyclin B1, Cdc2, survivin and Actin as labeled. (C) PC-3 and C4-2 cells were treated with DMSO or APX2009 (9 and 14 μM, respectively) for 48 hrs and then collected and stained with RNAse/PI wash. Flow Cytometry was then performed. n =3, *-denoting p < 0.05 by unpaired Student’s t-test.
APX2009 reduces survivin mRNA expression and perturbs NFκB activity
Based on the observation that inhibition of APE1/Ref-1 reduces survivin protein levels, we sought to determine the mechanism by which APE1/Ref-1 regulates survivin expression, and ultimately, cell growth. We hypothesized that APE1/Ref-1’s redox control of transcription factors like NFκB would decrease survivin transcript levels. C4-2 cells were treated with vehicle or APX2009 IC50 (14 μM) for 12 hours. RNA was collected and RT-qPCR was performed using a primer/probe set for survivin (BIRC5) and HPRT1 for the reference gene (Figure 6A) using the conditions suggested by the SuperScript III Platinum One-Step qRT-PCR System (Invitrogen). Survivin mRNA was significantly reduced upon treatment with the relative quantity (RQ) value of ˂ 0.5. Survivin has been shown in other cancers to be regulated by NFκB, and NFĸB is regulated by APE1/Ref-1 redox signaling [37–40]. Therefore, we evaluated the ability of these two proteins to interact physically with each other. In Figure 6B, we demonstrate via co-immunoprecipitation that APE1/Ref-1 interacts with NFκB subunit p65 when using an APE1/Ref-1 antibody and in reverse experiments using a p65 antibody. To determine if NFκB signaling is responsible for cell growth and regulated by APE1/Ref-1 redox activity, we treated C4-2 cells with increasing concentrations of APX2009 and NFκB inhibitor ammonium pyrrolidinedithiocarbamate (PDTC) to determine the respective growth inhibition (Figure 6C). We then determined NFκB activity in the presence of these two drugs and found a significant two-fold decrease in NFκB-driven luciferase activity (Figure 6D). To further confirm a role of NFκB in regulating survivin protein levels, we treated C4-2 cells with 14 μM APX2009 and 100 μM PDTC for 48 hours and observed a significant 95% and 67% reduction in survivin protein levels, respectively (Figure 6E). In addition, we assessed the cellular localization of both NFκB and APE1/Ref-1 upon treatment with APX2009 (Supplementary Figure 3). p65 and APE1/Ref-1 were found to be co-localized in the nucleus however upon treatment with APX2009, p65 nuclear localization was diminished suggesting altered NFκB protein trafficking.
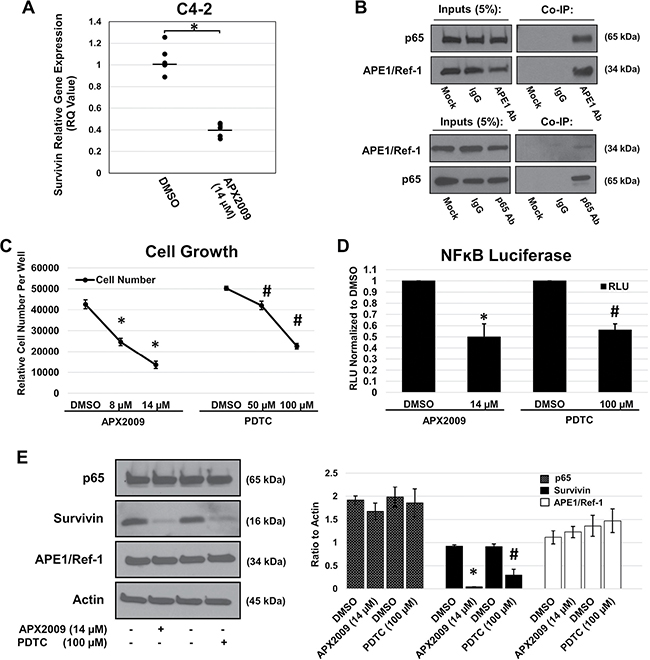
Figure 6: APE1/Ref-1 redox inhibition decreases survivin protein levels via NFĸB. (A) C4-2 cell line was treated with DMSO or APX2009 (14 μM) for 12 hours. RNA was isolated and RT-PCR for survivin was performed with HPRT1 as the reference gene. n = 6, *-denoting p < 0.05 by unpaired Student’s t-test. (B) Immunoblot validation of APE1/Ref-1 and p65 Co-Immunoprecipitation (Co-IP) reactions. A 5% sample of the total input of each reaction (Input) and the total IP reaction (IP) were loaded for each reaction. Beads lacking a conjugated antibody (Mock) and generic IgG (IgG) were used as negative controls for each IP experimental reaction, APE1 antibody (top blots) and p65 (bottom blots). (C) C4-2 cell line was treated with DMSO, APX2009 (8 and 14 μM) or PDTC (50 and 100 μM) for 72 hours and cells were fixed and methylene blue was performed. n = 3, *-denoting p < 0.05 (DMSO vs 8 and 14 μM APX2009) and #-denoting p < 0.05 (DMSO vs. 50 and 100 μM PDTC) as assessed by ANOVA. (D) C4-2 cells were transfected with NFκB–Luc construct and co-transfected with a Renilla vector, pRL-TK. After 16 hours, cells were treated with growth inhibitor IC50 concentrations of APX2009 and PDTC for 24 hours, and Firefly and Renilla luciferase activities were assayed using Renilla luciferase activity for normalization. All transfection experiments were performed in triplicate and repeated 3 times in independent experiments. Data are expressed as Relative Luciferase Units (RLU) normalized to DMSO showing the mean ± SEM. n = 3, *-denoting p < 0.05 (DMSO vs. 14 μM APX2009) and #-denoting p < 0.05 (DMSO vs. 100 μM PDTC) within unpaired Students t-test. (E) C4-2 cell line was treated with APX2009 (14 μM) and NFĸB-selective inhibitor PDTC (100 μM) for 48 hours. Immunoblotting was performed with antibodies for survivin, p65, APE1/Ref-1 and Actin as labeled. Data presented are representative of three determinations with densitometry quantification, n = 33, *-denoting p < 0.05 (DMSO vs. 14 μM APX2009) and #-denoting p < 0.05 (DMSO vs. 100 μM) as assessed by unpaired Student’s t-test.
