
INTRODUCTION
Human trisomy 21, the most common human aneuploidy compatible with postnatal survival, occurs in approximately one out of 700–1000 live births [1, 2]. This chromosomal anomaly causes a constellation of developmental abnormalities, classified as Down syndrome (DS). The major clinical manifestations of DS include cardiovascular malformations, craniofacial abnormalities, developmental cognitive deficits, hematopoietic abnormalities, and Alzheimer-type neurodegeneration with variable penetrance and onset [3, 4].
Among hematopoietic abnormalities, transient myeloproliferative disorder (TMD) occurs in a substantial percentage of children with DS, and many of these patients later develop acute megakaryoblastic leukemia (AMKL) [5]. It has been demonstrated that children with DS who develop TMD and AMKL have acquired somatic mutations in exon 2 of the GATA1 gene on the X chromosome, which lead to the generation of a mutant protein, GATA1s [6, 7]. The GATA1s germline mutation is associated with impaired erythropoiesis [8, 9].
To unravel the mechanism underlying development of myeloproliferative disorder in DS, genetic models with phenotypes mimicking DS at the molecular, cellular and organ levels are necessary. The mouse has been the most important model organism for DS due to evolutionary conservation between the human and mouse genomes. Regions on human chromosome 21 (Hsa21) are syntenically conserved with regions on mouse chromosome 10 (Mmu10), Mmu16 and Mmu17, which contain 41, 115, and 19 Hsa21 gene orthologs, respectively (Figure 1). DS is a contiguous gene syndrome [10] and evidence indicates that, for many DS phenotypes, more than one triplicated gene contributes through direct actions and/or interactions of the triplicated genes [11–17]. Therefore, in a mouse model, triplication of more Hsa21 gene orthologs increases the probability that all the direct actions and/or interactions that have a significant effect on a specific DS phenotype are mimicked.
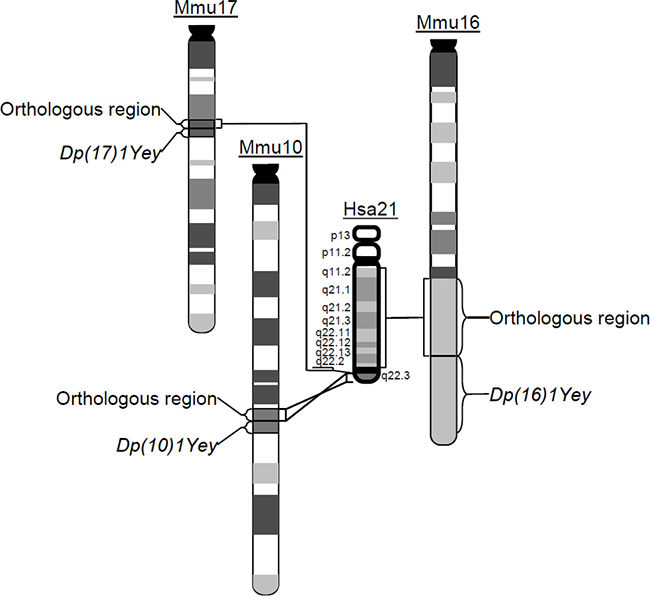
Figure 1: The schematic representation of the chromosomal alterations in Dp(10)1;Dp(16)1;Dp(17)1 mice. Generated using Cre/loxP-mediated chromosome engineering, Dp(10)1;Dp(16)1;Dp(17)1 mice contain three duplications spanning the entire Hsa21 orthologous regions on Mmu10, Mmu16 and Mmu17, respectively.
Myeloproliferative disorder has been observed in several mouse models of DS [18–21]. Among them, Ts65Dn is the first viable trisomic mouse model of DS. This mutant carries Ts(1716)65Dn, an unbalanced derivative of a balanced chromosomal translocation, which was randomly induced by irradiation at Muriel Davisson’s laboratory [22, 23]. Tc1 is another important mouse model of DS, developed by introducing Hsa21 into mouse embryonic stem (ES) cells using microcell-mediated chromosome transfer [24–27]. The hematopoietic phenotype has been extensively characterized in the Ts65Dn and Tc1 mouse models [18, 19]. However, a substantial number of Hsa21 gene orthologs are not triplicated in either model. To include those missed Hsa21 orthologs, we have generated Dp(10)1Yey/+ [i.e. Dp(10)1], Dp(16)1Yey/+ [i.e. Dp(16)1], and Dp(17)1Yey/+ [i.e. Dp(17)1] mice, by chromosome engineering which carry duplications spanning the entire Hsa21 orthologous regions on Mmu10, Mmu16, and Mmu17, respectively; thus all the DS-associated gene dosage alterations are mimicked (Figure 1) [15, 28]. We have shown that Dp(10)1;Dp(16)1;Dp(17)1 and Dp(16)1 mice exhibited DS-related heart defects, cognitive behavioral deficits, and impaired hippocampal long-term potentiation [15]. Extensive studies are also being performed on these mutant mice by many other laboratories [10, 29–38]. In this study, we extended the exploration of the impact of these engineered gene dosage alterations to DS-related hematopoiesis.
RESULTS
Generation of Gata1Yeym2 mice to delete exon 2 of the Gata1 gene
To assess the effect of GATA1s in DS-associated hematopoietic abnormalities, we used gene targeting to delete exon 2 of the Gata1 gene in mouse ES cells. First, we generated ES cell lines that carried the Gata1Yeym1 mutant allele using the replacement vector pTVGata1. Recombination between pTVGata1 and the Gata1 locus in ES cells led to the replacement of a genomic region containing exon 2 of the gene with a neomycin-resistance gene cassette flanked by two loxP sites (Figure 2A). We then expressed the Cre recombinase transiently in two Gata1Yeym1 ES cell clones and the neomycin-resistance gene cassette was removed by Cre/loxP-mediated recombination. The excised allele was designated as Gata1Yeym2 (Figure 2A). Germline transmission was established for four ES cell lines that carried Gata1Yeym2. Southern blot analysis was performed to confirm the deletion of the exon 2 region (Figure 2B). Expression of the mutant allele in the Gata1Yeym2 mice was confirmed by RT-PCR (Figure 2C).
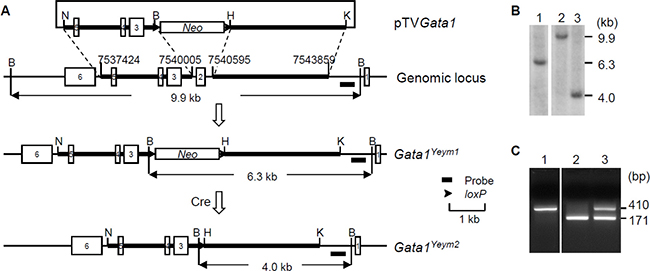
Figure 2: Development of Gata1Yeym2 mutant mouse strain. (A) Targeting strategy for generating the Gata1Yeym2 allele. Neo, neomycin-resistance gene cassette. B, BamHI; H, HindIII; K, KpnI; N, NotI. The genome coordinates of the homology arms were marked. (B) Southern blot analysis of BamHI-digested ES cell DNA. Lane 1, An ES cell clone containing the Gata1Yeym1 allele; lane 2, the wild-type ES cells; lane 3, the ES cell clone containing the Gata1Yeym2 allele. (C) RT-PCR analysis of total RNA isolated from the fetal livers of E14.5 embryos, showing the PCR products from the cDNA of wild type Gata1 and Gata1Yeym2. MW, molecular-weight size marker, TrackIt™ 50 bp DNA Ladder (Invitrogen); lane 1, a wild-type embryo; lane 2, a male Gata1Yeym2 embryo; lane 3, a heterozygous female Gata1Yeym2 embryo.
