
INTRODUCTION
The KRAS gene, a Kirsten ras oncogene homolog from the mammalian ras gene family[1], encodes a protein that is a member of the small GTPase superfamily[2]. The KRAS protein exists in an active form (KRAS-ATP) and in an inactive form (KRAS-ADP), both of which are tightly controlled by the guanine nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs)[2-4]. KRAS is a key intracellular protein that activates multiple pathways, including the MAPK and PI3K pathways[5].
Mutant KRAS proteins are GAP-insensitive; therefore, the protein is constitutively GTP-bound, which leads to persistent and independent activation of the downstream effectors[4]. In addition, certain tumor types are associated with specific KRAS mutations, which differ in their carcinogenic potential[6-10]. The high frequency (up to 30%) of RAS mutation in human cancers[11] has stimulated the development of targeted agents against KRAS. Farnesyltransferase inhibitors, which were the first agents used to block the binding of RAS isoforms to the cancer cell membrane, had disappointing results[12-14]. In recent years, the focus has shifted to inhibiting the downstream signaling pathways of RAS. Clinical trials with MEK inhibitors as single agents and/or in combination with cytotoxic agents have been completed[15-19].
Mutations in the KRAS gene, which occur commonly in codons 12, 13, and 61, encode for a single amino-acid substitution. Emerging data demonstrate that each amino-acid substitution may affect the activity of KRAS differently, leading to a different affinity for the various downstream molecules[6]. For example, the G12D mutation encodes for a protein with high affinity for PI3K, while the G12C and G12V mutations activate RAS-like GTPase.
The clinical outcomes of patients with advanced cancer and KRAS mutations by type of codon and amino-acid substitution have not been systematically explored. Therefore, we retrospectively reviewed data from patients with advanced cancer and KRAS mutations who were treated in the Phase I Clinical Trials Program at The University of Texas MD Anderson Cancer Center. Our objectives were to assess the clinical factors, including type of KRAS mutation and treatment, of patients with tumor KRAS mutations and their association with treatment outcomes.
Results
Patient characteristics
Of the 1,781 patients, 365 (20.5%) were found to have a KRAS mutation. The clinical and demographic characteristics and tumor types of the patients with and without a KRAS mutation are shown in Table 1. Briefly, there was no difference in age (median, 59; p=0.62) or sex (male, 47.3% vs. 46.3%, respectively; p= 0.71) between the patients with wild-type and mutated KRAS. However, there was a significant difference in the percentage of patients with KRAS-mutated disease by race (African American 29%, Asian 22%, White 20%; p=0.03). The occurrence of KRAS mutations varied by tumor type, as summarized in Table 1 and Figure 1a. The most commonly analyzed tumor types were colorectal, lung, and ovarian cancer, reflecting the pattern of referrals to the Phase I Clinic (Figure 1b). Molecular analyses were performed on tumor samples from the primary site in 198 (54%) patients and from a metastatic site in 167 (46%) patients.
Table 1: Baseline characteristics of patients with advanced cancer (any tumor type) who were tested for a KRAS mutation
Total Patients, N=1781 |
Wild-type KRAS, N=1416 (%) |
Mutant KRAS, N=365 (%) |
p |
|
Age at Phase I Program presentation (median, range) |
59 (4-90) |
59 (4-90) |
59 (20-84) |
0.62 |
Sex |
0.71 |
|||
Male |
840 |
671 (80) |
169 (20) |
|
Female |
941 |
745 (79) |
196 (21) |
|
Race |
0.03 |
|||
White |
1339 |
1079 (80) |
260 (20) |
|
African American |
174 |
124 (71) |
50 (29) |
|
Hispanic |
162 |
127 (78) |
35 (22) |
|
Asian |
6 |
5 (83) |
1 (17) |
|
Other |
100 |
81 (81) |
19 (19) |
|
Type of cancer |
NA |
|||
Colorectal |
427 |
206 (48) |
221 (52) |
|
Lung |
188 |
140 (74) |
48 (26) |
|
Pancreatic |
47 |
19 (40) |
28 (60) |
|
Endometrial |
54 |
47 (87) |
7 (13) |
|
Ovarian |
156 |
144 (92) |
12 (8) |
|
Other GI* |
131 |
118 (90) |
13 (10) |
|
Other GYN¤ |
85 |
76 (89) |
9 (11) |
|
Thyroid |
36 |
34 (94) |
2 (6) |
|
Other |
657 |
632(96) |
25(4) |
|
Number of metastatic sites |
||||
>2 |
518 |
386 (75) |
132 (25) |
0.0008 |
Number of prior therapies |
||||
Median (range) |
4 (0-15) |
4 (0-15) |
3 (0-12) |
0.53 |
* Gastrointestinal
¤ Gynecological
Patients with KRAS mutations were more likely to have more than two metastatic sites than patients with wild-type KRAS (36% vs. 27%, respectively; p=0.0008). No difference in the number of prior therapies was noted between patients with wild-type and mutated KRAS (p=0.53).