APE1/Ref-1 redox inhibition decreases survivin protein levels and cell proliferation in vivo
Based on the strong in vitro data demonstrating the regulation of survivin levels following APE1/Ref-1 inhibition, we confirmed our studies in vivo using C4-2 subcutaneous xenografts. The data in Figure 7 demonstrates that APE1/Ref-1 redox activity also plays a role in cell proliferation and survivin protein levels in vivo. Animals were treated with either APX2009 (25 mg/kg BID) or Vehicle for 5 days and then tumors were harvested. Total survivin protein via immunoblotting was significantly reduced (Figure 7A) when compared to control tumors. Survivin and APE1/Ref-1 localization via immunofluorescence remained nuclear with survivin co-localizing with the chromatin during mitosis (Figure 7B). Furthermore, BrdU incorporation was significantly reduced from 8.2% to 5.1% in the treatment group demonstrating that inhibition of APE1/Ref-1 redox activity reduces tumor cell proliferation (Figure 7C).
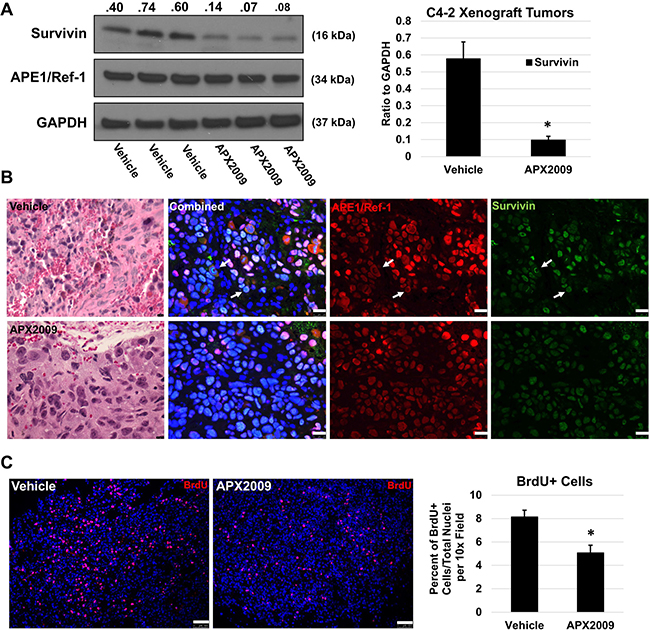
Figure 7: In vivo treatment with APX2009 reduces survivin protein levels and BrdU incorporation in C4-2 xenograft tumors. C4-2 xenograft tumors were treated with Vehicle (Propylene Glycol Kolliphor HS15 Tween 80 (PKT)) or APX2009 (25 mg/kg, IP bid) for 5 days (n = 3). Tumors were removed and processed for either immunofluorescence or immunoblotting. (A) APE1/Ref-1 and survivin protein levels were measured using immunoblotting as labeled (Left). Data was presented graphically (Right), *-denoting p < 0.05 by unpaired Student’s t-Test. (B) Hematoxylin and Eosin staining and immunofluorescence were performed using APE1/Ref-1 (red) and survivin (green) specific antibodies on vehicle and APX2009 groups. Representative images were taken. White arrows are depicting survivin nuclear staining patterns. Scale bar H&E = 10 μM. Scale bar immunofluorescence = 25 μm. (C) Mice were injected with BrdU 2 hours prior to sacrifice and tumors were collected and stained for BrdU incorporation (red). Scale bar = 100 μm. ImageJ Nucleus Counter was used to quantify number of BrdU+ nuclei and total nuclei per image. n = 3, *-denoting p < 0.05 by unpaired Student’s t-test.
DISCUSSION
Prostate cancer is one of the leading causes of cancer-related death in American men, and challenges remain in targeting key drivers of the aggressive phenotype despite recent advances in prostate cancer treatment. Androgen deprivation therapies and microtubule-targeting agents prolong survival but resistance to these therapeutics is inevitable. It is thought that this resistance is driven in part by aberrant survival signaling and the induction of survival proteins which allows for the cancer to evade cell death [41–42]. Survivin is a bifunctional protein that has been shown to be overexpressed in a number of different cancers including prostate cancer. Survivin has anti-apoptotic and pro-proliferative functions in cancer cells. Inhibition of survivin is a logical therapeutic strategy, however directly targeting survivin has been difficult. In this study, we took a novel approach to survivin targeting; we provide evidence that targeting the redox-signaling regulator APE1/Ref-1 with small molecule inhibitors effectively suppresses survivin protein levels and inhibits cell proliferation (Figure 8).
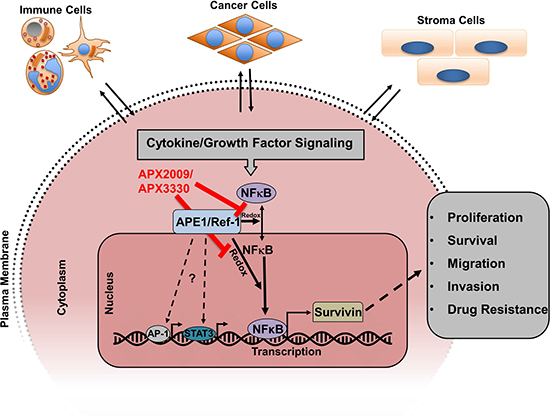
Figure 8: Model showing how cytokine/growth factor signaling induces survivin protein expression and where APX2009/APX3330 inhibits.