Triplications of all Hsa21 orthologous regions in mice result in peripheral blood abnormalities
Dp(10)1;Dp(16)1;Dp(17)1 and Dp(10)1;Dp(16)1;Dp(17)1;Gata1Yeym2 mice were generated as described in Materials and Methods. Peripheral complete blood counts (CBCs) of Dp(10)1;Dp(16)1;Dp(17)1 mice, Dp(10)1;Dp (16)1;Dp(17)1;Gata1Yeym2 mice and their wild-type controls were measured every 3 months until 15 months of age.
CBCs revealed reduced counts of red blood cells (RBCs) starting at 3 months of age (p < 0.001) and lowered hemoglobin concentrations (HGB) starting at 9 months of age (p < 0.05) in both Dp(10)1;Dp(16)1;Dp(17)1 mice and Dp(10)1;Dp(16)1;Dp(17)1;Gata1Yeym2 mice (Figure 3A–3B), which indicated that both mutants developed anemia. Dp(10)1;Dp(16)1;Dp(17)1;Gata1Yeym2 mice showed much lower RBC counts (p < 0.01) and HGB concentrations (p < 0.01) than Dp(10)1;Dp(16)1;Dp(17)1 mice (Figure 3A–3B), suggesting that the Gata1s mutation causes more severe anemia by further reducing the RBC counts and HGB concentrations.
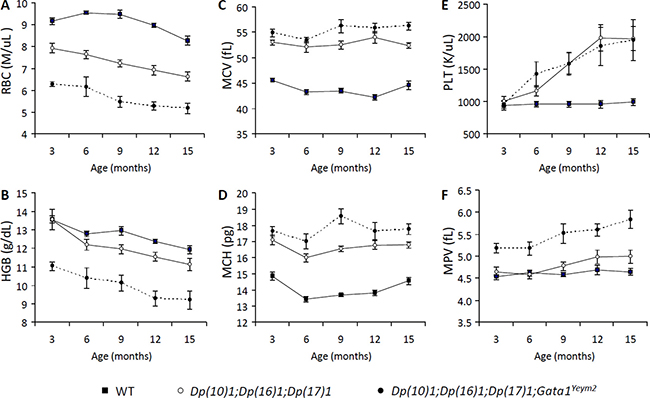
Figure 3: Complete blood counts (CBC) of Dp(10)1;Dp(16)1;Dp(17)1, Dp(10)1;Dp(16)1;Dp(17)1;Gata1Yeym2 mice and wild-type controls. (A) Red blood cells (RBC) (M/μL; millions per microliter ); (B) Hemoglobin (HGB) (g/dL, grams per deciliter); (C) Mean corpuscular volume (MCV) (fL, fentoliters); (D) Mean corpuscular hemoglobin (MCH) (pg, picograms); (E) Platelets (PLT) (K/μL; thousands per microliter); (F) Mean platelet volume (MPV). Closed square, the wild-type controls (n = 14, 14, 9, 15, 21 at 3, 6, 9, 12, 15 months of age, respectively); open circle, Dp(10)1;Dp(16)1;Dp(17)1 mice (n = 6, 13, 9, 14, 14 at 3, 6, 9, 12, 15 months of age, respectively); closed circle, Dp(10)1;Dp(16)1;Dp(17)1;Gata1Yeym2 mice (n = 8, 5, 5, 6, 5 at 3, 6, 9, 12, 15 months of age, respectively).
CBCs also showed increased mean corpuscular volume (MCV) (p < 0.0001) and mean corpuscular hemoglobin (MCH) (p < 0.0001) in both Dp(10)1;Dp(16)1;Dp(17)1 mice and Dp(10)1;Dp(16)1;Dp(17)1;Gata1Yeym2 mice starting at 3 months of age (Figure 3C–3D), indicating that both mutants developed macrocytosis. However, MCV and MCH were similar in Dp(10)1;Dp(16)1;Dp(17)1 mice and Dp(10)1;Dp(16)1;Dp(17)1;Gata1Yeym2 mice (Figure 3C–3D), indicating that the Gata1s mutation did not contribute to increases in the volume of red blood cells.
In addition, platelet counts were elevated in both Dp(10)1;Dp(16)1;Dp(17)1 mice and Dp(10)1;Dp(16)1;Dp(17)1;Gata1Yeym2 mice starting at 6 months of age (p < 0.05), indicating thrombocytosis. There was no significant difference in the magnitude of the thrombocytosis between the mutants (Figure 3E), suggesting that the Gata1s mutation did not significantly alter the severity of this phenotype.
Mean platelet volume was significantly increased in Dp(10)1;Dp(16)1;Dp(17)1;Gata1Yeym2 mice starting at 3 months of age (p < 0.001), but not in Dp(10)1; Dp(16)1;Dp(17)1 mice (Figure 3F). This result suggests that the Gata1s mutation, but not the triplications of the Hsa21 orthologous regions, underlies the expanded platelet volume.
Triplications of all Hsa21 orthologous regions in mice result in abnormalities in spleen and bone marrow
To investigate the hematopoietic features of Dp(10)1;Dp(16)1;Dp(17)1 and Dp(10)1;Dp(16)1;Dp(17)1;Gata1Yeym2 mice, 15 to 24 month-old mutant mice and the wild-type littermates were euthanized and their spleens and bone marrows were harvested for histology and flow cytometry analysis. The average weights of the spleens, after normalization to body weight, accounted for 0.91 ± 0.16%, 1.18 ± 0.32% and 0.32 ± 0.08% of the total body weights in Dp(10)1;Dp(16)1;Dp(17)1, Dp(10)1;Dp(16)1;Dp(17)1;Gata1Yeym2 and the wild-type control mice, respectively (Figure 4). These data indicate that both mutants exhibit splenomegaly (p < 0.01) but there is no significant difference between the weights of the spleens isolated from the two mutants (p > 0.05).
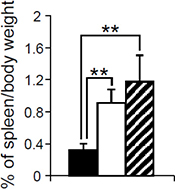
Figure 4: Relative weights of the spleens from mice with different genotypes. The percentage of spleen weight over body weight in the wild-type controls (n = 6, solid bar); Dp(10)1;Dp(16)1;Dp(17)1 mice (n = 6, open bar) and Dp(10)1;Dp(16)1;Dp(17)1;Gata1Yeym2 mice (n = 5, diagonal-lined bar). **p < 0.01.
Histological sections showed disruption of red and white pulp architecture in the spleens from Dp(10)1;Dp(16)1;Dp(17)1 mice and Dp(10)1;Dp(16) 1;Dp(17)1;Gata1Yeym2 mice (Figure 5). The structural distortions of the spleens are apparently due to severe megakaryocyte infiltration (Figure 6). The bone marrows of both Dp(10)1;Dp(16)1;Dp(17)1 mice and Dp(10)1;Dp(16)1;Dp(17)1;Gata1Yeym2 mice were also infiltrated with large numbers of megakaryocytes (Figure 6).