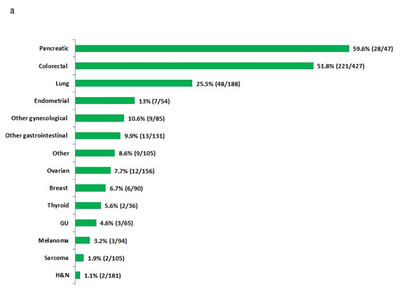
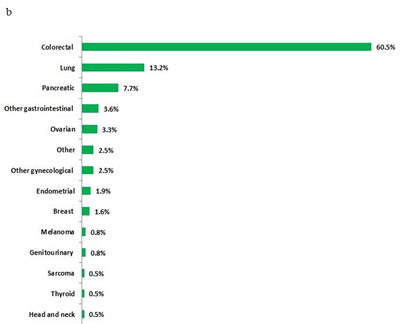
Figure 1: a. Proportion of KRAS mutations by tumor type in patients tested for KRAS mutations. b. Distribution of tumor types in patients with KRAS mutations (n=365)
Specific KRAS mutations and other alterations
The distribution of specific KRAS mutations by tumor type is summarized in Table 2. Overall, mutations in codons 12 and 13 were the most common. The G12D mutation was the most common mutation overall (29%) in all tumor types except for lung cancer, in which G12C and G12V were more frequent. No G12C or codon 13 (G13D, G13C, G13R, and G13V) mutations were noted in the 28 patients with pancreatic cancer.
Table 2: Distribution of KRAS mutations by tumor type
KRAS Mutation |
CRC* (%) |
Lung (%) |
Pancreatic (%) |
Other (%) |
Total |
G12D |
68 (31) |
8 (17) |
12 (43) |
18 (26) |
106 (29) |
G12V |
51 (23) |
10 (21) |
6 (21) |
13 (19) |
80 (22) |
G12C |
13 (6) |
19 (40) |
0 |
9 (13) |
41 (11) |
G13D |
29 (13) |
2 (4) |
0 |
9 (13) |
40 (11) |
G12A |
15 (7) |
4 (8) |
0 |
6 (9) |
25 (7) |
G12S |
14 (7) |
1 (2) |
0 |
0 |
15 (4) |
Q61H |
6 (3) |
2 (4) |
1 (4) |
5 (7) |
14 (4) |
G13C |
2 (1) |
1 (2) |
0 |
1 (1) |
4 (1) |
Other |
16 (7) |
0 |
8 (29) |
7 (10) |
40 (11) |
Unknown |
7(3) |
1 (2) |
1 (4) |
0 |
9 (2) |
Total |
221 |
48 |
28 |
68 |
365 |
*Colorectal cancer
Among the 365 patients with KRAS mutations, 256 (70%) were found to have tumors harboring KRAS mutations without any other molecular alterations. Alternatively, 109 (30%) patients were found to have tumors with ≥1 additional molecular alterations. The distribution of the various additional molecular alterations in both mutated and wild-type KRAS is summarized in Table 3. The number of additional molecular alterations was higher in mutated KRAS than in wild-type KRAS. The most frequently tested alterations were PI3KCA, BRAF, and EGFR; the most common alterations found in both wild-type and mutant KRAS tumors were PI3KCA, p53, and PTEN. A higher percentage of patients with mutated KRAS had PIK3CA mutations compared to patients with wild-type KRAS (24% vs. 10%, respectively; p<0.0001). In contrast, more patients with wild-type KRAS had BRAF mutations compared with patients with mutated KRAS (7% vs. 0.4% respectively; p=0.0002).
Table 3: Distribution of other molecular alterations by KRAS mutational status
Total, n= 1781 |
Wild-type KRAS, n= 1416 |
Mutant KRAS, n= 365 |
P |
|
No. of associated alterations |
||||
Median (range) |
0 (0-9) |
0(0-9) |
1(1-9) |
<0.0001 |
>2 |
118 |
81(6%) |
37 (10%) |
0.0025 |
Type of associated alterations |
||||
PI3KCA |
162/1386 (12%) |
118/1203 (10%) |
44/183 (24%) |
<0.0001 |
P53 |
184/510 (36%) |
155/427 (36%) |
29/83 (35%) |
0.87 |
PTEN |
132/1040 (13%) |
111/881 (13%) |
21/159 (13%) |
0.83 |
BRAF |
77/1370 (6%) |
76/1139 (7%) |
1/231 (0.4%) |
0.0002 |
EGFR |
54/1205 (5%) |
49/1022 (5%) |
5/183 (3%)* |
0.21 |
MET |
41/951(4%) |
32/786 (4%) |
9/165 (5.5%) |
0.43 |
* Four patients had an EGFR mutation and one patient had EGFR overexpression
Outcomes of patients with KRAS mutations
Best response by RECIST, PFS, and survival by specific KRAS mutation, tumor histology, and type of therapy are shown in Table 4. The clinical outcomes varied with the specific mutation (for instance, patients with the G12A mutation appeared to have poorer outcomes than those with other mutations), but the differences were not statistically significant (p=0.07) (Figure 2a).