APE1/Ref-1 is a multifunctional protein that was initially discovered as an enzyme in the base excision repair (BER) pathway, but has also emerged as a redox-signaling regulator of a number of transcription factors known to be involved in cancer, namely NFĸB, AP-1, HIF1a, and STAT3 [43]. These transcription factors have been shown to be important in the initiation and progression of prostate cancer, as well as other cancers. [44–46] In this way, inhibiting the redox activity of APE1/Ref-1 effectively targets multiple different pathways at once and may therefore represent an advantageous therapeutic strategy [47].
The data presented in our studies further support the rationale for APE1/Ref-1 as a viable target in prostate cancer. Our results indicate that APE1/Ref-1 and survivin are expressed in human primary and metastatic tumors as previously reported by Kelley et al. [48]. APE1/Ref-1 and survivin was found to be primarily nuclear localized but cytoplasmic staining was present in the tumors. This pattern was also exhibited in the prostatic cell lines with the cancerous cell lines being positive for APE1/Ref-1 and survivin cytoplasmic localization. Survivin cytoplasmic expression has been associated with poor prognosis in breast, lymphoma, non-small cell lung, live, gastric, ovarian and colorectal cancer [49]. In lung, ovarian, thyroid, and breast cancers, APE1/Ref-1 cytoplasmic distribution has been associated with a higher aggressiveness of the tumor [50].
Similar to other cancer cell lines, we found that APE1/Ref-1 siRNA knockdown decreased cell proliferation and survivin protein levels. Additionally, we demonstrate that inhibition of APE1/Ref-1 redox activity halts prostate cancer cell growth and induces G1 cell arrest in prostate cancer, consistent with recent reports in other cancers. APE1/Ref-1 is crucial in moving cells from G1 to S, and redox inhibition induces key cyclin-dependent kinase inhibitors (CDKi’s) like p21 and p27 [51–52]. This is a translationally relevant finding, as the first-generation APE1/Ref-1 small molecule inhibitor APX3330 used in this study is now approved for phase 1 clinical trials [Investigational New Drug (IND) application number 125360]. APX3330, and the second generation molecule APX2009, is known to bind to APE1/Ref-1 in the redox active region of the protein, cause unfolding of the APE1/Ref-1 protein and block the redox active cysteine 65 from functioning, thus effectively inhibiting its transcriptional regulatory activity of growth signaling pathways [53–56]. APX3330 has been shown to decrease cell proliferation in other cancers including pancreatic and ovarian, and here we show it has similar affects in prostate cancer. Even though cell cycle arrest is what we primarily observed, cell death may occur at later time points and more studies are needed to determine this.
A noteworthy point of these findings is that benign prostate cells also respond to redox-selective APE1/Ref-1 inhibition. This may suggest that benign cells of the prostate could also be affected by Ref-1 inhibition, however even though the E7-transformed cells are considered noncancerous, they still contain a defect in cell cycle regulation due to immortalization via the E7 gene transfection. In other organ systems, normal differentiated cells have not been responsive to Ref-1 inhibition. [57] In fact, APX3330 and APX2009 show a protective effect in neurons, the mechanism of which is still under active investigation [57–59]. While we do not know what effect these inhibitors would have on normal cells in the prostate, in vivo, given all the previous preclinical and clinical toxicology studies showing no effects on normal cells, we would not anticipate normal prostate cells to be any different. APX3330 was previously developed for a hepatitis indication and used in over 400 patients with no toxicity. The levels obtained by Eisai in their clinical trials were between 50 and 150 uM in blood such that our dosing is at or below the obtainable level. The proposed APX3330 Phase 1 clinical trial in cancer patients which will be initiating soon will start at a dose that is above levels used in these studies.
Survivin is known to be differentially regulated in various tissues and in response to external stimuli [60]. Survivin is transcriptionally regulated by a number of transcriptional activators including STAT3 and NFĸB. NFĸB-driven survivin protein expression was interrogated here due to our observation that APE1/Ref-1 inhibition is effective in PC-3 cells despite their lacking the gene coding for STAT3. All four cell lines express functional NFĸB signaling [61]. We provide evidence that survivin is at least partially transcriptionally regulated by NFĸB. Upon APE1/Ref-1 redox inhibition, survivin mRNA levels are reduced and IP experiments demonstrate a strong interaction between APE1/Ref-1 and NFĸB subunit p65. Following modulation of APE1/Ref-1 signaling with APX2009 and NFĸB inhibitor PDTC, NFĸB signaling was decreased by 2-fold as assessed by NFĸB-driven luciferase activity. In addition to this decrease in NFkB activity, we also demonstrated that survivin protein levels are reduced by APX2009 (95% reduction) and PDTC (67% reduction) [62]. This reduction in survivin protein levels could occur due to diminished paracrine signaling factors such as, IL-6 or IL-8, which activate NFĸB. It is also possible that APX2009 and PDTC disrupt p65 nuclear trafficking/DNA binding [63]. Further experiments are needed to elucidate this. Nevertheless, our data do not preclude that multiple transcription factors could be contributing to survivin protein levels, and future studies will be directed at carefully assessing the role of each potential transcriptional activator in APE1/Ref-1-mediated prostate cancer cell growth and survivin expression.