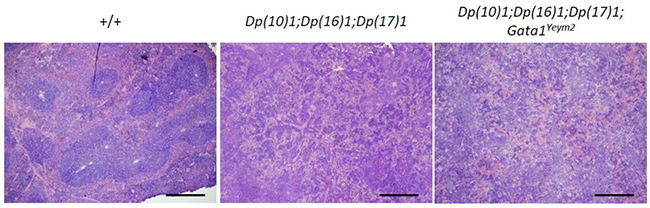
Figure 5: Histological sections of the spleens from different genotypes at lower magnification. The spleen structure of wild-type controls, Dp(10)1;Dp(16)1;Dp(17)1 mice and Dp(10)1;Dp(16)1;Dp(17)1;Gata1Yeym2 mice under hematoxylin and eosin staining. Scale bar, 40 μm.
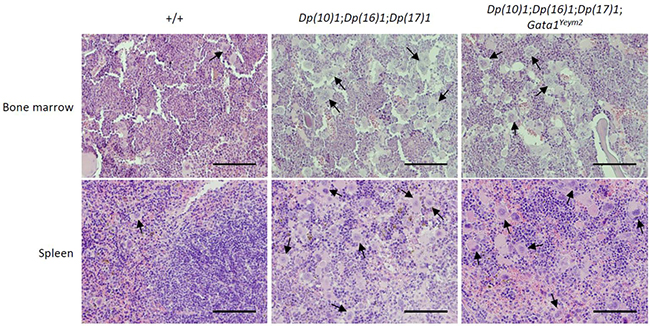
Figure 6: Histological sections of the bone marrow and spleens from different genotypes at higher magnification. Hematoxylin and eosin staining of bone marrow and spleen sections in wild-type controls, Dp(10)1;Dp(16)1;Dp(17)1 mice and Dp(10)1;Dp(16)1;Dp(17)1;Gata1Yeym2 mice. Scale bar, 10 μm. Arrows point to the megakaryocytes in the sections.
To further characterize the cells from bone marrow and spleen of the mouse mutants, we performed flow cytometric analysis. Consistent with the histological analysis, our flow cytometric data indicate an increase of cells from CD41+ megakaryocyte lineage in bone marrow and spleen of both Dp(10)1;Dp(16)1; Dp(17)1 and Dp(10)1;Dp(16)1;Dp(17)1;Gata1Yeym2 mice (Figure 7A). These data support the conclusion that both Dp(10)1;Dp(16)1;Dp(17)1 and Dp(10)1;Dp(16)1;Dp(17)1;Gata1Yeym2 mice developed megakaryocytosis. There is no significant difference in the percentages of CD41+ megakaryocyte lineage cells between Dp(10)1;Dp(16)1;Dp(17)1 and Dp(10)1;Dp(16)1;Dp(17)1;Gata1Yeym2 mice (Figure 7A). In addition, Ter119+ erythrocyte lineage cells were reduced in the bone marrows of both Dp(10)1;Dp(16)1;Dp(17)1 mice and Dp(10)1;Dp(16)1;Dp(17)1;Gata1Yeym2 mice (Figure 7B). Ter119+ erythrocyte lineage cells were increased in the spleens of both Dp(10)1;Dp(16)1;Dp(17)1 mice and Dp(10)1;Dp(16)1;Dp(17)1;Gata1Yeym2 mice, and such increases are likely associated with splenomegaly.
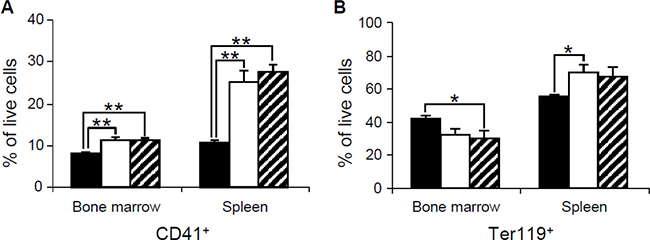
Figure 7: Flow cytometric analysis of bone marrow and spleen cells in wild-type controls, Dp(10)1;Dp(16)1;Dp(17)1 mice and Dp(10)1;Dp(16)1;Dp(17)1;Gata1Yeym2 mice. (A) CD41+ megakaryocyte lineage cells; (B) Ter119+ erythrocyte lineage cells. Solid bar, the wild-type controls (n = 6); open bar, Dp(10)1;Dp(16)1;Dp(17)1 mice (n = 6) and diagonal-lined bar, Dp(10)1;Dp(16)1; Dp(17)1;Gata1Yeym2 mice (n = 5). *p < 0.05; **p < 0.01.
Triplications of all Hsa21 orthologous regions in mice result in perturbations of hematopoietic stem cells as well as expansion of megakaryocyte-erythroid progenitors and reduction of granulocyte-macrophage progenitors
To further determine whether the abnormalities in the peripheral blood, spleen and bone marrow were associated with the changes in hematopoietic stem cells or progenitor populations in bone marrow, flow cytometry was performed to examine the lineage-Sca1+c-Kit+ (LSK) hematopoietic stem cells and myeloid progenitors in the bone marrow of 15-month-old Dp(10)1;Dp(16)1;Dp(17)1 mice, Dp(10)1;Dp(16)1;Dp (17)1;Gata1Yeym2 mice, and wild-type controls. Our results showed that the LSK stem cells were expanded in both Dp(10)1;Dp(16)1;Dp(17)1 mice and Dp(10)1;Dp(16)1; Dp(17)1;Gata1Yeym2 mice compared to the wild-type controls (Figure 8A). In myeloid progenitor compartments, the common myeloid progenitors (CMPs) (lineage–, Sca1–, c-Kit+, CD34+, FcγR–) remained unchanged, while the megakaryocyte-erythrocyte progenitors (MEPs) (lineage–, Sca1–, c-Kit+, CD34–, FcγR–) and the granulocyte-monocyte progenitors (GMPs) (lineage–, Sca1–, c-Kit+, CD34+, FcγR+) were increased and decreased, respectively, in both Dp(10)1;Dp(16)1;Dp(17)1 mice and Dp(10)1;Dp(16)1;Dp(17)1;Gata1Yeym2 mice when compared to the wild-type controls (Figure 8B). There were no significant differences in the LSK stem cells, MEPs, GMPs and CMPs between Dp(10)1;Dp(16)1;Dp(17)1 mice and Dp(10)1;Dp(16)1;Dp(17)1;Gata1Yeym2 mice (Figure 8).
