
INTRODUCTION
Cervical carcinoma ranks the third most common cancer in young women worldwide. It is an important cause of cancer mortalities [1–3]. The application of HPV-based screening and combination therapy have improved the overall survival of cervical carcinoma [1–3]. Yet, for the patients with advanced, metastatic or recurrent cervical carcinoma, the prognosis is still extremely poor [4]. Groups all over the world are developing novel and molecule-based anti-cervical carcinoma agents [5, 6].
Sphingosine kinase (SphK) is a oncotarget protein for human cervical carcinoma [7] and many other malignancies [8]. SphK is critical for the balance of cellular sphingolipids [9–11]. SphK activation leads to production of sphingosine-1-phosphate (S1P), which is a well-known pro-cancerous lipid molecule. S1P promotes cell survival, cell growth and angiogenesis [12]. Inhibition, depletion or mutation of SphK shall cause production of pro-apoptotic sphingosine and ceramide [13]. Two SphK isoforms, SphK1 and SphK2, have been characterized [14]. The oncogenic function of SphK1 has been well-documented [15]. Literatures have also focused on the potential activity of SphK2 in human malignancies [16–21].
ABC294640 is a novel, specific and potent SphK2 small-molecule inhibitor [16, 17, 22, 23]. Preclinical cancer studies have demonstrated that ABC294640 could possibly inhibit growth of different cancer cells [20, 22, 24]. ABC294640 is a competitive SphK2 inhibitor, which displays chemotherapeutic efficacy in the in vivo cancer studies [20, 22, 24]. Currently, ABC294640 is under phase II clinical trials [22]. The potential anti-cancer activity of ABC294640 against human cervical carcinoma cells is tested here.
RESULTS
ABC294640 inhibits human cervical carcinoma cell growth
First, we tested the expression of SphK2 in human cervical carcinoma cells. In the current study, two lines of primary human cervical carcinoma cells (“P1” and “P2”) as well as two lines of primary human cervical epithelial cells (“E1” and “E2”) were established. C33A is well-established human cervical carcinoma cell line [25]. The quantitative real-time PCR assay results in Figure 1A confirmed that SphK2 mRNA expression level was high in both primary (“P1” and “P2”) and established (C33A) human cervical carcinoma cells, and its level was relatively low in the primary epithelial cells (Figure 1A). Meanwhile, SphK2 protein was also upregulated in the cancerous cells, as compared to the epithelial cells (Figure 1B).
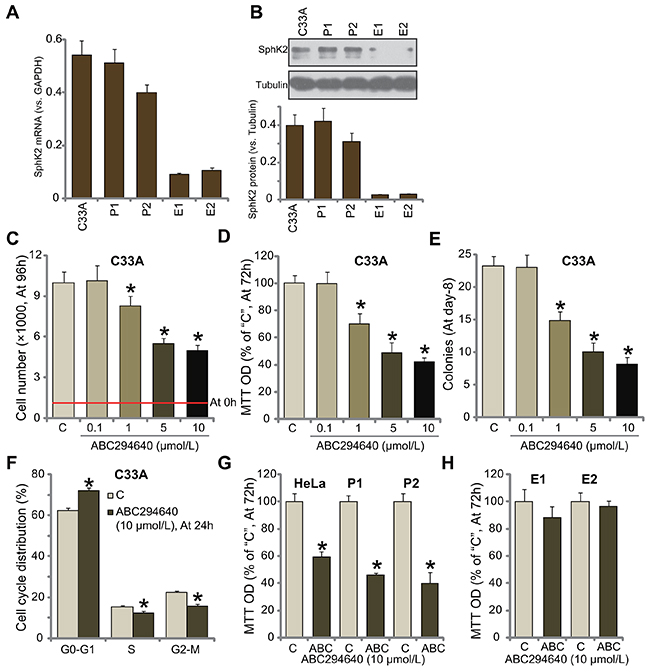
Figure 1: ABC294640 inhibits human cervical carcinoma cell growth. mRNA and protein expressions of SphK2 in the established human cervical carcinoma cells (C33A/HeLa), two primary human cervical carcinoma cells (“P1” and “P2”), or primary human cervical epithelial cells (“E1” and “E2”) were shown (A and B). The above cells were either left untreated (“C”) or treated with ABC294640 at designated concentrations (0.1-10 μmol/L), cells were further cultured for applied time; cell growth was tested by the listed assays (C-E, G and H); cell cycle progression was also tested (F, for C33A cells). Data were shown as mean (n=5) ± standard deviation (SD). *p<0.05 vs. “C” cells. Experiments in this figure were repeated three times, and similar results were obtained.
Cultured C33A cells were treated with ABC294640 at various concentrations (0.1-10 μmol/L). Simple cell counting assay results in Figure 1C demonstrated that ABC294640 inhibited C33A cell growth in a dose-dependent manner. The number of viable C33A cells (at 96-hour) was significantly decreased following ABC294640 (1-10 μmol/L) treatment (Figure 1C). ABC294640, at 1-10 μmol/L, dramatically inhibited MTT optic density (OD) value of C33A cells (Figure 1D). The number of C33A cell colonies was also decreased after ABC294640 (1-10 μmol/L) treatment (Figure 1E). These results suggest that ABC294640 inhibits C33A cell growth.
Results in Figure 1F showed that treatment with ABC294640 (10 μmol/L) in C33A cells also disrupted cell cycle progression, causing G1-S arrest. The potential effect of ABC294640 on other cervical carcinoma cells was also tested. MTT assay results in Figure 1G showed that ABC294640 treatment (10 μmol/L, 72 hours) significantly inhibited growth of primary (two lines, “P1” and “P2”) and the other established HeLa cervical carcinoma cells. On the other hand, the very same ABC294640 treatment failed to inhibit growth (MTT OD) of human cervical epithelial cells (“E1” and “E2”) (Figure 1H). Together, these results suggest that ABC294640 inhibits human cervical carcinoma cell growth in vitro.