In summary, our data indicate that APE1/Ref-1’s redox function plays a role in regulating the proliferative capacity of PCa cells by perturbation of NFĸB transcriptional activity and survivin protein levels in human prostate cancer cell lines and in vivo in tumors. Survivin plays an important role in prostate cancer survival, progression and therapeutic resistance. Thus, inhibition of APE1/Ref-1’s redox function in combination with the current therapeutics like docetaxel or cabazataxel may prove to be novel treatment strategy in advanced prostate cancer.
MATERIALS AND METHODS
Ethics statement
Investigation has been conducted in accordance with the ethical standards and according to the Declaration of Helsinki and according to national and international guidelines and has been approved by the authors’ institutional review board.
Cell lines
PC-3, LNCaP and C4-2 prostate cancer cell lines were purchased from and authenticated by the ATCC (Manassas, VA). E7 prostate epithelial cells were generated in the lab from human benign prostate tissue using the method published by Schwarze et al., Dr. David Jarrard, Department of Urology, University of Wisconsin Madison [64]. All cell lines were maintained at 37ºC in 5% CO2 and grown in RPMI (Corning: Manassas, VA) with 5% Fetal Bovine Serum (HyClone: Logan, UT).
Drugs
APX3330, which is also called E3330 [61], was synthesized as previously described [65]. APX2009 was a kind gift from Apexian Pharmaceuticals LLC (Indianapolis, IN). Synthesis and description of APX2009 and RN7-58 has been previously described [65–66]. PDTC (Ammonium pyrrolidinedithiocarbamate) (ab141406) was obtained from Dr. Tao Lu (Indianapolis, IN) who purchased it from Abcam (Cambridge, MA).
Immunofluorescence
Human prostate specimens or C4-2 xenograft tumors were fixed in 10% formalin, processed routinely, embedded in paraffin, and serially sectioned at 5 μm via microtome. Tissues were subjected to heat-induced antigen retrieval in 10 mM citrate buffer (citrate buffer stock solution of monohydrate-free acid citric acid, sodium citrate dehydrate, pH 6.0) for 10 minutes followed by 10 minute rest. Sections were blocked at room temperature with a bovine serum albumin (BSA)-Donkey serum mixture for 2 hours and incubated with primary antibody overnight at 4 C. Primary antibodies and dilutions included rabbit survivin (1:100, Cell Signaling Technologies), mouse APE1/Ref-1 (1:200, Novus Biologicals), rabbit BrdU (1:200, Cell Signaling Technologies), and mouse PanCK (1:200, Cell Signaling Technologies). Sections were washed with 1X PBS (Phosphate-buffered saline)-Tween and incubated with IgG Alexa 488 and IgG Alexa 594-conjugated secondary antibody against rabbit or mouse for 1 hour at room temperature (1:200, Invitrogen), followed by 10 minutes incubation with Hoechst 33258 nuclear stain (1μg/ml). Tissues were washed with 1× PBS-Tween and water and then covered with an aqueous medium/glass coverslips. The sections were analyzed by immunofluorescence.
Human specimens
Human prostate specimens (n = 12) were obtained with appropriate minimal risk institutional review board approval according to the approval and guidelines at Indiana University School of Medicine. Sections were cut from pre-existing paraffin-embedded human prostate tissues obtained as part of a prostatectomy or from prostate specimens removed collaterally from bladder cancer patients undergoing cystoprostatectomy as control human specimens. These controls were age-matched to the prostate cancer specimens and were verified by record to be naïve for pretreatment with Bacillus Calmette-Guérin (BCG) because these patients had presented first with muscle invasive bladder cancer. The controls were verified by pathology to be void of prostate cancer, BPH, or prostatitis. All human specimens were stained with survivin and APE1/Ref-1 antibodies for immunofluorescence, as described above.
Immunoblotting
Prostate cells were homogenized in lysis buffer containing protease inhibitor (150 mM NaCl, 10 mM tris, 1 mM EDTA, 1 mM benzenesulfonyl fluoride, and 10 μg/ml each of aprotinin, bestatin, L-luecine, and pepstatin A) and 1% Triton X-100. Total protein concentration was determined by BCA (bicinchoninic acid) assay (Pierce, Rockford, IL). 10 μg/well of Protein were resolved by electrophoresis in 4–15% gradient polyacrylamide gels (Bio-Rad Laboratories). Proteins were transferred to polyvinylidene difluoride (PVDF) membranes, blocked for 24 hours [(10% Dry milk, 5% BSA, .05% NaN3) in 1xPBS( 2.7 mM KCl, 1.5 mM KH2PO4, 136 mM NaCl, 8 mM Na2HPO4)-Tween 20] and incubated overnight with one of the following primary antibodies: mouse β-actin (1:2500, ThermoFisher Scientific), mouse APE1/Ref-1 (1:1000, Novus Biologicals), rabbit survivin (1:500, Cell Signaling Technologies), rabbit Bcl-2 (1:500, Cell Signaling Technologies), rabbit Mcl-1 (1:500, Cell Signaling Technologies), rabbit Cleaved Caspase 3 (1:250, Cell Signaling Technologies), rabbit Total Caspase 3 (1:1000, Cell Signaling Technologies), rabbit Cyclin B1 (1:500, Cell Signaling Technologies), Cdc2 (1:1000, Cell Signaling Technologies) and rabbit GAPDH (1:1000, Cell Signaling Technologies). After blots were washed 6 times with PBS-Tween, blots were incubated with donkey antibody against rabbit or mouse immunoglobulin G conjugated to horseradish peroxidase for 1 hour (1:10,000 dilution, Pierce) in nonfat dry milk, 1× PBS, and .05% Tween 20. Peroxidase activity was detected via Pico/ West-Femto chemiluminescence reagent (Pierce). Photo images were analyzed by densitometry.