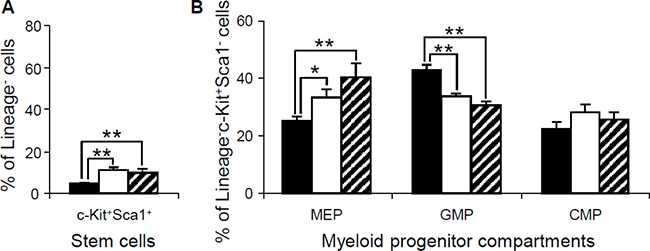
Figure 8: Flow cytometric analysis of the stem cells and myeloid progenitor compartments in bone marrow of wild-type controls, Dp(10)1;Dp(16)1;Dp(17)1 mice and Dp(10)1;Dp(16)1;Dp(17)1;Gata1Yeym2 mice. (A) Stem cells (lineage–, Sca1+, c-Kit+) (LSK); (B) Myeloid progenitor compartments: MEPs (lineage–, Sca1–, c-Kit+, CD34–, FcγR–), GMPs (lineage–, Sca1–, c-Kit+, CD34+, FcγR+) and CMPs (lineage–, Sca1–, c-Kit+, CD34+, FcγR–). Solid bar, the wild-type controls (n = 6); open bar, Dp(10)1;Dp(16)1;Dp(17)1 mice (n = 6) and diagonal-lined bar, Dp(10)1;Dp(16)1;Dp(17)1;Gata1Yeym2 mice (n = 5). *p < 0.05; **p < 0.01.
Triplication of the Hsa21 orthologous region on Mmu16 results in expansion of MEPs and reduction of GMPs
In contrast to Dp(10)1;Dp(16)1;Dp(17)1 mice, Ts65Dn mice have repeatedly shown reduction of MEPs and expansion of GMPs [19, 39]. Ts65Dn and Dp(16)1 carry 3 copies of the identical set of 100 Hsa21 gene orthologs on Mmu16, so we speculated that Dp(16)1 mice would likely also show reduction of MEPs and expansion of GMPs. We further speculated that expansion of MEPs and reduction of GMPs in Dp(10)1;Dp(16)1;Dp(17)1 mice may be caused by Dp(10)1 and/or Dp(17)1. To test these possibilities, we carried out phenotypic analysis of Dp(16)1 mice. To our surprise, the hematopoietic phenotypes of Dp(16)1 mice are almost identical to those of Dp(10)1;Dp(16)1;Dp(17)1 mice, including expansion of MEPs and reduction of GMPs (Figure 9 and Supplementary Figures 1–4). These results suggest that the causative gene(s) responsible for expansion of MEPs and reduction of GMPs are located in the Hsa21 orthologous region on Mmu16. These results also suggest that triplicated genes which differ between Dp(16)1 and Ts65Dn mice are responsible for diametrically opposite effect on MEPs and GMPs.
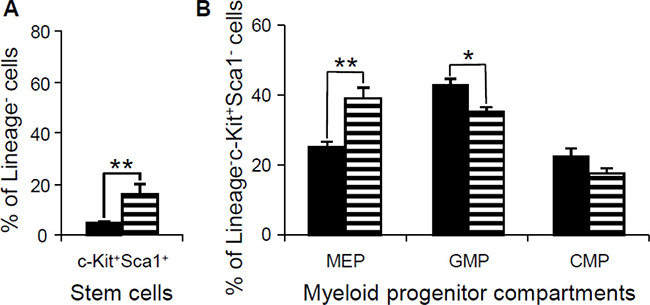
Figure 9: Flow cytometric analysis of the stem cells and myeloid progenitor compartments in bone marrow of wild-type controls and Dp(16)1 mice. (A) Stem cells (lineage–, Sca1+, c-Kit+) (LSK); (B) Myeloid progenitor compartments: MEPs (lineage–, Sca1–, c-Kit+, CD34–, FcγR–), GMPs (lineage–, Sca1–, c-Kit+, CD34+, FcγR+) and CMPs (lineage–, Sca1–, c-Kit+, CD34+, FcγR–). Solid bar, the wild-type controls (n = 6); horizontal-lined bar, Dp(16)1 mice (n = 5). *p < 0.05; **p < 0.01.
DISCUSSION
In this study, we analyzed the hematopoietic phenotype of the Dp(10)1;Dp(16)1;Dp(17)1 mouse model of DS, which contains duplications spanning the entire Hsa21 orthologous regions on Mmu10, Mmu16, and Mmu17 [15, 28]. These mice exhibited anemia, macrocytosis, thrombocytosis, megakaryocytosis, splenomegaly, and perturbed hematopoietic stem cells and progenitor cell compartments including expanded MEP and reduced GMP populations (Figure 10). The subsequent analysis of Dp(16)1 mice indicated that these phenotypes are the consequences of the triplication of Hsa21 gene orthologs on Mmu16.
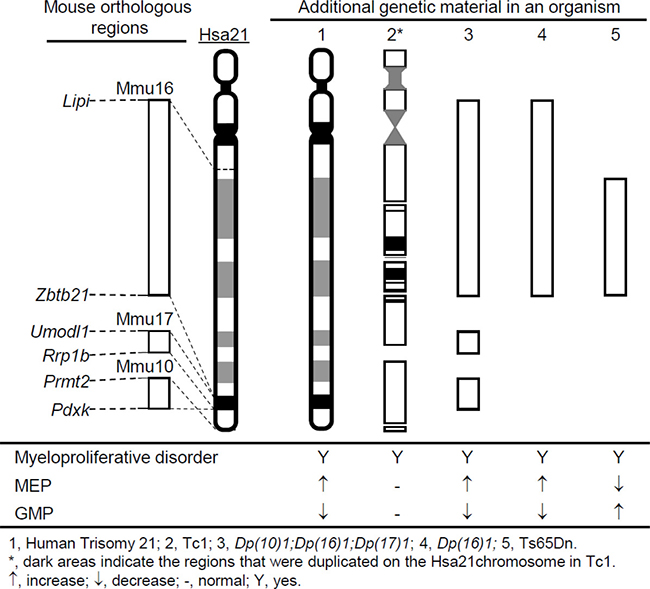
Figure 10: The status of the myeloid progenitors in the major segmental chromosomal alteration mouse models of DS showing myeloproliferative disorder.