ABC294640 induces apoptosis activation in human cervical carcinoma cells
SphKs are known anti-apoptosis proteins [14, 26]. Inhibition of SphK could thus induce cell apoptosis [14, 15, 26, 27]. The potential effect of ABC294640 on cervical carcinoma cell apoptosis was tested. As shown in Figure 2A, treatment with ABC294640 in C33A cells dose-dependently increased the Histone DNA apoptosis ELISA OD. Meanwhile, the number of C33A cells with positive TUNEL staining was increased following ABC294640 (1-10 μmol/L) treatment (Figure 2B). These results suggest that ABC294640 provoked apoptosis in C33A cells. In the other established (HeLa) and primary human cervical carcinoma cells (“P1” and “P2”), treatment with ABC294640 (10 μmol/L) similarly induced significant apoptosis activation (TUNEL assay, Figure 2C). Yet, same ABC294640 treatment failed to provoke apoptosis in the primary cervical epithelial cells (“E1” and “E2”) (Figure 2D). These results demonstrate that ABC294640 provokes apoptosis in human cervical carcinoma cells.
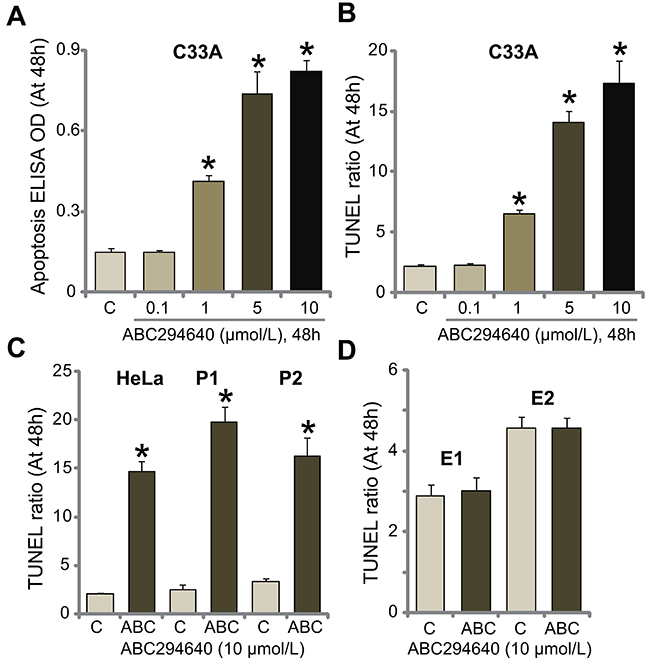
Figure 2: ABC294640 induces apoptosis activation in human cervical carcinoma cells. The established human cervical carcinoma cells (C33A and HeLa), the primary human cervical carcinoma cells (“P1” and “P2”), or the primary human cervical epithelial cells (“E1” and “E2”) were either left untreated (“C”) or treated with ABC294640 at designated concentrations (0.1-10 μmol/L), cells were further cultured for applied time; cell apoptosis was tested by the listed assays (A-D). Data were shown as mean (n=5) ± standard deviation (SD). *p<0.05 vs. “C” cells. Experiments in this figure were repeated three times, and similar results were obtained.
ABC294640 inactivates SphK and STAT3 in C33A cells
ABC294640 is the novel and specific SphK2 inhibitor [16, 17, 22, 23], the potential effect of ABC294640 on SphK activity was tested. Using the method described, we showed that treatment with ABC294640 at 1-10 μmol/L significantly inhibited SphK activity in C33A cells (Figure 3A). Consequently, cellular content of sphingosine-1-phosphate (S1P), the pro-cancerous sphingosine [12, 28], was reduced (Figure 3B). On the other hand, the level of pro-apoptotic ceramide was significantly increased (Figure 3C). ABC294640 displayed dose-dependent effect in inhibiting SphK in C33A cells (Figure 3A-3C). At a low concentration (0.1 μmol/L), ABC294640 failed to change SphK activation (Figure 3A) nor S1P/ceramide production (Figure 3B and 3C).
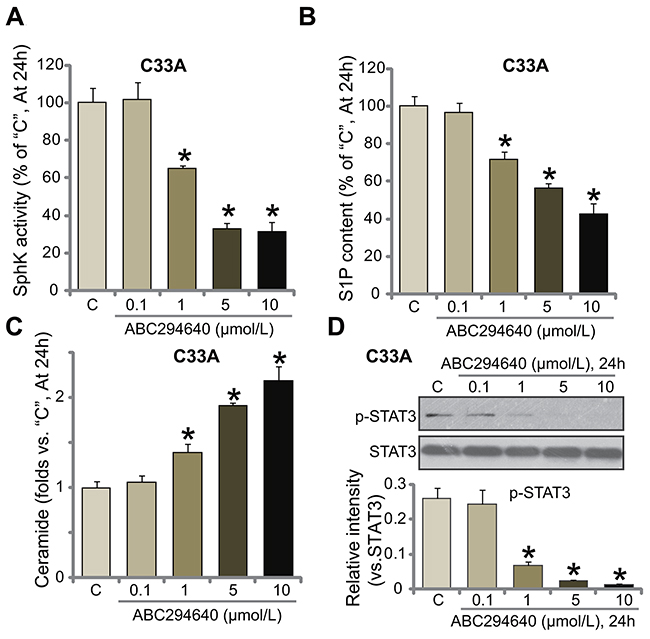
Figure 3: ABC294640 inactivates SphK and STAT3 in C33A cells. C33A cells were either left untreated (“C”) or treated with ABC294640 at designated concentrations (0.1-10 μmol/L), cells were further cultured for 24 hours; relative SphK activity (A), S1P content (B) and ceramide level (C) as well as STAT3 activation (D) were examined. p-STAT3 (vs. total STAT3) was quantified (D). Data were shown as mean (n=5) ± standard deviation (SD). *p<0.05 vs. “C” cells. Experiments in this figure were repeated three times, and similar results were obtained.