Table 4: Clinical outcomes of patients with KRAS mutations
Therapy |
Evaluable |
PR+SD≥6 Months (RECIST) |
P |
Median PFS, months |
P |
Median Survival, months |
P |
|
KRAS mutation |
0.60 |
0.57 |
0.07 |
|||||
G12A |
Any |
10 |
0 (0%) |
1.9 |
4.4 |
|||
G12C |
Any |
20 |
2 (10%) |
2.6 |
8.4 |
|||
G12D |
Any |
44 |
7 (16%) |
2.2 |
8.0 |
|||
G12V |
Any |
35 |
7 (20%) |
2.3 |
8.6 |
|||
G13D |
Any |
20 |
3 (15%) |
2.2 |
7.0 |
|||
Other |
Any |
23 |
2 (9%) |
2.1 |
6.8 |
|||
Patients with KRAS mutation alone |
||||||||
Colorectal |
MEK-containing |
12 |
1 (8%) |
0.54 |
1.9 |
.94 |
5.7 |
0.06 |
Other |
73 |
4 (5%) |
2.1 |
7.5 |
||||
Lung |
MEK-containing |
23 |
4 (17%) |
1.00 |
3.2 |
.71 |
7.9 |
0.13 |
Other |
10 |
2 (20%) |
3.2 |
17.6 |
||||
Pancreas |
MEK-containing |
5 |
2 (40%) |
0.07 |
11.0 |
.08 |
14.4 |
0.27 |
Other |
12 |
0 (0%) |
1.9 |
4.5 |
||||
Other tumors |
MEK-containing |
7 |
4 (57%) |
0.34 |
7.7 |
.44 |
12.1 |
0.54 |
Other |
15 |
4 (27%) |
2.2 |
9.8 |
||||
Total KRAS mutation alone |
MEK-containing Other |
47 110 |
11 (23%) 10 (9%) |
0.02 |
2.8 2.1 |
.10 |
8.0 7.5 |
0.59 |
Patients with KRAS and other molecular alterations§ |
0.044 |
0.09 |
0.02 |
|||||
MEK-containing |
9 |
2 (22%) |
3.6 |
Not reached |
||||
Targeted therapy matched the other alterations¤ |
24 |
0 |
2.0 |
4.4 |
||||
Other, non-matched |
33 |
5 (15%) |
2.8 |
6.8 |
||||
Total KRAS* mutation |
MEK-containing vs. Other |
56 167 |
13 (23%) 15 (9%) |
0.005 |
3.3 2.2 |
0.09 |
8.4 7.0 |
0.38 |
§ The total number of patients with KRAS and other molecular alterations was 69 (66 were evaluable for response; all 69 patients were evaluable for PFS and OS).
¤ All the targeted therapy matching the other alterations were inhibitors of the PI3K/AKT/mTOR pathway.
* The total number of patients who received MEK was 60 (56 patients were evaluable for response; however, all 60 patients had data for PFS and OS). The total number of patients who received other therapy was 177 (167 had clinical benefit data; all 176 had data for PFS and OS).
Univariate and multivariate analyses of the prognostic significance of baseline characteristics of patients with KRAS mutations are summarized in Supplemental Table 1. Briefly, previously reported prognostic covariates (lactate dehydrogenase level, serum albumin level, number of metastatic sites, and ECOG performance status) for patients with advanced cancer treated on phase I clinical trials were confirmed in this analysis. Furthermore, the presence of additional mutations was found to be a good prognostic feature (HR: 0.7; p=0.03). Interestingly, after adjustment for other variables, G12V was found to be associated with longer survival compared to G12A (p=0.008; Supplemental Table 1).