Methylene blue assay (cell proliferation)
Prostate cells were seeded 1,000-5000 per well (cell line/experiment-dependent) and treated with either APX3330, APX2009 or RN7-58 for 5 days. Media was then removed and cells were fixed with methanol for 10 minutes and stained with 100 μL of 0.05% of methylene blue (LC16920-1 diluted in 1× PBS) for 1 hour. The cells were then washed 3× with water and allowed to air dry overnight. 100 μL’s of 0.5N HCl was added to each well to dissolve the methylene blue stain and absorbance (@630nm) was measured via spectrophotometry. The percent viabilities, normalized to DMSO control, were graphed and IC50 concentrations determined. DMSO control was not significantly different from media alone cells.
Reverse transcription-polymerase chain reaction (RT-PCR)
RNA isolation was performed using RNeasy Mini Kit (Qiagen). 10 nanograms of total RNA was reverse transcribed using Superscript III One-Step RT-PCR System (ThermoFisher Scientific). Real-time PCR was performed using the TaqMan Gene Expression Assay (BIRC5 (Hs04194392_s1) and HPRT1 (Hs02800695_m1), ThermoFisher Scientific) and Applied Biosystems 7500 Fast Real-Time PCR System.
Co-immunoprecipitation
Samples were co-immunoprecipitated using the Pierce Co-IP kit (Thermo Scientific). Additionally, the cells were washed twice with 1× PBS and the proteins were cross-linked using DTBP (Thermo Scientific, 5 mm, for 30 min on ice). DTBP was quenched by washing with cold inactivation buffer (100 mm Tris-HCl, pH 8, 150 mm NaCl) and 1XPBS. Cells were then lysed and the lysates added to columns and after extensive washing, the bound proteins were eluted and prepared for immunoblot analysis.
Luciferase assay
C4-2 cells were co-transfected with constructs containing luciferase driven by NFκB (pLuc-MCS with NFκB responsive promoter; P0athDetect cis-Reporting Systems, Stratagene, La Jolla, Ca) and a Renilla luciferase control reporter vector pRL-TK (Promega Corp., Madison, WI) at a 20:1 ratio by using Effectene Transfection Reagent (Qiagen; Valencia, CA). After 16 hours, cells were treated with increasing concentrations of APX2009 or PDTC in serum free media for 24 hours. Firefly and Renilla luciferase activities were assessed by using the Dual Luciferase Reporter Assay System (Promega Corp.). Renilla luciferase activity was used for normalization and all transfection experiments were performed in triplicate and repeated 3 times in independent experiments.
Propidium iodide (PI) cell cycle analysis
PC-3 and C4-2 cells were treated with APX2009 (9 and 14 μM, respectively) for 48 hours. 500,000 cells were then aliquoted for cell cycle analysis and 0.1 mg/ml Propidium Iodide and 0.6% NP-40 PBS stain wash was added to the tubes. The cells were then centrifuged at 1900 rpms for 10 minutes with the brake on low and then decanted and blotted. RNAase and stain wash were added and cells incubated on ice for 30 minutes. Propidium Iodide intensity was measured via flow cytometry.
In vivo subcutaneous tumor
2 × 106 C4-2 cells were subcutaneously implanted in the hind flank of male athymic nude mice using a 100μl volume of 50:50 solution of Matrigel: RPMI medium. When tumor volumes reached 150 –200 mm3, the animals were treated with 25 mg/kg IP APX2009 or vehicle (Propylene Glycol Kolliphor HS15 Tween 80 (PKT)) every 12 hours for 5 days. BrdU was injected into the animals 2 hours prior to sacrifice and tumor tissues were analyzed for survivin levels (immunofluorescence and immunoblotting) and BrdU incorporation (immunofluorescence).
siRNA transfection
All siRNA transfections were performed using the HiPerfect Transfection Reagent (Qiagen) protocol. Post-transfection C4-2 cells (1,000 per well) and PC-3 cells (1,500 per well) were replated in a 96 well plate and fixed daily up to 6 days and methylene blue assay was performed. Samples for immunoblotting were collected 72 hours post transfection of cancer cells with APE1/Ref-1 siRNA and scrambled siRNA control. Prevalidated APE1/Ref-1 siRNA (siAPE1 #2) was purchased from LifeTech (#s1446).
Statistics
Summary statistics are presented using the mean, median, and SD. Either a Student’s t-test or ANOVA test was performed to compare the groups as appropriate. Statistical significance was assessed at the p < 0.05.
Author contributions
All authors contributed extensively to the work presented in this paper; David W. McIlwain: Designed and Performed Experiments; Wrote Paper; Melissa L. Fishel: Supervised the Project; Wrote Paper; Designed and Provided Drug for In Vivo Experiment; Mark R. Kelley: Supervised the Project; Wrote Paper; Designed and Provided Drug for In Vivo Experiment; Travis J. Jerde: Designed and Performed Experiments; Wrote Paper; Supervised the Project.
ACKNOWLEDGMENTS
We would like to acknowledge Dr. Tao Lu for providing the drug PDTC.
CONFLICTS OF INTEREST
Mark R. Kelley has licensed APX3330 through Indiana University Research and Technology Corporation to Apexian Pharmaceuticals LLC. APX2009 is a second generation compound from Apexian Pharmaceuticals. Apexian Pharmaceuticals had neither control nor oversight of the studies, interpretation, or presentation of the data in this manuscript.
FUNDING
This work was funded by NIH-NIDDK: DK092366-01A1, the DOD-PCRP- W81XWH-13-PCRP-IDA and the Indiana Clinical and Translational Sciences Institute (CTSI) (DWM). Financial support for this work was also provided by the National Cancer Institute [CA122298 (M.L. Fishel), CA138798 (M.L. Fishel and M.R. Kelley) CA205166 (Kelley) and the National Institutes of Health, [NS091667 (M.R. Kelley)]. Additional financial support was provided by Ralph W. and Grace M. Showalter Research Trust Fund (M.L. Fishel), the Earl and Betty Herr Professor in Pediatric Oncology Research, Jeff Gordon Children’s Foundation and the Riley Children’s Foundation (M.R. Kelley).