S1P could serve as a key upstream signal of STAT3 (signal transducer and activator of transcription 3) [29, 30], the latter is a key participant in cervical carcinoma progression [31, 32]. In the current study, we showed that STAT3 activation, tested by p-STAT3 at Tyr705, was largely inhibited in ABC294640-treated C33A cells (Figure 3D). Collectively, these results show that ABC294640 inactivates SphK and STAT3 in C33A cells.
Inhibition or silence of Bcl-2 sensitizes ABC294640 in C33A cells
One important objective of this study is to identify possible ABC294640’s resistance factors. We focused on Bcl-2. Bcl-2 is well-established anti-apoptosis protein [33–35. Bcl-2 has been recognized as a key chemo-resistance factor [33–35]. Third, Bcl-2 inhibition, mutation or depletion could significantly sensitize the anti-cancer activity by a number of molecule-targeted agents [36-40]. Bcl-2 pharmacological inhibitors were then applied, including the specific Bcl-2 inhibitor GDC-0199 (also known as ABT-199) [41, 42] and the pan Bcl-2 inhibitor ABT-737 [37, 40, 43]. As shown in Figure 4A, ABC294640 (5 μmol/L)-induced C33A cell growth inhibition (MTT OD reduction) was largely potentiated with co-treatment of the Bcl-2 inhibitors, which also dramatically facilitated ABC294640-induced cell apoptosis (TUNEL assay, Figure 4B). These results imply that Bcl-2 inhibition could possibly sensitize ABC294640 in C33A cells.
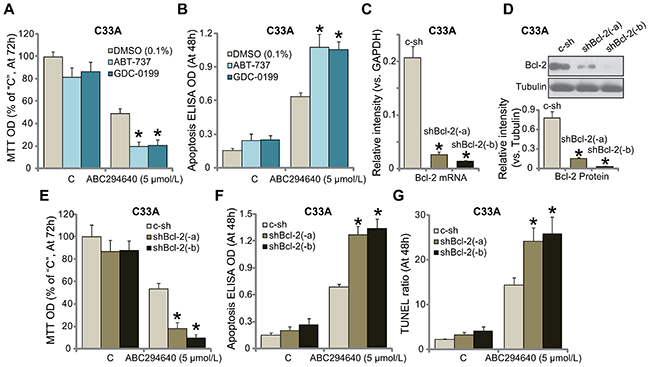
Figure 4: Inhibition or silence of Bcl-2 sensitizes ABC294640 in C33A cells. C33A cells were treated with ABC294640 (5 μmol/L), together with/out ABT-737 (200 nM) or GDC-0199 (200 nM), cells were further cultured for indicated time, cell growth (MTT assay, (A) and apoptosis (Histone DNA ELISA assay, (B) were tested. mRNA (C) and protein (D) expression of Bcl-2 in stable C33A cells, expressing Bcl-2 shRNA (“shBcl-2-a/-b”, with non-overlapping sequence) or scramble non-sense control shRNA (“c-sh”), were shown; cells were also treated with ABC294640 (5 μmol/L) for indicate time; cell growth (MTT assay, (E) and apoptosis (F and G) were tested. Data were shown as mean (n=5) ± standard deviation (SD). *p<0.05 vs. DMSO (0.1%) cells (A and B). *p<0.05 vs. “c-sh” cells (C-G). Experiments in this figure were repeated three times, and similar results were obtained.
To exclude the off-target effect of the above inhibitors, i.e. ABT-737 [37, 40, 43], we utilized shRNA method to selectively knockdown Bcl-2 in C33A cells. Two distinct Bcl-2 shRNAs (“-a/-b”), with non-overlapping sequences, were applied. Quantitative real-time PCR (“qRT-PCR”) assay results in Figure 4C demonstrated that the applied Bcl-2 shRNA dramatically silenced Bcl-2 mRNA in C33A cells. Consequently, Bcl-2 protein expression was also silenced in stable cells with Bcl-2 shRNA (Figure 4D). Remarkably, knockdown of Bcl-2 significantly sensitized ABC294640 (5 μmol/L)-induced cytotoxicity against C33A cells, causing substantial growth inhibition (MTT OD reduction, Figure 4E) and apoptosis (Apoptosis ELISA OD/TUNEL ratio increase, Figure 4F and 4G). Collectively, these results confirm that inhibition or silence of Bcl-2 sensitizes ABC294640 in C33A cells.
Exogenous over-expression of Bcl-2 de-sensitizes ABC294640 in C33A cells
Next, the Bcl-2 expression vector (a gift from Dr. Sun [38]) was introduced to C33A cells, and two stable cell lines (“L1” and “L2”) expressing the construct were established. As compared to vector control cells, Bcl-2 mRNA level was significantly increased in the two stable lines (Figure 5A). Bcl-2 protein was also over-expressed (Figure 5B). Notably, the exogenous Bcl-2 was tagged with Flag (Figure 5B). Remarkably, ABC294640-induced growth inhibition (MTT OD reduction, Figure 5C) and apoptosis (Histone DNA ELISA assay, Figure 5D) were largely attenuated in Bcl-2-over-expressed C33A cells. Thus, exogenous over-expression of Bcl-2 could significantly attenuate ABC294640’s cytotoxicity against C33A cells, further confirming the chemo-resistance function of Bcl-2 against ABC294640.