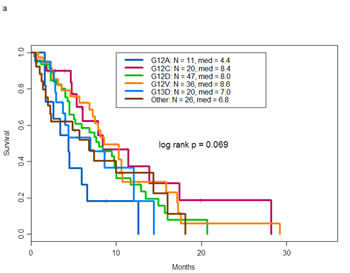
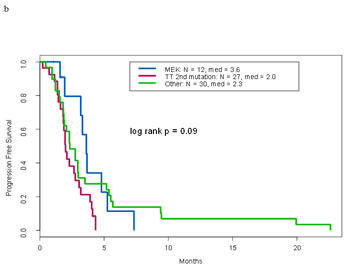
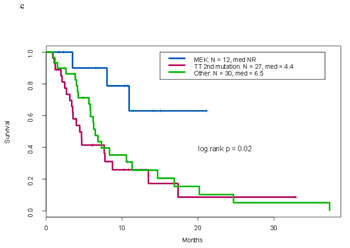
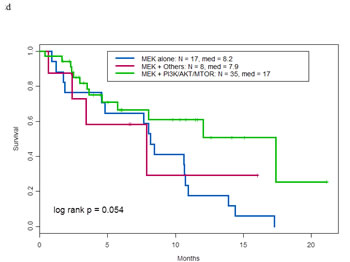
Figure 2: a. Overall survival by KRAS mutation types . b. Progression free survival of patients with KRAS and other molecular aberrations by treatment type. c. Overall survival of patients with KRAS and other molecular aberrations by treatment type. d. Overall survival of patients treated with MEK inhibitor containing trials by type of therapy
Outcomes of patients with KRAS mutations by therapy
The median number of phase I trials that patients with a KRAS mutation were treated on was one (range, 1-5). Clinical outcomes, including clinical benefit (partial response and stable disease ≥ 6 months per RECIST), PFS, and OS, are summarized in Table 4. Of 365 patients with KRAS mutations, 223 were evaluable for response. The other patients were excluded for the following reasons: loco-regional therapy (n=54), decline in performance status (n=28), consent withdrawal (n= 14), loss of follow-up (n=11), early toxicity (n=8), or other (n=27).
Of 157 patients with a KRAS mutation and no additional known alterations, 47 received a MEK inhibitor--containing therapy and 110 received non-MEK inhibitor therapy including another targeted agent, chemotherapy, or both. In subset analyses by tumor type, no significant difference was noted in outcomes between patients treated with a MEK inhibitor--containing therapy and those treated with other therapies. However, a trend towards a higher rate of clinical benefit was noted in patients with pancreatic cancer treated with MEK inhibitor--containing therapy compared to other therapies (p=0.07). When the total number of patients with a KRAS mutation was analyzed regardless of tumor type, the rate of clinical benefit of patients treated with a MEK inhibitor--containing therapy was higher than that of those treated with other therapies (p=0.02).
Of 66 patients with KRAS mutations and other molecular alterations, nine received a MEK inhibitor--containing therapy, 24 received therapies matched with other alterations (all targeted against alterations in the PI3K/AKT/mTOR pathway), and 33 received non-matched therapies. The overall clinical benefit rates were 22%, 0%, and 15%, respectively (p=0.04). In addition, patients who received a MEK inhibitor--containing therapy had longer PFS (p=0.09) and longer overall survival (p=0.02) compared to the other treatment groups (Figure 2b and 2c).
When all 223 patients with a KRAS mutation with or without an additional alteration were included in the analysis, MEK inhibitor-containing therapy had a statistically significantly higher rate of clinical benefit compared to other therapies (23% vs. 9%, respectively; p=0.005). A trend towards longer PFS was also noted in the MEK inhibitor group compared to others (p=0.09). No difference in overall survival was noted (p=0.38).
Of the 60 patients who received a MEK inhibitor--containing therapy, 35 also received a PI3K/AKT/mTOR pathway inhibitor, 17 received a MEK inhibitor only, and eight also received other targeted or cytotoxic therapy. Their respective median OS were 17, 8.2 and 7.9 months (p=0.054) (Figure 2d).
Discussion
To our knowledge, this is the first systematic analysis of the clinical outcomes of patients with KRAS mutations treated on phase I clinical trials. We found that KRAS mutations vary by tumor type. This heterogeneity in amino acid substitution is thought to be associated with differences in the clinical course of the disease. The use of MEK inhibitors in patients with KRAS-mutated advanced cancer was associated with higher clinical benefit rates compared to other therapies.
In our series, the proportion of tested patients with pancreatic cancer with KRAS mutation was similar to that reported in the COSMIC database[20]. However, the proportions of tested patients with colon and lung cancers with KRAS mutation were slightly higher than published data, perhaps in part reflecting the referral pattern in our Phase I Program, which may be affected by the availability of MEK inhibitor trials. This discrepancy can also be explained by differences in the detection method, histology subtypes, disease stage, and other factors.