Hematopoietic phenotypes have been characterized in two other major mouse models of DS, Ts65Dn and Tc1 [18, 19]. Ts65Dn mice carry a triplication of a genomic fragment of approximately 13.5 Mb extending from miR-155 to the telomere on Mmu16 and encompassing 100 Hsa21 gene orthologs, which accounts for approximately 57% of the Hsa21 gene orthologs (Figure 10) [22, 40, 41]. Ts65Dn mice also carry a triplicated genomic fragment of approximately 10 Mb extending from the centromere to 1700010I14Rik on Mmu17 which contains 36 Hsa6 gene orthologs [40, 41]. Ts65Dn mice, Dp(10)1;Dp(16)1;Dp(17)1 mice and Dp(16)1 mice share some major features of the hematopoietic phenotype, such as anemia, macrocytosis and myeloproliferative disease characterized by thrombocytosis, megakaryocytic hyperplasia and a distorted hematopoietic stem cell compartment [19]. However, these three models also exhibit important differences in the hematopoietic features. Compared with the wild-type controls, the percentages of MEPs and GMPs in the myeloid progenitor compartments are increased and decreased, respectively, in both Dp(10)1;Dp(16)1;Dp(17)1 and Dp(16)1 mice (Figures 8B, 9B). In contrast, the percentages of MEPs and GMPs are decreased and increased, respectively, in Ts65Dn mice (Figure 10) [19]. Because MEPs are the progenitors of megakaryocytes, this specific observation in Dp(10)1;Dp(16)1;Dp(17)1 and Dp(16)1 mice may better reflect the abnormal hematopoiesis underlying DS-associated myeloproliferative disorder. Such reasoning is also supported by the observation that the percentages of MEPs and GMPs are increased and decreased, respectively, in the livers of fetuses carrying human trisomy 21 [42–44]. Tc1 mice carry a Hsa21 with three major deletions and other genomic alterations, including duplications and gene mutations, which are likely caused by irradiation of Hsa21 during microcell-mediated chromosome transfer [26, 45]. The random loss of the transferred human chromosome during mouse development resulted in variable levels of mosaicism of the extra chromosome in different tissues [26]. Tc1 mice, Dp(10)1;Dp(16)1;Dp(17)1 and Dp(16)1 mice share some important features of the hematopoietic phenotype, such as anemia, macrocytosis, splenomegaly, and an increase in the numbers of megakaryocytes in the spleen [18]. However, unlike Dp(10)1;Dp(16)1;Dp(17)1 and Dp(16)1 mice, Tc1 mice did not exhibit significant abnormalities in hematopoietic stem cell and myeloid progenitor compartments, such as MEPs and GMPs [18]. The differences in these phenotypic features could be caused by the Hsa21 genes deleted or mutated on the transchromosome, by transchromosome mosaicism, and/or by species-specific differences between the proteins encoded by human and mouse chromosomes. Therefore, Dp(10)1;Dp(16)1;Dp(17)1 mice exhibit more important DS-patient-associated hematopoietic abnormalities than other major mouse models of DS carrying an additional copy of Hsa21 orthologous regions, particularly expansion of MEPs and reduction of GMPs. These mice will provide a more desirable system for a better understanding of disease processes and the underlying pathogenic mechanism.
It is a surprise that Dp(16)1 and Ts65Dn exhibit opposing MEP and GMP phenotypes since they share the same 100 triplicated genes. However, there are additional triplicated genes which differ between the two models: Dp(16)1 carries a triplication of 15 additional Hsa21 gene orthologs on Mmu16, while Ts65Dn carries a triplication of 36 additional Hsa6 gene orthologs on Mmu17. Therefore, these genetic differences may underlie the opposite phenotypes related to MEPs and GMPs.
MEPs are the myeloid progenitors of megakaryocytes, and therefore the expansion of MEPs observed in human DS and Dp(10)1;Dp(16)1;Dp(17)1 and Dp(16)1 mice could be a major logical cellular event leading to myeloproliferative disorder in humans and mouse models (Figure 10). However, Ts65Dn mice also exhibit myeloproliferative disorder (Figure 10) [19] despite exhibiting a reduction of MEPs. One possible reason for this is that differentiation of MEPs to megakaryoblasts may be further accelerated in Ts65Dn, which may then lead to a relative reduction in the number of MEPs. Regardless of the possible reasons, reduction of MEPs detected in Ts65Dn mice deviates from the hematopoietic abnormality observed in human DS. Thus, unraveling why Dp(16)1 and Ts65Dn are different in MEP- and GMP-related phenotypes may help to improve our understanding of the mechanism underlying myeloproliferative disorder in DS.
Expansion of MEPs and reduction of GMPs were also observed in compound transgenic mice which carry a human ERG transgene and a germline Gata1s mutation [46]. The ERG transgene was driven by regulatory elements from the mouse vav gene. There are several potential reasons why this compound mutant may not accurately model the related pathogenic process in human DS. First, the cell- and temporal-specific expression patterns of vav and Erg may be different, so the expression pattern of the ERG transgene may deviate from that of the endogenous Erg. Second, the levels of expression between the wild-type allele of Erg and the transgenic ERG may be also different. A more desirable approach to assess the contribution of an individual Hsa21 gene ortholog to a phenotype is to use a “subtractive strategy”, which involves normalizing the dosage of a Hsa21 gene ortholog by compounding its nonfunctional allele in a segmental chromosomal alteration mouse model of DS. When this strategy was applied to Erg in Ts65Dn mice, reducing Erg from 3 copies to 2 copies actually led to a significant expansion of MEPs [39], which indicates that the increased Erg copy number in Ts65Dn mice is responsible for the reduction of MEPs. This result suggests the ERG/Gata1s transgenic mice described above may not be appropriate to model the MEP expansion in human DS.
In normal human hematopoietic cells, both full length GATA1 and GATA1s protein are expressed [47]. Germline and somatic mutations have been detected in exon 2 of the GATA1 gene, which resulted in expression of only the GATA1s protein without GATA1. The somatic mutations are associated with DS-TMD and AMKL in children with DS [6, 8, 48–51]. The human germline GATA1s mutation has been shown to cause anemia in males by affecting erythrogenesis [8]. This mutation-associated effect is also reflected in Dp(10)1;Dp(16)1;Dp(17)1;Gata1Yeym2 mice, since they exhibited more severe anemia than Dp(10)1;Dp(16)1;Dp(17)1 mice (Figures 3A, 3B). In humans, the germline GATA1s mutation affects the morphology of platelets but not their number in the peripheral blood [8]. The impact of the Gata1s mutation on the platelets of our mouse models is reminiscent of that seen in humans; there is no significant difference in the number of platelets in the CBC between Dp(10)1;Dp(16)1;Dp(17)1 and Dp(10)1;Dp(16)1;Dp(17)1;Gata1Yeym2 mice, but the mean platelet volume is expanded in Dp(10)1;Dp(16)1;Dp(17)1;Gata1Yeym2 mice when compared to Dp(10)1;Dp(16)1;Dp(17)1 mice (Figures 3E, 3F).
In this study, we demonstrated expansion of MEPs and reduction of GMPs in major mouse models of DS with human trisomy 21-associated segmental genomic alterations for the first time, mirroring the corresponding changes of these myeloid progenitors in DS. Thus, further study of these models may lead to identification of the causative gene(s) for expansion of MEPs, which will be the key to a better understanding of the pathogenic mechanism of DS-associated myeloproliferative disorder. In particular, these models may provide further insight into how trisomy 21 leads to expansion of MEPs and how the quantitative alteration of MEPs contributes to the development of myeloproliferative disorder in DS.
MATERIALS AND METHODS
Generation of the Gata1Yeym2 mutant mice
The mutant mouse strain was generated using gene-targeting in ES cell to delete exon 2 of the Gata1 gene which resulted in the expression of a truncated mutant protein, GATA1s. The targeting vector, pTVGata1, was constructed by inserting two homologous regions into a Bluescript KS plasmid that contained a PGKneobpA (loxP) cassette in the polylinker (a gift from Dr. Richard Behringer). The first homology region was amplified by primers N1033 (5′-TGCGGCCGCTGGCCCTGATCTCAGCTCAGAATA-3′) and B3614 (5′-TGGATCCTACCGCCCCATTTGTACCAATCCT-3′) to produce a 2598-bp fragment in between intron 2 and intron 5. After cloning the first fragment into pCR-XL-TOPO® using TOPO®XL PCR Cloning Kit (Invitrogen), the homology arm was excised out by digesting with NotI and BamHI. The second homology region was amplified by primers H4204 (5′-TTAAGCTTCCACCACCGCCTGGCTCTTC-3′) and K7468 (5′-TGGTACCAGTGCTGGGTTTAAAGGTGTGC-3′) to produce a 3286-bp fragment inside the first intron. After cloning the second fragment into pCR-XL-TOPO®, the homologous arm was excised out by digesting with HindIII and KpnI. The excised fragments were then sequentially inserted between NotI and BamHI site as well as between HindIII and KpnI sites of the ploylinkers on either side of the PGKneobpA(loxP) cassette. The resulting targeting vector, pTVGata1, was linerized with KpnI and electroporated into AB2.2 ES cells. The targeted allele, Gata1Yeym1, was obtained by G418 selection as described previously [52, 53]. Cre/loxP-mediated marker excision [54] was carried out and the final excised allele, Gata1Yeym2, was identified by Southern blot analysis with BamHI digested DNA. The probe used in the Southern blot hybridization was amplified by primers Probe-U (5′-GGGGGACATTGGGACAGAATAGTT-3′) and Probe-L (5′-ATTCCCCCACGCTCCTATCTCAT-3′), which is mapped to the genomic region downstream of the homologous region in Intron 1 (Figure 2).