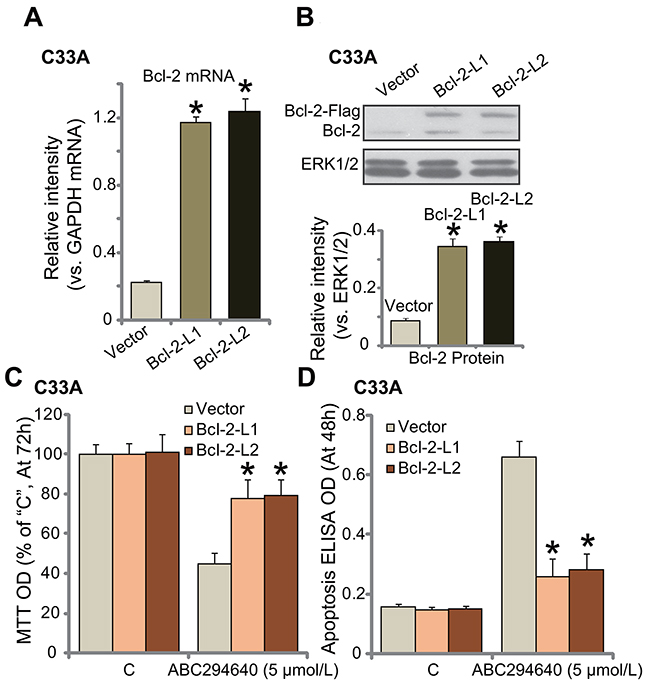
Figure 5: Exogenous over-expression of Bcl-2 de-sensitizes ABC294640 in C33A cells. mRNA (A) and protein (B) expression of Bcl-2 (endogenous and exogenous) in stable C33A cells, expressing the Bcl-2 construct (two lines, “Bcl-2-L1/L2”) or empty vector (Ad-GFP, “Vector”) were shown; cells were also treated with ABC294640 (5 μmol/L) for indicate time; cell growth (MTT assay, (C) and apoptosis (Histone DNA ELISA assay, (D) were tested. Data were shown as mean (n=5) ± standard deviation (SD). *p<0.05 vs. “Vector” cells. Experiments in this figure were repeated three times, and similar results were obtained.
ABC294640 inhibits C33A tumor growth in nude mice, sensitized with co-administration of GDC-0199
At last, the anti-cervical carcinoma activity of ABC294640 in vivo was tested. As described, C33A cells were s.c. injected to the nude mice. Within 2-3 weeks, when the tumors were established (with 100 mm3 in volume), mice were randomly assigned into four groups, with 10 mice per group. Tumor growth curve results in Figure 6A demonstrated that oral administration of ABC294640 (20 mg/kg, daily, p.o., for 21 days) [17] inhibited C33A tumor growth in mice. The tumor volumes in ABC294640-treated mice were significantly lower than those of vehicle control mice (Figure 6A). Remarkably, co-administration of the Bcl-2 inhibitor GDC-0199 (25 mg/kg, daily, i.p., for 21 days) [41, 42] strikingly potentiated the anti-cancer activity by ABC294640, leading to profound inhibition of C33A tumors (Figure 6A). The combined activity was dramatically more potent than ABC294640 single treatment in inhibiting C33A tumors (Figure 6A). The weight of tumors was also lowest in the combination treatment group (Figure 6B). Although, ABC294640 single treatment also decreased the weight of C33A tumors (At week-7, Figure 6B). Notably, treatment with GDC-0199 alone failed to induce significant inhibition on C33A tumor growth (Figure 6A and 6B). Mice body weight, the major indicator of animal health, was not significantly changed by the single or combined treatment (Figure 6C). We also failed to notice any signs of apparent toxicities during the experimental periods. Thus, these nude mice were obviously well-tolerated to the treatment regimens here.
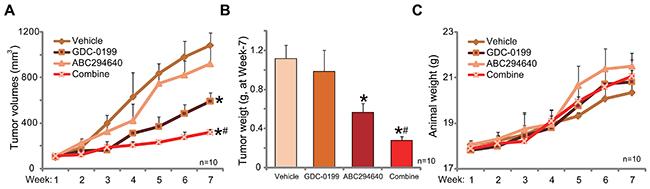
Figure 6: ABC294640 inhibits C33A tumor growth in nude mice, sensitized by co-administration of GDC-0199. C33A tumor-bearing mice were administrated withABC294640 (20 mg/kg, daily, p.o., for 21 days) and/or GDC-0199 (25 mg/kg, daily, i.p., for 21 days); estimated tumor volume (A) and mice body weight (deducting tumor weight, (C) were recorded weekly for total six weeks. At the end of experiment (week-7), tumors of the each group were isolated and weighted (B). Data were shown as mean (n=10) ± standard deviation (SD). *p<0.05 vs. “Vehicle” (0.375% Polysorbate-80) treatment. #p<0.05 vs. ABC294640 only treatment.
DISCUSSIONS
The results of this current study proposed that SphK2 could be a novel oncotarget protein and rational therapeutic target for human cervical carcinoma. We demonstrated that SphK2 is over-expressed in primary and established human cervical carcinoma cells. ABC294640, the novel, specific and competitive SphK2 inhibitor, in-activated SphK in cervical carcinoma cells, leading to S1P depletion, ceramide production and STAT3 inhibition. Treatment with ABC294640 induced growth inhibition, G1-S arrest and apoptosis in a panel of established human cervical carcinoma cells. In vivo, ABC294640 oral administration at a well-tolerated dose potently inhibited C33A xenograft in nude mice. These results suggest that targeting of SphK2 by ABC294640 could inhibit human cervical carcinoma cells in vitro and in vivo.