Our analysis showed that KRAS mutation by specific amino acid substitution varies by tumor type. The specific KRAS mutations in colorectal cancer were similar to those previously reported in a smaller series, in which G12D and G12V were the two most commonly seen mutations [21]. Furthermore, the various specific KRAS mutations seen in lung cancer in our patient group were similar to those previously reported [22].
One of the intriguing findings of our analysis was the association of the KRAS G12V mutation with longer survival compared with the KRAS G12A mutation (p=0.008; Supplemental Table 1). This appears to be in line with preclinical and clinical studies demonstrating that specific KRAS mutations are associated with heterogeneous outcomes [22-24].
In our analysis, the use of MEK inhibitors was associated with an improved clinical benefit rate compared with other therapies in patients with a KRAS mutation (23% vs. 9%; p=0.005) (Table 4). This better disease control with MEK inhibitor-containing trials translated into a trend toward longer PFS in the MEK inhibitor group compared with others (3.3 vs. 2.2 months; p=0.09). However, no statistical difference in overall survival was noted between the two groups (8.4 vs. 7 months, p=0.38), perhaps because patients were treated with subsequent therapies. In addition to the relatively small number of patients, the short PFS seen in our analysis may be explained by the poor-prognosis patient population, the development of resistance to MEK inhibitors, or the development of other driver alterations by the time the patients started on treatment (archival tissue was used for molecular profiling in most patients). Our results are compatible with emerging data demonstrating that MEK inhibitors have promising antitumor activity in patients with KRAS-mutated cancer[18].
We also found that the presence of other molecular alterations in addition to KRAS alterations was associated with longer survival (Supplemental Table 1). The reason is unknown, but the numbers of patients with a KRAS mutation and 1 or ≥2 additional molecular alterations were too small compared to patients with a sole KRAS mutation (72, 37, and 256 patients, respectively) to draw meaningful conclusions. Clinical studies should explore the clinical significance of additional alterations in patients with tumor KRAS mutations. For instance, additional alterations may partially negate the effects of the KRAS mutation.
These results are hypothesis-generating, since they were derived from a retrospective analysis and not from a randomized trial. Therefore, one cannot distinguish between the prognostic value and the predictive value of certain types of KRAS mutations.
Our data also demonstrated that patients with KRAS mutations had a higher proportion of PI3KCA alterations than patients with wild-type KRAS (24% vs. 10%, respectively; p<0.0001) and that patients with wild-type KRAS had a higher proportion of BRAF mutations than patients with KRAS mutations (7% vs. 0.4%, p=0.0002) (Table 3). The coexistence of PI3K and KRAS mutations and the mutual exclusion of KRAS and BRAF mutations had been previously reported in a subset of patients with colorectal cancer [25, 26]. Furthermore, KRAS and EGFR mutations coexisted in 4 patients (2 patients with pancreatic cancer, 1 with CRC and 1 with NSCLC). Various reports showed that KRAS and EGFR mutations are mutually exclusive in patients with lung cancer[27-30]; however, some cases were previously reported to harbor both EGFR and KRAS mutations[31-33].
In both preclinical and clinical settings, the co-existence of both KRAS and PI3KCA alterations was reported to be a negative predictor of response to PI3K inhibitors[26, 34-37] and MEK inhibitors[38]. Both PI3K/AKT/mTOR and RAS/RAF/MEK/ERK are downstream pathways of the KRAS protein[39, 40]. Our data showed that treatment with a MEK inhibitor resulted in better outcomes than treatment with PI3K pathway inhibitors. Other investigators have demonstrated that tumors in xenograft models with both PI3K and KRAS molecular alterations regressed upon MEK inhibition and had a less-pronounced response to PI3K inhibition[41], perhaps suggesting that KRAS mutation may represent the driver mutation. Whether the combination of MEK and PI3K inhibitors is associated with better outcomes than either inhibitor alone is currently being investigated in clinical trials.
Our analysis has several limitations, which are typical for retrospective analyses for patients with any tumor type treated with targeted therapy based on genetic alterations. These limitations include relatively small numbers of patients with specific targetable alterations per tumor type, not including alterations other than KRAS mutations that can activate the MAPK pathway (as such alterations were not tested routinely in CLIA-certified laboratories), and various dose levels and various drugs used in the analyses, resulting in large tested hypotheses that cannot rule out false positive conclusions.
Prospective studies assessing the prognostic significance of various KRAS mutations will help elucidate the role of KRAS in human carcinogenesis. The development of novel agents for treating patients with KRAS mutations and therapeutic strategies that include novel agents combined with other targeted therapies, chemotherapy and/or immunotherapy is urgently needed. In this direction, the National Cancer Institute is launching a large-scale project to develop therapeutic strategies against cancers driven by RAS with the contribution of national scientific leaders and using core facilities and modern technology.