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016; 66:7–30.
2. Weiner AB, Matulewicz RS, Eggener SE, Schaeffer EM. Increasing incidence of metastatic prostate cancer in the United States (2004–2013). Prostate Cancer Prostatic Dis. 2016; 19:395–397.
3. Konety BR, Bird VY, Deorah S, Dahmoush L. Comparison of the incidence of latent prostate cancer detected at autopsy before and after the prostate specific antigen era. J Urol. 2005; 174:1785–1788.
4. Sakr WA, Haas GP, Cassin BF, Pontes JE, Crissman JD. The frequency of carcinoma and intraepithelial neoplasia of the prostate in young male patients. J Urol. 1993; 150:379–385.
5. Carter HB, Kettermann A, Warlick C, Metter EJ, Landis P, Walsh PC, Epstein JI. Expectant management of prostate cancer with curative intent: an update of the Johns Hopkins experience. J Urol. 2007; 178:2359–2364.
6. Soloway MS, Hardeman SW, Hickey D, Todd B, Soloway S, Raymond J, Moinuddin M. Stratification of patients with metastatic prostate cancer based on extent of disease on initial bone scan. Cancer. 1988; 61:195–202.
7. Higano CS. Understanding treatments for bone loss and bone metastases in patients with prostate cancer: a practical review and guide for the clinician. Urol Clin North Am. 2004; 31:331–352.
8. Coleman RE. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin Cancer Res. 2006; 12:6243s–6249s.
9. Madan RA, Pal SK, Sartor O, Dahut WL. Overcoming chemotherapy resistance in prostate cancer. Clin Cancer Res. 2011; 17:3892–3902.
10. Abidi A. Cabazitaxel: A novel taxane for metastatic castration-resistant prostate cancer-current implications and future prospects. J Pharmacol Pharmacother. 2013; 4:230–237.
11. Trachootham D, Lu W, Ogasawara MA, Valle NR, Huang P. Redox Regulation of Cell Survival. Antioxid Redox Signal. 2008; 10:1343–1274.
12. Acharya A, Das I, Chandhok D, Sahacorresponding T. Redox regulation in cancer: a double-edged sword with therapeutic potential. Oxid Med Cell Longev. 2010; 3:23–34.
13. Jorgenson TC, Zhong W, Oberley TD. Redox imbalance and biochemical changes in cancer. Cancer Res. 2013; 73:6118–6123.
14. Wilson DM 3rd, Barsky D. The major human abasic endonuclease: formation, consequences and repair of abasic lesions in DNA. Mutat Res. 2001; 485:283–307.
15. Xanthoudakis S, Curran T. Identification and characterization of Ref-1, a nuclear protein that facilitates AP-1 DNA-binding activity. EMBO J. 1992; 11:653–665.
16. Bapat A, Fishel ML, Kelley MR. Going ape as an approach to cancer therapeutics. Antioxid Redox Signal. 2009; 11:651–658.
17. Tell G, Quadrifoglio F, Tiribelli C, Kelley MR. The Many Functions of APE1/Ref-1: Not Only a DNA Repair Enzyme. Antioxid Redox Signal. 2009; 11:601–619.
18. Kelley MR, Georgiadis MM, Fishel ML. APE1/Ref-1 role in redox signaling: translational applications of targeting the redox function of the DNA repair/redox protein APE1/Ref-1. Curr Mol Pharmacol. 2012; 5:36–53.
19. Thakur S, Sarkar B, Cholia RP, Gautam N, Dhiman M, Mantha A. APE1/Ref-1 as an emerging therapeutic target for various human diseases: phytochemical modulation of its functions. Exp Mol Med. 2014; 46:e106.
20. Zou GM, Maitra A. Small-molecule inhibitor of the AP endonuclease 1/REF-1 E3330 inhibits pancreatic cancer cell growth and migration. Mol Cancer Res. 2008; 7:2012–2021
21. Cesaratto L, Codarin E, Vascotto C, Leonardi A, Kelley MR, Tiribelli C, Tell G. Specific inhibition of the redox activity of ape1/ref-1 by e3330 blocks tnf-α-induced activation of IL-8 production in liver cancer cell lines. PloS One. 2013; 8:e70909.
22. Biswas A, Khanna S, Roy S, Pan X, Sen CK, Gordillo GM. Endothelial cell tumor growth is Ape/ref-1 dependent. Am J Physiol Cell Physiol. 2015; 309:C296–307.
23. Logsdon DP, Grimard M, Luo M, Shahda S, Jiang Y, Tong Y, Yu Z, Zyromski N, Schipani E, Carta F, Supuran CT, Korc M, Ivan M, et al. Regulation of HIF1α under Hypoxia by APE1/Ref-1 Impacts CA9 Expression: Dual Targeting in Patient-Derived 3D Pancreatic Cancer Models. Mol Cancer Res. 2016; 15:2722–2732.
24. Guan Z, Basi D, Li Q, Mariash A, Xia YF, Geng JG, Kao E, Hall JL. Loss of Redox Factor 1 Decreases NF-κB Activity and Increases Susceptibility of Endothelial Cells to Apoptosis. Arterioscler Thromb Vasc Biol. 2005; 25:96–101.
25. Montaldi AP, Godoy PR, Sakamoto-Hojo ET. APE1/REF-1 down-regulation enhances the cytotoxic effects of temozolomide in a resistant glioblastoma cell line. Mutat Res Genet Toxicol Environ Mutagen. 2015; 793:19–29.