Signal transducers and activators of transcription (STAT) transcription factor family proteins are vital in the regulation a number of key cancerous behaviors, including cell survival, growth, apoptosis-resistance, migration, and angiogenesis [44, 45]. Constitutively-activated STAT3 is often detected in cervical carcinoma, which is associated with tumorigenesis, cancer progression and poor prognosis [44, 45]. It has been previously shown that S1P is essential for production of NF-κB-regulated cytokine IL-6, and subsequent activation of STAT3 [29]. On the other hand, S1P depletion could significantly inhibit STAT3 activation. In the current study, we showed that ABC294640 inhibited SphK activity, causing S1P reduction and downstream STAT3 inactivation. This could be an important reason of the superior anti-cervical carcinoma activity by ABC294640. The underlying mechanisms of STAT3 inhibition by ABC294640 may warrant further investigations.
Another novel finding of this study is that Bcl-2 could be a major resistance factor of ABC294640 in cervical carcinoma cells. Pharmacological inhibition (by adding ABT-737 or GDC-0199) or silence of Bcl-2 sensitized ABC294640-induced anti-C33A cell activity. On the other hand, exogenous over-expression of Bcl-2 de-sensitized ABC294640 in C33A cells. In vivo, co-administration of the Bcl-2 inhibitor GDC-0199 significantly potentiated ABC294640-induced anti-C33A tumor activity in nude mice. Therefore, targeting Bcl-2 should sensitize ABC294640’s anti-cancer efficiency. Further studies will be needed to explore the mechanism of ABC294640’s resistance by Bcl-2. It will be interesting to test this theory in other cancer cells.
Together, we suggest that targeting SphK2 by ABC294640 inhibits human cervical carcinoma cell growth in vitro and in vivo. Bcl-2 inhibition could further sensitize ABC294640’s activity against cervical carcinoma cells. ABC294640 might have translational value for the treatment of cervical carcinoma.
MATERIALS AND METHODS
Reagents
ABC294640 was provided by DC Chemicals (Shanghai, China). ABT-737 and GDC-0199 were obtained from Selleck (Shanghai, China). Puromycin was purchased from Sigma Aldrich (St. Louis, MO). The antibodies utilized in this study were from Cell Signaling Tech (Beverly, MA).
Cell culture
The established human cervical carcinoma cell lines, C33A and HeLa were provided by the Cell Bank of Shanghai Institute of Biological Science (Shanghai, China). Cells were maintained in the DMEM/F12 medium with 10% FBS. Two cervical carcinoma patients administrated at the Minhang Branch, Zhongshan Hospital were consent to provide the tissues. The cervical carcinoma and para-carcinoma epithelial tissues were obtained at the time of surgery. The tissues were separated carefully and were digested for 10 hours at 4°C in dispase. Tissues were cut into 1 mm2 pieces and digested in 0.25% trypsin for 15 min at 37°C. Trypsin was then neutralized by FBS (10%). The cells were collected by low speed centrifugation. Primary cancer cells were cultured in the DMEM plus 10% FBS medium. Cultures with 95% epithelial cells were maintained in keratinocyte serum-free medium (KSFM, Invitrogen, Carlsbad, CA). A total of two lines of primary human cervical carcinoma cells, namely “P1” and “P2”, as well as two lines of primary human cervical epithelial cells (“E1” and “E2”), were established. The protocols were approved by the Institutional Review Board and Ethics Committee of Fudan University, and experiments were conducted according to the principles of Declaration of Helsinki.
MTT assay of cell growth
Cell growth was examined by routine 3-[4, 5-dimethylthiazol-2-yl]-2, 5 diphenyltetrazolium bromide (MTT) assay. Briefly, 5,000 cells per well were plated onto the 96-well plates. After the applied treatment, MTT (5 mg/mL, 20 μL per well, Sigma) was added to the medium for another 2-3 hours. Absorbance at 490 nm of MTT was measured by a microplate reader (Bio-Rad, Basel, Switzerland).
Clonogenicity assay
C33A cells were initially plated at 10,000 cells/well onto the six-well plate, and were treated with designated concentration of ABC294640. Cells were further incubated for another eight days. The number of colonies was then manually counted.
Cell cycle distribution analysis
C33Acells with applied ABC294640 treatment were fixed in ice-cold ethanol, which were then washed and stained with 10 μg/mL propidium iodide (PI, Invitrogen) and100 μg/mL RNase (Invitrogen). DNA content was analyzed with a flow cytometer (BD Biosciences, Franklin Lakes, NJ). Cell cycle distribution (G0-G1, S and G2-M) percentage was recorded.
Fragmented DNA detection by ELISA
Histone-bound DNA was tested via a specific two-site ELISA kit (Roche, Shanghai, China). Histone DNA ELISA OD was recorded at 450 nm was tested.
TUNEL assay of apoptosis
Following the ABC294640 treatment, cells were subjected to the TUNEL dye assay (Invitrogen). For each condition, at least 200 cells in five random views were counted. TUNEL percentage (vs. total cell number) was recorded.
Western blotting assay
Lysate proteins were separated by 8-12% of SDS-PAGE gels, which were then transferred to PVDF blots (Millipore, Wuxi, China). After blocked in 10% milk, the blots were incubated with the designated primary and corresponding secondary antibodies. The protein signals were visualized via the ECL detection kit. Quantification of the signal was performed via the Image J software.
Bcl-2 shRNA
Six distinct lentiviral shRNAs against non-overlapping sequence of human Bcl-2 were designed and synthesized by Genepharm (Nanjing, China). The lentiviral shRNA (10 μL/mL) was added to cultured C33A cells for 24 hours. Afterwards, puromycin (2.5 μg/mL, Sigma) was utilized to select stable cells, which could last for another 8-12 days. Bcl-2 knockdown in stable cells was verified by qRT-PCR assay and Western blotting assay. Of the five tested Bcl-2 shRNAs, two of them efficiently downregulated Bcl-2. Control cells were infected with lentiviral scramble control shRNA (Santa Cruz Biotech).