Methods
Patients
We identified 1,781 consecutive patients with advanced cancer who were referred to the Phase I Clinical Trials Program at The University of Texas MD Anderson Cancer between November 2006 and March 2013 and who had undergone tumor molecular analysis for KRAS mutations as part of molecular profiling. Patients referred to the Phase I Clinic were of various ages, had advanced/metastatic cancer that was refractory to standard therapy or had relapsed after standard therapy, or had a tumor for which there was no standard therapy available. All protocols in the Phase I Clinical Trials Program required that participants have evidence of evaluable or measurable disease according to Response Evaluation Criteria in Solid Tumors (RECIST) guidelines[42, 43] and an Eastern Cooperative Oncology Group (ECOG) performance status of 0-2. Additional eligibility criteria varied according to the protocol on which the patient was enrolled. All patients provided written informed consent prior to enrollment onto a trial. All trials, as well as this analysis, were performed with the approval of and in accordance with the guidelines of the MD Anderson Cancer Center Institutional Review Board.
Analysis of molecular alterations
Molecular profiling had been performed in a Clinical Laboratory Improvement Amendments (CLIA)-certified Molecular Diagnostics Laboratory, as previously described[44] or at Foundation Medicine, Inc.
Therapy
Treatment was selected as previously described[44]. Briefly, patients whose tumors had a molecular aberration were preferably treated on a clinical trial with a matched targeted agent, when available. The allocation of patients to investigational treatment varied according to protocol availability, eligibility criteria, histologic diagnosis, the patient’s prior response to therapy, potential toxicity, insurance coverage, and patient preference or physician choice. Patients treated with regional therapy were excluded from the outcome analysis.
The patients with tumors harboring KRAS mutations were treated with MEK inhibitor-containing therapy when a clinical trial was available. Patients were treated in the following clinical trials if the eligibility criteria were met: NCT01138085, NCT00454090, NCT01378377, NCT00687622, NCT01155453, and NCT01337765. If ≥ 2 molecular alterations were present, patients were treated in a matched trial that targeted both alterations, if available.
The types of treatment were categorized as (a) MEK inhibitor--containing therapy or (b) other therapies (targeted therapy with or without cytotoxic agents). If an additional alteration to the KRAS mutation was present, the treatment was categorized as follows: (a) MEK inhibitor--containing therapy, (b) targeted therapy for the additional alteration, when applicable, or (c) other therapies.
Endpoints and statistical methods
Best response was assessed using imaging studies performed every two cycles (1 cycle = 3-4 weeks, depending on the protocol) by an MD Anderson radiologist. Tumor measurements were confirmed independently by a physician in the response assessment clinic within our department using RECIST guidelines applicable at the time of the patient’s response assessment. Clinical benefit (CB) from any therapy was defined as complete response (CR) or partial response (PR) or stable disease (SD) for ≥ 6 months. Progression-free survival (PFS) was measured from the first day of treatment on a clinical trial until the patient came off study because of disease progression or death, whichever came first. Overall survival (OS) was measured from initiation of treatment on a clinical trial until death from any cause or last follow-up.
Patients’ characteristics at the time of presentation in the Phase I Clinic were analyzed using descriptive statistics. Categorical data were described using contingency tables, including counts and percentages. Continuously scaled measures were summarized by median and range. The association between two categorical variables was examined using the chi-squared test. Survival and PFS were estimated using the Kaplan-Meier method, and survival was compared between groups using the two-sided log-rank test. Survival and PFS were analyzed based on the specific type of KRAS mutation and the type of treatment. Statistical analyses were carried out using TIBCO Spotfire S+ 8.2 for Windows (TIBCO Software, Inc.).
Conflict of Interest
No conflicts to disclose: R. Said, Y. Ye, G. S. Falchook, A. Naing, R. Zinner, S. Fu, D. S. Hong, S. Piha-Paul, J. J. Wheler, K. R. Hess, R. Kurzrock, A. M. Tsimberidou
Potential Conflicts
G. R. Blumenschein reports receiving research funding through Exelixis, Genentech, Mediumune, Bayer, GSK, Immunogen, Xcovery, Merck, BMS, and Novartis. G. Palmer reports salary compensation from Foundation Medicine. F. Janku reports receiving research funding through Trovagene, Transgenomic.
References
1. Kirsten WH, Mayer LA. Morphologic responses to a murine erythroblastosis virus. J Natl Cancer Inst. 1967;39:311-35.
2. Vigil D, Cherfils J, Rossman KL, Der CJ. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat Rev Cancer. 2010;10:842-57.
