
INTRODUCTION
Epithelial ovarian carcinomas (EOCs) are composed of a group of heterogeneous subtypes classified by their histology and the degree of epithelial proliferation and invasion [1]. Among these subtypes, clear cells carcinoma (CCC) and endometrioid carcinoma (EC) share many similarities in their tumor behavior, clinical features, and pathology. Endometriosis (ES) is found in 15%-20% of CCC and EC, and is associated with 2-3 fold increase of EOC incidence [2] [3]. The atypical ES, characterized by large nuclei and increased nuclear-cytoplasmic ratio, composes 8% of ES [4] and is found in 36% and 23% in CCC and EC, respectively [5]. Atypical ES was shown direct continuity with CCC and EC and is considered to be a precancerous transformation process of CCC and EC [6]. These clinical observations indicate a close relationship between ES and CCC/EC, and support the hypothesis of endometriosis associated ovarian carcinoma (EAOC). Recent genomic studies have greatly increased our understanding of the molecular landscape of EOC [7] [8]. However, the molecular pathogenesis involving in the malignant transformation from ES to EAOC is still unclear.
The Sampson’s theory of retrograde menstruation is the most widely accepted theory on the pathogenesis of ES [9]. However, there exists a paradox: although retrograde menstruation is widely encountered among reproductive women, the incidence of ES is relatively uncommon compared with the manifestation of retrograde menstruation experienced by most of the women in the same group [10]. One hypothesis is that in comparison to women without ES, the women that develop ES have a defective immune system unable to recognize the endometrial fragments within the pelvic cavity. Inflammatory responses play key roles at different stages of tumor development, including initiation, promotion, malignant conversion, invasion, and metastasis. Inflammation also disturbs immune surveillance and tumor responses to therapy. Immune cells that infiltrate tumors involve in a dynamic crosstalk with cancer cells and some of the molecular consequences that mediate this dialog have been identified [11].
The Gene Ontology (GO) [2] is the primary tool to annotate the gene products and enable the functional interpretation of the genomic data. It defines relatively comprehensive human functionome like biological processes, molecular functions and cellular components. This gene set regularity (GSR) model has been successfully utilized to demonstrate the dualistic model of ovarian carcinogenesis [12], and to quantify the function deterioration of the FIGO staging I to IV for serous ovarian carcinoma [13]. In this study, we investigated the dysregulated functions involving in the malignant transformation from ES to EAOC with GSR model by analyzing the functionomes consisted of 5917 GO defined functions of ES, CCC and EC with the DNA microarray datasets downloaded from the publicly available database. The results demonstrated that the immune/inflammation related functions were crucial elements involving in the transformation of EAOC. Among these dysregulated immune/inflammation related functions, the inflammasome complex (G0:0061702) is noticeable because it is postulated to become activated during malignant transformation of tumorigenesis and plays diverse roles in cancer promotion [14]. To study the role of inflammasome complex in the malignant transformation from ES to EACO, we explored the expressions of the inflammasome related genes by carrying out an integrative analysis with the same DNA microarray expression datasets. The results revealed several inflammasome complex and inflammasome-related genes (NLR Family Pyrin Domain Containing 3 (NLRP3), Absent In Melanoma 2 (AIM2), PYD And CARD Domain Containing (PYCARD), NLR Family Apoptosis Inhibitory Protein (NAIP), Tumor Necrosis Factor (TNF), Toll Like Receptor 1 (TLR1), Toll Like Receptor 7 (TLR7), Toll Interacting Protein (TOLLIP), and NFKB Inhibitor Alpha (NFKBIA)) differentially expressed in ES, CCC and EC, and significantly correlating with poor progression-free survival. The expression levels of these identified genes were confirmed by immunohistochemistrical staining in ES, CCC and EC specimens. These findings are vital to clarify the role of inflammasome in EAOC carcinogenesis.
RESULTS
Workflow of the study
We utilize a two-stage strategy to discover the gene signatures involving in the transformation of EAOC, that is, starting with investigating the functionomes of ES, CCC and EC with the GSR model, and then followed by extracting the differentially expressed genes (DEGs) involving in these deregulated functions with integrative analysis. During the first stage, the GSR model was applied to find out the deregulated function related to the malignant transformation, it consisted with 4 steps as displayed on the left side of Figure 1A. First, extraction of expression profiles of gene set elements. The gene expression profiles for a given gene set were extracted from the publicly available microarray datasets according to the gene elements defined by each gene set. Second, computing GSR indices. The extracted gene expression profiles were converted to quantified functions based on the gene expression orderings of the gene elements in each gene set defined by the 5917 GO terms. This quantified function, i.e. the GSR index, is a measurement of the expression regularity of the genes in that gene set. The quantified functions range from 0 to 1; 1 represented unchanged regularity in a given gene set between the case and the most common gene expression orderings in the normal controls, while 0 represented the most chaotic state of the gene set regularity. Third, validating the functional regularity patterns. The informativeness of the functionome consisted of the 5917 GSR indices is evaluated by the accuracies of classification and prediction by the machine learning. Finally, investigation of EAOC pathogenesis. In this step, the key deregulated functions involving in the malignant transformation of ES to CCC or EC are investigated by a secession of analytic procedures. During the second stage (right side of Figure 1A), an integrative analysis of DNA microarray was applied to detect the differentially expressed genes. Then the principle genes involving in the malignant transformation of EAOC were filtered from those genes related to the deregulated functions detected by the first stage of analysis.