Exogenous Bcl-2 over-expression
The Bcl-2 expression vector was a gift from Dr. Sun [38]. The construct was transfected to C33A cells via the routine Lipofectamine 2000 protocol [46]. Puromycin (2.5 μg/mL, Sigma) was added again to select stable cells for another 8-12 days. Two stable C33A cell lines with the construct were established. Bcl-2 expression (endogenous and exogenous) in the stable cells was verified by qRT-PCR assay and Western blotting assay.
Quantitative RT-PCR
Trizol reagents (Sigma) were applied to extract total cellular RNA, and High Capacity cDNA Reverse Transcription Kit was utilized to synthesis cDNA from 0.5 μg mRNA (per treatment). Power SYBR Green RT-PCR Reagents Kit was used to perform the quantitative real-time PCR (“qRT-PCR”) via the ABI-7500 system (Applied Biosystems, Foster, CA). The primers for Bcl-2 mRNA were described previously [47, 48]. The primers GAPDH mRNA were also described early [49]. SphK2 mRNA primers were described previously [50]. Relative mRNA expression was carried out using 2−ΔΔCt method after normalization to GAPDH.
Assay of SphK activity and S1P content
Following the indicated ABC294640 treatment, 20 μg protein lysates (per treatment) were incubated with D-erythrosphingosine (5 μmol/L, dissolved in 0.1% Triton X-100, 2 mmol/L ATP, and [γ-32P] ATP) [17]. HCl (1 N) was added to stop the reaction, followed by adding 800 μL of chloroform/methanol/HCl (100:200:1, v/v). After vortex, phases were separated by centrifugation. Radio-labeled S1P was separated by 60 thin-layer chromatography (TLC) on silica gel G60-plates with the described solvent, and phosphate incorporation was visualized and quantified via the scintillation counter (LS-6500, Beckman, Shanghai, China) [51]. The SphK activity was valued as pmol/hour/g protein, which was always normalized to that of control group.
Ceramide detection
Autoradiography detection of cellular ceramide content was described in detail in previous studies [52, 53]. Ceramide content in the treatment group was always normalized to the control level.
C33A xenograft assay
All animal protocols in the study were approved by Fudan University’s Ethics Board. Animals were provided by the Animal Center of Fudan University (Shanghai, China) and were maintained in routine conditions. The female nude mice (6-8 weeks age, 17.5-18.8 g weight) were subcutaneously (s.c.) inoculated with 5 × 106 C33A cells (per mouse, in 0.2 mL DMEM/10 % FBS) into the right flanks. When C33A xenograft volumes reached around 100 mm3, mice were randomized into four groups, which were treated as describe in the text. The xenografted tumor volumes along with mice body weights were recorded every week for a total of six weeks. At the end of experiment (week-7), tumors of each group were isolated and weighted.
Statistics
Data were expressed as the mean ± SD (standard deviation). Comparisons between groups were performed via one-way ANOVA (SPSS 18.0). p values < 0.05 were considered statistically significant.
Author contributions
All authors carried out the experiments, participated in the design of the study and performed the statistical analysis, participated in its design and coordination and helped to draft the manuscript.
CONFLICTS OF INTEREST
The listed authors have no conflict of interests.
FUNDING
This study was supported in part by the Shanghai key medical college construction project (ZK2015B14), Wu Jieping medical foundation special fund for clinical research (320.6750.13152) and by the Minhang District Central Hospital Youth Foundation(2016MHJC06).
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017; 67: 7-30. https://doi.org/10.3322/caac.21387.
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016; 66: 7-30. https://doi.org/10.3322/caac.21332.
3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015; 65: 5-29. https://doi.org/10.3322/caac.21254.
4. de Azevedo CR, Thuler LC, de Mello MJ, Ferreira CG. Neoadjuvant chemotherapy followed by chemoradiation in cervical carcinoma: a review. Int J Gynecol Cancer. 2016; 26: 729-36. https://doi.org/10.1097/IGC.0000000000000663.
5. Duenas-Gonzalez A, Serrano-Olvera A, Cetina L, Coronel J. New molecular targets against cervical cancer. Int J Womens Health. 2014; 6: 1023-31. https://doi.org/10.2147/IJWH.S49471.
6. Banno K, Iida M, Yanokura M, Kisu I, Iwata T, Tominaga E, Tanaka K, Aoki D. MicroRNA in cervical cancer: oncomiRs and tumor suppressor miRs in diagnosis and treatment. ScientificWorldJournal. 2014; 2014: 178075. https://doi.org/10.1155/2014/178075.
7. Kim HS, Yoon G, Ryu JY, Cho YJ, Choi JJ, Lee YY, Kim TJ, Choi CH, Song SY, Kim BG, Bae DS, Lee JW. Sphingosine kinase 1 is a reliable prognostic factor and a novel therapeutic target for uterine cervical cancer. Oncotarget. 2015; 6: 26746-56. https://doi.org/10.18632/oncotarget.4818.
8. Pyne S, Bittman R, Pyne NJ. Sphingosine kinase inhibitors and cancer: seeking the golden sword of Hercules. Cancer Res. 2011; 71: 6576-82. https://doi.org/10.1158/0008-5472.CAN-11-2364.
9. Young MM, Kester M, Wang HG. Sphingolipids: regulators of crosstalk between apoptosis and autophagy. J Lipid Res. 2013; 54: 5-19. https://doi.org/10.1194/jlr.R031278.
10. Ogretmen B, Hannun YA. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat Rev Cancer. 2004; 4: 604-16. https://doi.org/10.1038/nrc1411.
11. Gangoiti P, Granado MH, Alonso A, Goni FM, Gomez-Munoz A. Implication of ceramide, ceramide 1-phosphate and sphingosine 1-phosphate in tumorigenesis. Transl Oncogenomics. 2008; 3: 81-98.
12. Maceyka M, Harikumar KB, Milstien S, Spiegel S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012; 22: 50-60. doi: S0962-8924(11)00177-2.