3. Mitin N, Rossman KL, Der CJ. Signaling interplay in Ras superfamily function. Curr Biol. 2005;15:R563-74.
4. Roberts PJ, Stinchcombe TE. KRAS mutation: should we test for it, and does it matter? J Clin Oncol. 2013;31:1112-21.
5. Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291-310.
6. Ihle NT, Byers LA, Kim ES, Saintigny P, Lee JJ, Blumenschein GR, et al. Effect of KRAS oncogene substitutions on protein behavior: implications for signaling and clinical outcome. J Natl Cancer Inst. 2012;104:228-39.
7. Span M, Moerkerk PT, De Goeij AF, Arends JW. A detailed analysis of K-ras point mutations in relation to tumor progression and survival in colorectal cancer patients. Int J Cancer. 1996;69:241-5.
8. Al-Mulla F, Milner-White EJ, Going JJ, Birnie GD. Structural differences between valine-12 and aspartate-12 Ras proteins may modify carcinoma aggression. J Pathol. 1999;187:433-8.
9. Al-Mulla F, MacKenzie EM. Differences in in vitro invasive capacity induced by differences in Ki-Ras protein mutations. J Pathol. 2001;195:549-56.
10. Winder T, Mundlein A, Rhomberg S, Dirschmid K, Hartmann BL, Knauer M, et al. Different types of K-Ras mutations are conversely associated with overall survival in patients with colorectal cancer. Oncol Rep. 2009;21:1283-7.
11. Fernandez-Medarde A, Santos E. Ras in cancer and developmental diseases. Genes Cancer. 2011;2:344-58.
12. Cohen SJ, Ho L, Ranganathan S, Abbruzzese JL, Alpaugh RK, Beard M, et al. Phase II and pharmacodynamic study of the farnesyltransferase inhibitor R115777 as initial therapy in patients with metastatic pancreatic adenocarcinoma. J Clin Oncol. 2003;21:1301-6.
13. Van Cutsem E, van de Velde H, Karasek P, Oettle H, Vervenne WL, Szawlowski A, et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J Clin Oncol. 2004;22:1430-8.
14. Macdonald JS, McCoy S, Whitehead RP, Iqbal S, Wade JL, 3rd, Giguere JK, et al. A phase II study of farnesyl transferase inhibitor R115777 in pancreatic cancer: a Southwest oncology group (SWOG 9924) study. Invest New Drugs. 2005;23:485-7.
15. Adjei AA, Cohen RB, Franklin W, Morris C, Wilson D, Molina JR, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol. 2008;26:2139-46.
16. Banerji U, Camidge DR, Verheul HM, Agarwal R, Sarker D, Kaye SB, et al. The first-in-human study of the hydrogen sulfate (Hyd-sulfate) capsule of the MEK1/2 inhibitor AZD6244 (ARRY-142886): a phase I open-label multicenter trial in patients with advanced cancer. Clin Cancer Res. 2010;16:1613-23.
17. Hainsworth JD, Cebotaru CL, Kanarev V, Ciuleanu TE, Damyanov D, Stella P, et al. A phase II, open-label, randomized study to assess the efficacy and safety of AZD6244 (ARRY-142886) versus pemetrexed in patients with non-small cell lung cancer who have failed one or two prior chemotherapeutic regimens. J Thorac Oncol. 2010;5:1630-6.
18. Janne PA, Shaw AT, Pereira JR, Jeannin G, Vansteenkiste J, Barrios C, et al. Selumetinib plus docetaxel for KRAS-mutant advanced non-small-cell lung cancer: a randomised, multicentre, placebo-controlled, phase 2 study. Lancet Oncol. 2013;14:38-47.
19. Falchook GS, Lewis KD, Infante JR, Gordon MS, Vogelzang NJ, DeMarini DJ, et al. Activity of the oral MEK inhibitor trametinib in patients with advanced melanoma: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13:782-9.
20. http://cancer.sanger.ac.uk/cosmic/gene/overview?search=&ln=KRAS.
21. Dienstmann R, Serpico D, Rodon J, Saura C, Macarulla T, Elez E, et al. Molecular profiling of patients with colorectal cancer and matched targeted therapy in phase I clinical trials. Mol Cancer Ther. 2012;11:2062-71.
22. Garassino MC, Marabese M, Rusconi P, Rulli E, Martelli O, Farina G, et al. Different types of K-Ras mutations could affect drug sensitivity and tumour behaviour in non-small-cell lung cancer. Ann Oncol. 2011;22:235-7.
23. Janakiraman M, Vakiani E, Zeng Z, Pratilas CA, Taylor BS, Chitale D, et al. Genomic and biological characterization of exon 4 KRAS mutations in human cancer. Cancer Res. 2010;70:5901-11.