Mice
Gata1Yeym2 mice were first established in a 129SvEvxC57BL/6JF1 strain background and then backcrossed to C57BL/6J mice for five generations. Dp(10)1, Dp(16)1, and Dp(17)1 mice were generated using recombinase-mediated genome engineering, which carry the duplications spanning the entire Hsa21 orthologous regions on Mmu10, Mmu16 and Mmu17, respectively [15, 28]. Dp(10)1, Dp(16)1 and Dp(17)1 mice were backcrossed to C57BL/6J mice for five generations before being used to generate Dp(10)1;Dp(16)1;Dp(17)1 mice. The latter was crossed to Gata1Yeym2 mice to establish Dp(10)1;Dp(16)1;Dp(17)1;Gata1Yeym2 mice. All mice were maintained at a temperature- and humidity-controlled animal facility. Gata1 is located on X chromosome. To facilitate appropriate interpretation, we have used only male mice with different genotypes in all the experimental procedures of this study, so only single copy of the Gata1 or Gata1s allele is present in all mice used in the procedures. All the experimental procedures performed were approved by the Institutional Animal Care and Use Committee at Roswell Park Cancer Institute.
RT-PCR
Total RNA was extracted from the livers of E14.5 embryos using TRIzol Reagent and PureLink RNA Micro kit (Invitrogen Corp., Carlsbad, CA). One μg of the total RNA from each embryo was used to synthesize cDNA by Superscript version III reverse transcriptase (Invitrogen Corp., Carlsbad, CA). To detect the expression of Gata1 and Gata1s, cDNA was analyzed by PCR amplification with the primers GATA1SRTF1 (TCAGAGGCCAAGGCCAGTGAGGACT) and GATA1SRTR1 (TTGCCATAGGCCCAGCTAGCATAAGG). The lengths of the PCR products are 410- and 171-bp, reflecting to the wild-type and the Gata1Yeym2 alleles, respectively (Figure 2C).
Complete blood counts
Blood samples (~100 μL) were collected from the retro-orbital sinus using heparinized capillary tubes (Fisher Scientific, Pittsburgh, PA) into Mutlivette 600 K3E vials (SARSTEDT AG & Co., Nümbrecht, Germany) and analyzed on a Hemavet 850 complete blood counter (Drew Scientific, Waterbury, CT).
Histology
Bones from femur or sternum and spleen were fixed in 10% buffered formalin and were further processed by the Roswell Park Cancer Institute Pathology Core Facility. Bone marrow samples were decalcified prior to processing. Paraffin sections of spleen and bone marrow were stained with hematoxylin and eosin. Slides were photographed on an Olympus BX41 microscope with a DP70 camera and captured with DP Controller software version 1.2.1.108 (Olympus Optical Co., Japan).
Flow cytometry
Single-cell suspensions from bone marrow or spleen were prepared by passing the tissues through 70 μm cell strainers (BD Pharmingen, San Diego, CA) and red blood cells were lysed in RBC lysis buffer (150 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA). One million cells from each sample were then labeled with the following, anti-CD41-FITC (BD Pharmingen) or anti-TER119-APC-Cy7 (BD Pharmingen) for 30 minutes on ice. Labeled cells were washed in autoMACS Rinsing Solution (Miltenyi Biotec, Aubum, CA) with the addition of 0.5% BSA, fixed with 1% paraformaldehyde in PBS overnight and then analyzed on a LSRII cytometer (BD Biosciences, San Jose, CA). For analysis of Lineage-Sca1+c-Kit+ (LSK) cell and myeloid progenitor cells, ten million bone marrow cells were lineage depleted by using the Lineage Cell Depletion Kit (Miltenyi Biotec) according to the manufacturer’s protocol with the addition of the anti-IL-7R-biotin antibody (BD Pharmingen). After magnetic separation of the lineage cells through the MACS MS column, the lineage negative cells were incubated with streptavidin-PE-Cy5.5 (eBioscience, San Diego, CA), anti-Sca1-PE-Cy7 (BD Pharmingen), anti-c-Kit-APC (BD Pharmingen), anti-FcγR-PE (BD Pharmingen) and anti-CD34-FITC (BD Pharmingen) on ice for 30 minutes and analyzed on a LSRII cytometer after washing and fixation as described above.
Statistical analysis
Statistical analysis was performed using Student t-test and the non-parametric Mann-Whitney U-test. The level of statistical significance was set at p = 0.05. Data are presented as Mean ± SEM.
CONFLICTS OF INTEREST
None of the authors has any conflicts of interest.
FUNDING
This study was supported in part by grants from Roswell Park Alliance Foundation, Louis Sklarow Memorial Fund, American Cancer Society, Association for Research of Childhood Cancer, the Children’s Guild Foundation, the National Institutes of Health (R01HL91519, R01NS66072, R21GM114645, P30CA16056) and the National Natural Science Foundation of China (81370745).
REFERENCES
1. Parker SE, Mai CT, Canfield MA, Rickard R, Wang Y, Meyer RE, Anderson P, Mason CA, Collins JS, Kirby RS, Correa A, and National Birth Defects Prevention Network. Birth Defects Res A Clin Mol Teratol. 2010; 88:1008–1016.
2. Khoshnood B, Greenlees R, Loane M, Dolk H, and EUROCAT Project Management Committee, and EUROCAT Working Group. Paper 2: EUROCAT public health indicators for congenital anomalies in Europe. Birth Defects Res A Clin Mol Teratol. 2011; 91:S16–22.
3. Epstein CJ. The consequences of chromosome imbalance: principles, mechanism and models. New York: Cambridge University Press. 1986:486.
4. Roizen NJ, Patterson D. Down's syndrome. Lancet. 2003; 361:1281–1289.
5. Athale UH, Razzouk BI, Raimondi SC, Tong X, Behm FG, Head DR, Srivastava DK, Rubnitz JE, Bowman L, Pui CH, Ribeiro RC. Biology and outcome of childhood acute megakaryoblastic leukemia: a single institution's experience. Blood. 2001; 97:3727–3732.
6. Wechsler J, Greene M, McDevitt MA, Anastasi J, Karp JE, Le Beau MM, Crispino JD. Acquired mutations in GATA1 in the megakaryoblastic leukemia of Down syndrome. Nat Genet. 2002; 32:148–152.