13. Dimanche-Boitrel MT, Rebillard A, Gulbins E. Ceramide in chemotherapy of tumors. Recent Pat Anticancer Drug Discov. 2011; 6: 284-93. https://doi.org/10.2174/157489211796957838.
14. Maceyka M, Payne SG, Milstien S, Spiegel S. Sphingosine kinase, sphingosine-1-phosphate, and apoptosis. Biochim Biophys Acta. 2002; 1585: 193-201. doi: S1388198102003414.
15. Shida D, Takabe K, Kapitonov D, Milstien S, Spiegel S. Targeting SphK1 as a new strategy against cancer. Curr Drug Targets. 2008; 9: 662-73.
16. Yang J, Yang C, Zhang S, Mei Z, Shi M, Sun S, Shi L, Wang Z, Wang Y, Li Z, Xie C. ABC294640, a sphingosine kinase 2 inhibitor, enhances the antitumor effects of TRAIL in non-small cell lung cancer. Cancer Biol Ther. 2015; 16: 1194-204. https://doi.org/10.1080/15384047.2015.1056944.
17. Xun C, Chen MB, Qi L, Tie-Ning Z, Peng X, Ning L, Zhi-Xiao C, Li-Wei W. Targeting sphingosine kinase 2 (SphK2) by ABC294640 inhibits colorectal cancer cell growth in vitro and in vivo. J Exp Clin Cancer Res. 2015; 34: 94. https://doi.org/10.1186/s13046-015-0205-y.
18. Xiao M, Liu Y, Zou F. Sensitization of human colon cancer cells to sodium butyrate-induced apoptosis by modulation of sphingosine kinase 2 and protein kinase D. Exp Cell Res. 2012; 318: 43-52. doi: S0014-4827(11)00400-9.
19. Liu W, Ning J, Li C, Hu J, Meng Q, Lu H, Cai L. Overexpression of Sphk2 is associated with gefitinib resistance in non-small cell lung cancer. Tumour Biol. 2016; 37: 6331-6. https://doi.org/10.1007/s13277-015-4480-1.
20. Gestaut MM, Antoon JW, Burow ME, Beckman BS. Inhibition of sphingosine kinase-2 ablates androgen resistant prostate cancer proliferation and survival. Pharmacol Rep. 2014; 66: 174-8. https://doi.org/10.1016/j.pharep.2013.08.014.
21. Antoon JW, White MD, Slaughter EM, Driver JL, Khalili HS, Elliott S, Smith CD, Burow ME, Beckman BS. Targeting NFkB mediated breast cancer chemoresistance through selective inhibition of sphingosine kinase-2. Cancer Biol Ther. 2011; 11: 678-89.
22. French KJ, Zhuang Y, Maines LW, Gao P, Wang W, Beljanski V, Upson JJ, Green CL, Keller SN, Smith CD. Pharmacology and antitumor activity of ABC294640, a selective inhibitor of sphingosine kinase-2. J Pharmacol Exp Ther. 2010; 333: 129-39. https://doi.org/10.1124/jpet.109.163444.
23. Antoon JW, White MD, Meacham WD, Slaughter EM, Muir SE, Elliott S, Rhodes LV, Ashe HB, Wiese TE, Smith CD, Burow ME, Beckman BS. Antiestrogenic effects of the novel sphingosine kinase-2 inhibitor ABC294640. Endocrinology. 2010; 151: 5124-35. https://doi.org/10.1210/en.2010-0420.
24. Gao P, Peterson YK, Smith RA, Smith CD. Characterization of isoenzyme-selective inhibitors of human sphingosine kinases. PLoS One. 2012; 7: e44543. https://doi.org/10.1371/journal.pone.0044543.
25. Chung IH, Wu TI, Liao CJ, Hu JY, Lin YH, Tai PJ, Lai CH, Lin KH. Overexpression of lipocalin 2 in human cervical cancer enhances tumor invasion. Oncotarget. 2016; 7: 11113-26. https://doi.org/10.18632/oncotarget.7096.
26. Vadas M, Xia P, McCaughan G, Gamble J. The role of sphingosine kinase 1 in cancer: oncogene or non-oncogene addiction? Biochim Biophys Acta. 2008; 1781: 442-7. doi: S1388-1981(08)00123-6.
27. Alemany R, van Koppen CJ, Danneberg K, Ter Braak M, Meyer Zu Heringdorf D. Regulation and functional roles of sphingosine kinases. Naunyn Schmiedebergs Arch Pharmacol. 2007; 374: 413-28. https://doi.org/10.1007/s00210-007-0132-3.
28. Kunkel GT, Maceyka M, Milstien S, Spiegel S. Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat Rev Drug Discov. 2013; 12: 688-702. https://doi.org/10.1038/nrd4099.
29. Liang J, Nagahashi M, Kim EY, Harikumar KB, Yamada A, Huang WC, Hait NC, Allegood JC, Price MM, Avni D, Takabe K, Kordula T, Milstien S, et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell. 2013; 23: 107-20. https://doi.org/10.1016/j.ccr.2012.11.013.
30. Theiss AL. Sphingosine-1-phosphate: driver of NFkappaB and STAT3 persistent activation in chronic intestinal inflammation and colitis-associated cancer. JAKSTAT. 2013; 2: e24150. https://doi.org/10.4161/jkst.24150.
31. Wei LH, Kuo ML, Chen CA, Chou CH, Lai KB, Lee CN, Hsieh CY. Interleukin-6 promotes cervical tumor growth by VEGF-dependent angiogenesis via a STAT3 pathway. Oncogene. 2003; 22: 1517-27. https://doi.org/10.1038/sj.onc.1206226.
32. Page C, Huang M, Jin X, Cho K, Lilja J, Reynolds RK, Lin J. Elevated phosphorylation of AKT and Stat3 in prostate, breast, and cervical cancer cells. Int J Oncol. 2000; 17: 23-8.