26. Shariat SF, Lotan Y, Saboorian H, Khoddami SM, Roehrborn CG, Slawin KM, Ashfaq R. Survivin Expression Is Associated with Features of Biologically Aggressive Prostate Carcinoma. Cancer. 2004; 100:751–756.
27. Zhang M, Latham DE, Delaney MA, Chakravarti A. Survivin mediates resistance to antiandrogen therapy in prostate cancer. Oncogene. 2005; 24:2474–82.
28. Zhang M, Coen JJ, Suzuki Y, Siedow MR, Niemierko A, Khor LY, Pollack A, Zhang Y, Zietman AL, Shipley WU, Chakravarti A. Survivin is a potential mediator of prostate cancer metastasis. Int J Radiat Oncol Biol Phys. 2010; 78:1095–1103.
29. Sampath SC, Ohi R, Leismann O, Salic A, Pozniakovski A, Funabiki H. The chromosomal passenger complex is required for chromatin-induced microtubule stabilization and spindle assembly. Cell. 2004; 118:187–202.
30. McIlwain DW, Zoetemelk M, Myers JD, Edwards MT, Snider BM, Jerde TJ. Coordinated induction of cell survival signaling in the inflamed microenvironment of the prostate. Prostate. 2016; 76:722–734.
31. Cardoso AA, Jiang Y, Luo M, Reed AM, Shahda S, He Y, Maitra A, Kelley MR, Fishel ML. APE1/Ref-1 Regulates STAT3 Transcriptional Activity and APE1/Ref-1–STAT3 Dual-Targeting Effectively Inhibits Pancreatic Cancer Cell Survival. PLoS ONE. 2012; 7:e47462.
32. Kelley MR, Luo M, Reed A, Su D, Delaplane S, Borch RF, Nyland RL, Gross ML, Georgiadis MM. Functional Analysis of Novel Analogues of E3330 That Block the Redox Signaling Activity of the Multifunctional AP Endonuclease/Redox Signaling Enzyme APE1/Ref-1. Antioxid Redox Signal. 2011; 14:1387–1401.
33. Liu BB, Wang WH. Survivin and pancreatic cancer. World J Clin Oncol. 2011; 2:164–168.
34. Min L, Ji Y, Bakiri L, Qiu Z, Cen J, Chen X, Chen L, Scheuch H, Zheng H, Qin L, Zatloukal K, Hui L, Wagner EF. Liver cancer initiation is controlled by AP-1 through SIRT6-dependent inhibition of survivin. Nat Cell Biol. 2012; 14:1203–1211.
35. Fishel ML, He Y, Reed AM, Chin-Sinex H, Hutchins GD, Mendonca MS, Kelley MR. Knockdown of the DNA repair and redox signaling protein Ape1/Ref-1 blocks ovarian cancer cell and tumor growth. DNA Repair (Amst). 2008; 7:177–186.
36. Zou GM, Luo MH, Reed A, Kelley MR, Yoder MC. Ape1 regulates hematopoietic differentiation of embryonic stem cells through its redox functional domain. Blood. 2007; 109:1917–1922.
37. Kawakami H, Tomita M, Matsuda T, Ohta T, Tanaka Y, Fujii M, Hatano M, Tokuhisa T, Mori N. Transcriptional activation of survivin through the NF-kappaB pathway by human T-cell leukemia virus type I tax. Int J Cancer. 2005; 115:967–74
38. Zhang Y, Huang H, Zhou H, Du T, Zeng L, Cao Y, Chen J, Lai Y, Li J, Wang G, Guo Z. Activation of Nuclear Factor ĸB Pathway and Downstream Targets Survivin and Livin by SHARPIN Contributes to the Progression and Metastasis of Prostate Cancer. Cancer. 2014; 120:3208–3218.
39. Hiramoto M, Shimizu N, Sugimoto K, Tang J, Kawakami Y, Ito M, Aizawa S, Tanaka H, Makino I, Handa H. Nuclear targeted suppression of NF-kappa B activity by the novel quinone derivative E3330. J Immunol. 1998; 160:810–819.
40. Ando K, Hirao S, Kabe Y, Ogura Y, Sato I, Yamaguchi Y, Wada T, Handa H. A new APE1/Ref-1-dependent pathway leading to reduction of NF-κB and AP-1, and activation of their DNA-binding activity. Nucleic Acids Res. 2008; 36:4327–4336
41. Seruga B, Ocana A, Tannock IF. Drug resistance in metastatic castration-resistant prostate cancer. Nat Rev Clin Oncol. 2011; 8:12–23.
42. Semenas J, Allegrucci C, Boorjian SA, Mongan NP, Persson JL. Overcoming Drug Resistance and Treating Advanced Prostate Cancer. Curr Drug Targets. 2012; 13:1308–1323.
43. Di Maso V, Mediavilla MG, Vascotto C, Lupo F, Baccarani U, Avellini C, Tell G, Tiribelli C, Crocè LS. Transcriptional Up-Regulation of APE1/Ref-1 in Hepatic Tumor: Role in Hepatocytes Resistance to Oxidative Stress and Apoptosis. PloS One. 2015; 10:e0143289.
44. Grivennikov S, Karin M. Dangerous liaisons: STAT3 and NF-κB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010; 21:11–19.
45. Hoesel B, Schmid JA. The complexity of NF-κB signaling in inflammation and cancer. Mol Cancer. 2013; 12:86.
46. Kajanne R, Miettinen P, Tenhunen M, Leppä S. Transcription factor AP-1 promotes growth and radioresistance in prostate cancer cells. Int J Oncol. 2009; 35:1175–82.
47. Shah F, Logsdon D, Messmann RA, Fehrenbacher JC, Fishel ML, Kelley MR. Exploiting the APE1-Ref-1 node in cancer signaling and other diseases: from bench to clinic. NPJ Precis Oncol. 2017; 1:19.