24. Tejpar S, Celik I, Schlichting M, Sartorius U, Bokemeyer C, Van Cutsem E. Association of KRAS G13D tumor mutations with outcome in patients with metastatic colorectal cancer treated with first-line chemotherapy with or without cetuximab. J Clin Oncol. 2012;30:3570-7.
25. Li HT, Lu YY, An YX, Wang X, Zhao QC. KRAS, BRAF and PIK3CA mutations in human colorectal cancer: relationship with metastatic colorectal cancer. Oncol Rep. 2011;25:1691-7.
26. Garrido-Laguna I, Hong DS, Janku F, Nguyen LM, Falchook GS, Fu S, et al. KRASness and PIK3CAness in patients with advanced colorectal cancer: outcome after treatment with early-phase trials with targeted pathway inhibitors. PLoS One. 2012;7:e38033.
27. Pao W, Wang TY, Riely GJ, Miller VA, Pan Q, Ladanyi M, et al. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005;2:e17.
28. Kosaka T, Yatabe Y, Endoh H, Kuwano H, Takahashi T, Mitsudomi T. Mutations of the epidermal growth factor receptor gene in lung cancer: biological and clinical implications. Cancer Res. 2004;64:8919-23.
29. Shigematsu H, Lin L, Takahashi T, Nomura M, Suzuki M, Wistuba, II, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst. 2005;97:339-46.
30. Tam IY, Chung LP, Suen WS, Wang E, Wong MC, Ho KK, et al. Distinct epidermal growth factor receptor and KRAS mutation patterns in non-small cell lung cancer patients with different tobacco exposure and clinicopathologic features. Clin Cancer Res. 2006;12:1647-53.
31. Eberhard DA, Johnson BE, Amler LC, Goddard AD, Heldens SL, Herbst RS, et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol. 2005;23:5900-9.
32. Han SW, Kim TY, Jeon YK, Hwang PG, Im SA, Lee KH, et al. Optimization of patient selection for gefitinib in non-small cell lung cancer by combined analysis of epidermal growth factor receptor mutation, K-ras mutation, and Akt phosphorylation. Clin Cancer Res. 2006;12:2538-44.
33. Zhu CQ, da Cunha Santos G, Ding K, Sakurada A, Cutz JC, Liu N, et al. Role of KRAS and EGFR as biomarkers of response to erlotinib in National Cancer Institute of Canada Clinical Trials Group Study BR.21. J Clin Oncol. 2008;26:4268-75.
34. Di Nicolantonio F, Arena S, Tabernero J, Grosso S, Molinari F, Macarulla T, et al. Deregulation of the PI3K and KRAS signaling pathways in human cancer cells determines their response to everolimus. J Clin Invest. 2010;120:2858-66.
35. Kim A, Lee JE, Lee SS, Kim C, Lee SJ, Jang WS, et al. Coexistent mutations of KRAS and PIK3CA affect the efficacy of NVP-BEZ235, a dual PI3K/MTOR inhibitor, in regulating the PI3K/MTOR pathway in colorectal cancer. Int J Cancer. 2013;133:984-96.
36. Janku F, Tsimberidou AM, Garrido-Laguna I, Wang X, Luthra R, Hong DS, et al. PIK3CA mutations in patients with advanced cancers treated with PI3K/AKT/mTOR axis inhibitors. Mol Cancer Ther. 2011;10:558-65.
37. Janku F, Hong DS, Fu S, Piha-Paul SA, Naing A, Falchook GS, et al. Assessing PIK3CA and PTEN in Early-Phase Trials with PI3K/AKT/mTOR Inhibitors. Cell Rep. 2014;6:377-87.
38. Wee S, Jagani Z, Xiang KX, Loo A, Dorsch M, Yao YM, et al. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res. 2009;69:4286-93.
39. Saxena N, Lahiri SS, Hambarde S, Tripathi RP. RAS: target for cancer therapy. Cancer Invest. 2008;26:948-55.
40. Repasky GA, Chenette EJ, Der CJ. Renewing the conspiracy theory debate: does Raf function alone to mediate Ras oncogenesis? Trends Cell Biol. 2004;14:639-47.
41. Hofmann I, Weiss A, Elain G, Schwaederle M, Sterker D, Romanet V, et al. K-RAS mutant pancreatic tumors show higher sensitivity to MEK than to PI3K inhibition in vivo. PLoS One. 2012;7:e44146.
42. Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205-16.
43. Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228-47.
44. Tsimberidou AM, Iskander NG, Hong DS, Wheler JJ, Falchook GS, Fu S, et al. Personalized medicine in a phase I clinical trials program: the MD Anderson Cancer Center initiative. Clin Cancer Res. 2012;18:6373-83.