33. Kvansakul M, Hinds MG. The Bcl-2 family: structures, interactions and targets for drug discovery. Apoptosis. 2015; 20: 136-50. https://doi.org/10.1007/s10495-014-1051-7.
34. Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014; 15: 49-63. https://doi.org/10.1038/nrm3722.
35. Burton TR, Gibson SB. The role of Bcl-2 family member BNIP3 in cell death and disease: NIPping at the heels of cell death. Cell Death Differ. 2009; 16: 515-23. https://doi.org/10.1038/cdd.2008.185.
36. Zhao X, Sun B, Zhang J, Zhang R, Zhang Q. Short-chain C6 ceramide sensitizes AT406-induced anti-pancreatic cancer cell activity. Biochem Biophys Res Commun. 2016; 479: 166-72. https://doi.org/10.1016/j.bbrc.2016.08.121.
37. Shen J, Xu L, Zhao Q. Perifosine and ABT-737 synergistically inhibit lung cancer cells in vitro and in vivo. Biochem Biophys Res Commun. 2016; 473: 1170-6. https://doi.org/10.1016/j.bbrc.2016.04.035.
38. Li JP, Huang ZJ, Lu XS, Zhou YC, Shao Y, He XP, Chen SR, Wang DD, Qin LS, Sun WH. Pre-clinical characterization of PKC412, a multi-kinase inhibitor, against colorectal cancer cells. Oncotarget. 2016; 7: 77815-24. https://doi.org/10.18632/oncotarget.12802.
39. Jiang Y, Meng Q, Chen B, Shen H, Yan B, Sun B. The small-molecule IAP antagonist AT406 inhibits pancreatic cancer cells in vitro and in vivo. Biochem Biophys Res Commun. 2016; 478: 293-9. https://doi.org/10.1016/j.bbrc.2016.07.011.
40. Yu T, Chen C, Sun Y, Sun H, Li TH, Meng J, Shi X. ABT-737 sensitizes curcumin-induced anti-melanoma cell activity through facilitating mPTP death pathway. Biochem Biophys Res Commun. 2015; 464: 286-91. https://doi.org/10.1016/j.bbrc.2015.06.144.
41. Cang S, Iragavarapu C, Savooji J, Song Y, Liu D. ABT-199 (venetoclax) and BCL-2 inhibitors in clinical development. J Hematol Oncol. 2015; 8: 129. https://doi.org/10.1186/s13045-015-0224-3.
42. Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DC, Hymowitz SG, Jin S, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013; 19: 202-8. https://doi.org/10.1038/nm.3048.
43. Romani AA, Desenzani S, Morganti MM, Baroni MC, Borghetti AF, Soliani P. The BH3-mimetic ABT-737 targets the apoptotic machinery in cholangiocarcinoma cell lines resulting in synergistic interactions with zoledronic acid. Cancer Chemother Pharmacol. 2011; 67: 557-67. https://doi.org/10.1007/s00280-010-1345-6.
44. Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014; 14: 736-46. https://doi.org/10.1038/nrc381844.
45. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009; 9: 798-809. https://doi.org/10.1038/nrc2734.
46. Wang B, Wang XB, Chen LY, Huang L, Dong RZ. Belinostat-induced apoptosis and growth inhibition in pancreatic cancer cells involve activation of TAK1-AMPK signaling axis. Biochem Biophys Res Commun. 2013; 437: 1-6. https://doi.org/10.1016/j.bbrc.2013.05.090.
47. Zhang H, Cai X, Wang Y, Tang H, Tong D, Ji F. microRNA-143, down-regulated in osteosarcoma, promotes apoptosis and suppresses tumorigenicity by targeting Bcl-2. Oncol Rep. 2010; 24: 1363-9.
48. Chen G, Zhu W, Shi D, Lv L, Zhang C, Liu P, Hu W. MicroRNA-181a sensitizes human malignant glioma U87MG cells to radiation by targeting Bcl-2. Oncol Rep. 2010; 23: 997-1003.
49. Silverstein AM, Barrow CA, Davis AJ, Mumby MC. Actions of PP2A on the MAP kinase pathway and apoptosis are mediated by distinct regulatory subunits. Proc Natl Acad Sci U S A. 2002; 99: 4221-6. https://doi.org/10.1073/pnas.072071699.
50. Qiu W, Yang Z, Fan Y, Zheng Q. MicroRNA-613 inhibits cell growth, migration and invasion of papillary thyroid carcinoma by regulating SphK2. Oncotarget. 2016; 7: 39907-15. https://doi.org/10.18632/oncotarget.9530.
51. Altura BM, Shah NC, Shah GJ, Zhang A, Li W, Zheng T, Perez-Albela JL, Altura BT. Short-term Mg deficiency upregulates protein kinase C isoforms in cardiovascular tissues and cells; relation to NF-kB, cytokines, ceramide salvage sphingolipid pathway and PKC-zeta: hypothesis and review. Int J Clin Exp Med. 2014; 7: 1-21.
52. Yao C, Wu S, Li D, Ding H, Wang Z, Yang Y, Yan S, Gu Z. Co-administration phenoxodiol with doxorubicin synergistically inhibit the activity of sphingosine kinase-1 (SphK1), a potential oncogene of osteosarcoma, to suppress osteosarcoma cell growth both in vivo and in vitro. Mol Oncol. 2012; 6: 392-404. https://doi.org/10.1016/j.molonc.2012.04.002.
53. Zheng QY, Jin FS, Yao C, Zhang T, Zhang GH, Ai X. Ursolic acid-induced AMP-activated protein kinase (AMPK) activation contributes to growth inhibition and apoptosis in human bladder cancer T24 cells. Biochem Biophys Res Commun. 2012; 419: 741-7. https://doi.org/10.1016/j.bbrc.2012.02.093.