48. Kelley MR, Cheng L, Foster R, Tritt R, Jiang J, Broshears J, Koch M. Elevated and Altered Expression of the Multifunctional DNA Base Excision Repair and Redox Enzyme Ape1/ref-1 in Prostate Cancer. Clin Cancer Res. 2001; 7:824–830.
49. Kim SA, Hong R. Significance of intracellular localization of survivin in cervical squamous cell lesions: Correlation with disease progression. Oncol Lett. 2014; 7:1589–1593.
50. Di Maso V, Avellini C, Crocè LS, Rosso N, Quadrifoglio F, Cesaratto L, Codarin E, Bedogni G, Beltrami CA, Tell G, Tiribelli C. Subcellular Localization of APE1/Ref-1 in Human Hepatocellular Carcinoma: Possible Prognostic Significance. Mol Med. 2007;13:89–96.
51. Jiang Y, Zhou S, Sandusky GE, Kelley MR, Fishel ML. Reduced Expression of DNA Repair and Redox Signaling Protein APE1/Ref-1 Impairs Human Pancreatic Cancer Cell Survival, Proliferation, and Cell Cycle Progression. Cancer Invest. 2010; 28:885–895.
52. Sengupta S, Mitra S, Bhakat KK. Dual Regulatory Roles of Human AP-Endonuclease (APE1/Ref-1) in CDKN1A/p21 Expression. PloS One. 2013; 8:e68467.
53. Zhang J, Luo M, Marasco D, Logsdon D, LaFavers KA, Chen Q, Reed A, Kelley MR, Gross ML, Georgiadis MM. Inhibition of apurinic/apyrimidinic endonuclease I's redox activity revisited. Biochemistry. 2013; 52:2955–66.
54. Luo M, Zhang J, He H, Su D, Chen Q, Gross ML, Kelley MR, Georgiadis MM. Characterization of the redox activity and disulfide bond formation in apurinic/apyrimidinic endonuclease. Biochemistry. 2012; 51:695–705.
55. Su D, Delaplane S, Luo M, Rempel DL, Vu B, Kelley MR, Gross ML, Georgiadis MM. Interactions of apurinic/apyrimidinic endonuclease with a redox inhibitor: evidence for an alternate conformation of the enzyme. Biochemistry. 2011; 50:82–92.
56. Georgiadis MM, Luo M, Gaur RK, Delaplane S, Li X, Kelley MR. Evolution of the redox function in mammalian apurinic/apyrimidinic endonuclease. Mutat Res. 2008; 643:54–63.
57. Kelley MR, Jiang Y, Guo C, Reed A, Meng H, Vasko MR. Role of the DNA Base Excision Repair Protein, APE1 in Cisplatin, Oxaliplatin, or Carboplatin Induced Sensory Neuropathy. Kirchmair R, ed. PLoS One. 2014; 9:e106485. doi:10.1371/journal.pone.0106485.
58. Kelley MR, Wikel JH, Guo C, Pollok KE, Bailey BJ, Wireman R, Fishel ML, Vasko MR. Identification and Characterization of new chemical entities targeting apurinic/apyrimidinic endonuclease 1 for the prevention of chemotherapy-induced peripheral neuropathy. J Pharmacol Exp Ther. 2016; 359:300-309.
59. Kelley MR, Fehrenbacher JC. Challenges and opportunities identifying therapeutic targets for chemotherapy-induced peripheral neuropathy resulting from oxidative DNA damage. Neural Regen Res. 2017; 12:72–74.
60. Kishi H, Igawa M, Kikuno N, Yoshino T, Urakami S, Shiina H. Expression of the survivin gene in prostate cancer: correlation with clinicopathological characteristics, proliferative activity and apoptosis. J Urol. 2004; 171:1855–1860.
61. Clark J, Edwards S, Feber A, Flohr P, John M, Giddings I, Crossland S, Stratton MR, Wooster R, Campbell C, Cooper CS. Genome-wide screening for complete genetic loss in prostate cancer by comparative hybridization onto cDNA microarrays. Oncogene. 2003; 22:1247–1252.
62. Li Y, Liu X, Zhou T, Kelley MR, Edwards P, Gao H, Qiao X. Inhibition of APE1/Ref-1 redox activity rescues human retinal pigment epithelial cells from oxidative stress and reduces choroidal neovascularization. Redox Biol. 2014; 2:485–494.
63. Yan M, Xu Q, Zhang P, Zhou XJ, Zhang ZY, Chen WT. Correlation of NF-κB signal pathway with tumor metastasis of human head and neck squamous cell carcinoma. BMC Cancer. 2010; 10:437.
64. Schwarze SR, DePrimo SE, Grabert LM, Fu VX, Brooks JD, Jarrard DF. Novel pathways associated with bypassing cellular senescence in human prostate epithelial cells. J Biol Chem. 2002; 277:14877–83.
65. Luo M, Delaplane S, Jiang A, Reed A, He Y, Fishel ML, Nyland RL, Borch RF, Qiao X, Georgiadis MM, Kelley MR. Role of the Multifunctional DNA Repair and Redox Signaling Protein Ape1/Ref-1 in Cancer and Endothelial Cells: Small-Molecule Inhibition of the Redox Function of Ape1. Antioxid Redox Signal. 2008; 10:1853–1867.
66. Nyland RL, Luo M, Kelley MR, Borch RF. (2010). Design and Synthesis of Novel Quinone Inhibitors Targeted to the Redox Function of Apurinic/Apyrimidinic Endonuclease 1/Redox Enhancing Factor-1 (Ape1/Ref-1). J Med Chem. 2010; 53:1200–1210.