7. Yoshida K, Toki T, Okuno Y, Kanezaki R, Shiraishi Y, Sato-Otsubo A, Sanada M, Park MJ, Terui K, Suzuki H, Kon A, Nagata Y, Sato Y, et al. The landscape of somatic mutations in Down syndrome-related myeloid disorders. Nat Genet. 2013; 45:1293–1299.
8. Hollanda LM, Lima CS, Cunha AF, Albuquerque DM, Vassallo J, Ozelo MC, Joazeiro PP, Saad ST, Costa FF. An inherited mutation leading to production of only the short isoform of GATA-1 is associated with impaired erythropoiesis. Nat Genet. 2006; 38:807–812.
9. Sankaran VG, Ghazvinian R, Do R, Thiru P, Vergilio JA, Beggs AH, Sieff CA, Orkin SH, Nathan DG, Lander ES, Gazda HT. Exome sequencing identifies GATA1 mutations resulting in Diamond-Blackfan anemia. J Clin Invest. 2012; 122:2439–2443.
10. Gupta M, Dhanasekaran AR, Gardiner KJ. Mouse models of Down syndrome: gene content and consequences. Mamm Genome. 2016; 27:538–555.
11. Ahn KJ, Jeong HK, Choi HS, Ryoo SR, Kim YJ, Goo JS, Choi SY, Han JS, Ha I, Song WJ. DYRK1A BAC transgenic mice show altered synaptic plasticity with learning and memory defects. Neurobiol Dis. 2006; 22:463–472.
12. Jiang X, Liu C, Yu T, Zhang L, Meng K, Xing Z, Belichenko PV, Kleschevnikov AM, Pao A, Peresie J, Wie S, Mobley WC, Yu YE. Genetic dissection of the Down syndrome critical region. Hum Mol Genet. 2015; 24:6540–6551.
13. Marechal D, Lopes Pereira P, Duchon A, Herault Y. Dosage of the Abcg1-U2af1 region modifies locomotor and cognitive deficits observed in the Tc1 mouse model of Down syndrome. PLoS One. 2015; 10:e0115302.
14. Pereira PL, Magnol L, Sahun I, Brault V, Duchon A, Prandini P, Gruart A, Bizot JC, Chadefaux-Vekemans B, Deutsch S, Trovero F, Delgado-Garcia JM, Antonarakis SE, et al. A new mouse model for the trisomy of the Abcg1-U2af1 region reveals the complexity of the combinatorial genetic code of down syndrome. Hum Mol Genet. 2009; 18:4756–4769.
15. Yu T, Li Z, Jia Z, Clapcote SJ, Liu C, Li S, Asrar S, Pao A, Chen R, Fan N, Carattini-Rivera S, Bechard AR, Spring S, et al. A mouse model of Down syndrome trisomic for all human chromosome 21 syntenic regions. Hum Mol Genet. 2010; 19:2780–2791.
16. Zhang L, Meng K, Jiang X, Liu C, Pao A, Belichenko PV, Kleschevnikov AM, Josselyn S, Liang P, Ye P, Mobley WC, Yu YE. Human chromosome 21 orthologous region on mouse chromosome 17 is a major determinant of Down syndrome-related developmental cognitive deficits. Hum Mol Genet. 2014; 23:578–589.
17. Lana-Elola E, Watson-Scales S, Slender A, Gibbins D, Martineau A, Douglas C, Mohun T, Fisher EM, Tybulewicz VL. Genetic dissection of Down syndrome-associated congenital heart defects using a new mouse mapping panel. Elife. 2016; 5:e11614.
18. Alford KA, Slender A, Vanes L, Li Z, Fisher EM, Nizetic D, Orkin SH, Roberts I, Tybulewicz VL. Perturbed hematopoiesis in the Tc1 mouse model of Down syndrome. Blood. 2010; 115:2928–2937.
19. Kirsammer G, Jilani S, Liu H, Davis E, Gurbuxani S, Le Beau MM, Crispino JD. Highly penetrant myeloproliferative disease in the Ts65Dn mouse model of Down syndrome. Blood. 2008; 111:767–775.
20. Klusmann JH, Godinho FJ, Heitmann K, Maroz A, Koch ML, Reinhardt D, Orkin SH, Li Z. Developmental stage-specific interplay of GATA1 and IGF signaling in fetal megakaryopoiesis and leukemogenesis. Genes Dev. 2010; 24:1659–1672.
21. Malinge S, Bliss-Moreau M, Kirsammer G, Diebold L, Chlon T, Gurbuxani S, Crispino JD. Increased dosage of the chromosome 21 ortholog Dyrk1a promotes megakaryoblastic leukemia in a murine model of Down syndrome. J Clin Invest. 2012; 122:948–962.
22. Davisson MT, Schmidt C, Akeson EC. Segmental trisomy of murine chromosome 16: a new model system for studying Down syndrome. Prog Clin Biol Res. 1990; 360:263–280.
23. Reeves RH, Irving NG, Moran TH, Wohn A, Kitt C, Sisodia SS, Schmidt C, Bronson RT, Davisson MT. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat Genet. 1995; 11:177–184.
24. Hernandez D, Mee PJ, Martin JE, Tybulewicz VL, Fisher EM. Transchromosomal mouse embryonic stem cell lines and chimeric mice that contain freely segregating segments of human chromosome 21. Hum Mol Genet. 1999; 8:923–933.
25. Inoue T, Shinohara T, Takehara S, Inoue J, Kamino H, Kugoh H, Oshimura M. Specific impairment of cardiogenesis in mouse ES cells containing a human chromosome 21. Biochem Biophys Res Commun. 2000; 273:219–224.
26. O'Doherty A, Ruf S, Mulligan C, Hildreth V, Errington ML, Cooke S, Sesay A, Modino S, Vanes L, Hernandez D, Linehan JM, Sharpe PT, Brandner S, et al. An aneuploid mouse strain carrying human chromosome 21 with Down syndrome phenotypes. Science. 2005; 309:2033–2037.
27. Shinohara T, Tomizuka K, Miyabara S, Takehara S, Kazuki Y, Inoue J, Katoh M, Nakane H, Iino A, Ohguma A, Ikegami S, Inokuchi K, Ishida I, et al. Mice containing a human chromosome 21 model behavioral impairment and cardiac anomalies of Down's syndrome. Hum Mol Genet. 2001; 10:1163–1175.
28. Li Z, Yu T, Morishima M, Pao A, LaDuca J, Conroy J, Nowak N, Matsui S, Shiraishi I, Yu YE. Duplication of the entire 22.9-Mb human chromosome 21 syntenic region on mouse chromosome 16 causes cardiovascular and gastrointestinal abnormalities. Hum Mol Genet. 2007; 16:1359–1366.
29. Belichenko PV, Kleschevnikov AM, Becker A, Wagner GE, Lysenko LV, Yu YE, Mobley WC. Down Syndrome Cognitive Phenotypes Modeled in Mice Trisomic for All HSA 21 Homologues. PLoS One. 2015; 10:e0134861.
30. Bhutta MF, Cheeseman MT, Herault Y, Yu YE, Brown SD. Surveying the Down syndrome mouse model resource identifies critical regions responsible for chronic otitis media. Mamm Genome. 2013; 24:439–445.
31. Souchet B, Guedj F, Sahun I, Duchon A, Daubigney F, Badel A, Yanagawa Y, Barallobre MJ, Dierssen M, Yu E, Herault Y, Arbones M, Janel N, et al. Excitation/inhibition balance and learning are modified by Dyrk1a gene dosage. Neurobiol Dis. 2014; 69:65–75.
32. Starbuck JM, Dutka T, Ratliff TS, Reeves RH, Richtsmeier JT. Overlapping trisomies for human chromosome 21 orthologs produce similar effects on skull and brain morphology of Dp(16)1Yey and Ts65Dn mice. Am J Med Genet A. 2014; 164A:1981–1990.
33. Block A, Ahmed MM, Dhanasekaran AR, Tong S, Gardiner KJ. Sex differences in protein expression in the mouse brain and their perturbations in a model of Down syndrome. Biol Sex Differ. 2015; 6:24.
34. Guedj F, Pennings JL, Massingham LJ, Wick HC, Siegel AE, Tantravahi U, Bianchi DW. An Integrated Human/Murine Transcriptome and Pathway Approach To Identify Prenatal Treatments For Down Syndrome. Sci Rep. 2016; 6:32353.
35. Patel A, Yamashita N, Ascaño M, Bodmer D, Boehm E, Bodkin-Clarke C, Ryu YK, Kuruvilla R. RCAN1 links impaired neurotrophin trafficking to aberrant development of the sympathetic nervous system in Down syndrome. Nat Commun. 2015; 6:10119.
36. Peiris H, Duffield MD, Fadista J, Jessup CF, Kashmir V, Genders AJ, McGee SL, Martin AM, Saiedi M, Morton N, Carter R, Cousin MA, Kokotos AC, et al. A Syntenic Cross Species Aneuploidy Genetic Screen Links RCAN1 Expression to beta-Cell Mitochondrial Dysfunction in Type 2 Diabetes. PLoS Genet. 2016; 12:e1006033.
37. Raveau M, Nakahari T, Asada S, Ishihara K, Amano K, Shimohata A, Sago H, Yamakawa K. Brain ventriculomegaly in Down syndrome mice is caused by Pcp4 dose-dependent cilia dysfunction. Hum Mol Genet. 2017; 26:923–931.
38. Block A, Dhanasekaran AR, Ahmed M, Gardiner K. Abnormal protein profiles in hippocampus of mouse models of Down syndrome: similarities with Alzheimer’s disease. J Alzheimers Dis Parkinsonism. 2014; 4:138–149.
39. Ng AP, Hu Y, Metcalf D, Hyland CD, Ierino H, Phipson B, Wu D, Baldwin TM, Kauppi M, Kiu H, Di Rago L, Hilton DJ, Smyth GK, Alexander WS. Early lineage priming by trisomy of Erg leads to myeloproliferation in a Down syndrome model. PLoS Genet. 2015; 11:e1005211.
40. Duchon A, Raveau M, Chevalier C, Nalesso V, Sharp AJ, Herault Y. Identification of the translocation breakpoints in the Ts65Dn and Ts1Cje mouse lines: relevance for modeling Down syndrome. Mamm Genome. 2011; 22:674–684.
41. Reinholdt LG, Ding Y, Gilbert GJ, Czechanski A, Solzak JP, Roper RJ, Johnson MT, Donahue LR, Lutz C, Davisson MT. Molecular characterization of the translocation breakpoints in the Down syndrome mouse model Ts65Dn. Mamm Genome. 2011; 22:685–691.
42. Chou ST, Opalinska JB, Yao Y, Fernandes MA, Kalota A, Brooks JS, Choi JK, Gewirtz AM, Danet-Desnoyers GA, Nemiroff RL, Weiss MJ. Trisomy 21 enhances human fetal erythro-megakaryocytic development. Blood. 2008; 112:4503–4506.
43. Tunstall-Pedoe O, Roy A, Karadimitris A, de la Fuente J, Fisk NM, Bennett P, Norton A, Vyas P, Roberts I. Abnormalities in the myeloid progenitor compartment in Down syndrome fetal liver precede acquisition of GATA1 mutations. Blood. 2008; 112:4507–4511.
44. Roy A, Cowan G, Mead AJ, Filippi S, Bohn G, Chaidos A, Tunstall O, Chan JK, Choolani M, Bennett P, Kumar S, Atkinson D, Wyatt-Ashmead J, et al. Perturbation of fetal liver hematopoietic stem and progenitor cell development by trisomy 21. Proc Natl Acad Sci U S A. 2012; 109:17579–17584.
45. Gribble SM, Wiseman FK, Clayton S, Prigmore E, Langley E, Yang F, Maguire S, Fu B, Rajan D, Sheppard O, Scott C, Hauser H, Stephens PJ, et al. Massively parallel sequencing reveals the complex structure of an irradiated human chromosome on a mouse background in the Tc1 model of Down syndrome. PLoS One. 2013; 8:e60482.
46. Birger Y, Goldberg L, Chlon TM, Goldenson B, Muler I, Schiby G, Jacob-Hirsch J, Rechavi G, Crispino JD, Izraeli S. Perturbation of fetal hematopoiesis in a mouse model of Down syndrome's transient myeloproliferative disorder. Blood. 2013; 122:988–998.
47. Calligaris R, Bottardi S, Cogoi S, Apezteguia I, Santoro C. Alternative translation initiation site usage results in two functionally distinct forms of the GATA-1 transcription factor. Proc Natl Acad Sci U S A. 1995; 92:11598–11602.
48. Hitzler JK, Cheung J, Li Y, Scherer SW, Zipursky A. GATA1 mutations in transient leukemia and acute megakaryoblastic leukemia of Down syndrome. Blood. 2003; 101:4301–4304.
49. Mundschau G, Gurbuxani S, Gamis AS, Greene ME, Arceci RJ, Crispino JD. Mutagenesis of GATA1 is an initiating event in Down syndrome leukemogenesis. Blood. 2003; 101:4298–4300.
50. Rainis L, Bercovich D, Strehl S, Teigler-Schlegel A, Stark B, Trka J, Amariglio N, Biondi A, Muler I, Rechavi G, Kempski H, Haas OA, Izraeli S. Mutations in exon 2 of GATA1 are early events in megakaryocytic malignancies associated with trisomy 21. Blood. 2003; 102:981–986.
51. Xu G, Nagano M, Kanezaki R, Toki T, Hayashi Y, Taketani T, Taki T, Mitui T, Koike K, Kato K, Imaizumi M, Sekine I, Ikeda Y, et al. Frequent mutations in the GATA-1 gene in the transient myeloproliferative disorder of Down syndrome. Blood. 2003; 102:2960–2968.
52. Ramirez-Solis R, Davis AC, Bradley A. Gene targeting in embryonic stem cells. Methods Enzymol. 1993; 225:855–878.
53. Bradley A. Production and analysis of chimaeric mice. Teratocarcinomas and Embryonic Stem Cells - A Practical Approach, E. Robertson, Editor. IRL Press: Oxford. 1987: 113–151.
54. Abuin A, Bradley A. Recycling selectable markers in mouse embryonic stem cells. Mol Cell Biol. 1996; 16:1851–1856.