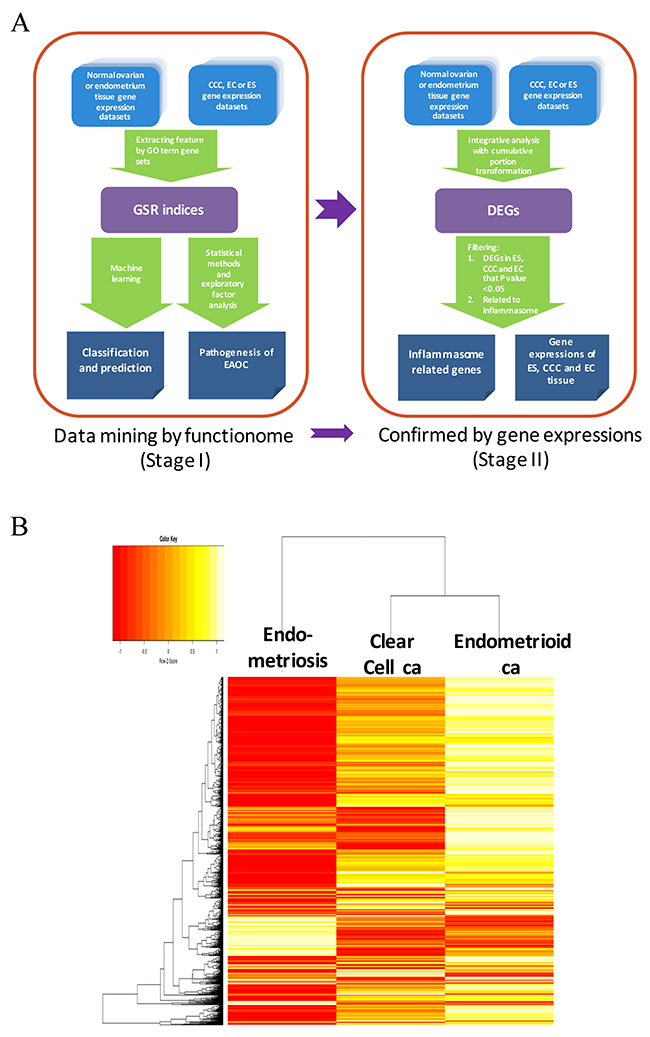
Figure 1: Work flow of the two-stage strategy to discover gene signatures for EAOC. (A) Workflow of the gene set regularity model. The gene set regularity (GSR) index was computed by converting the gene expression ordering of gene elements in a gene set through the Gene Ontology (GO) term or canonical pathway databases. The informativeness of the GSR index was assessed by the accuracy of recognition, classification, and prediction by machine learning using binary or multiclass classifications. Functionome analyses were carried out to investigate the pathogenesis of endometriosis (ES), clear cell carcinoma (CCC), endometrioid ca (EC) and endometriosis-associated ovarian carcinoma (EAOC) by statistical methods, hierarchical clustering, and exploratory factor analysis. (B) Heatmaps and dendrogram for the three diseases. The dendrogram (left side of the heatmap) showed the relationship of the three diseases. When displayed on the heatmap, each of the three diseases computed through either the GO term gene sets showed a distinct pattern. However, the patterns were more similar between CCC and EC.
The microarray gene expression profiles of ES, CCC, EC and the normal control samples were downloaded from the GEO database, including 107 ES, 156 normal endometrium controls, 85 CCC, 90 EC, and 136 normal ovarian tissue control samples (Table 1). These samples data were collected from 39 datasets containing 7 different DNA microarray platforms without missing data. The detailed sample information, including the DNA microarray platforms, dataset series, and accession number, were listed in Supplementary Table 1. Because different genes utilized in different microarray platforms, a total of 5905 common gene sets were utilized finally for the GSR model in this study. Table 1 displays the sample number, mean and standard deviation (SD) of the GSR indices for the three diseases and the normal tissue controls. The means of GSR indices for the three diseases were significantly lower than the controls, indicating that the functions are generally deteriorated in the ES, CCC or EC when comparing with the normal control group. The informativeness of the GSR indices was evaluated by the accuracies of classification and prediction for the functional regularity patterns of the three diseases. Supervised classification was performed by support vector machine (SVM) and the performance was assessed by the accuracies of the binary and multiclass classification of the GSR matrices computed from the total samples through 5905 GO term gene sets. The performance was tested by five-fold cross validation. The results showed up to 100% accuracies of binary classification (case vs control). The area under curves (AUCs) ranged from 0.98 to 1 (Table 2). The accuracies of multiclass classification among the three diseases and the normal control group were 98.68%. The high accuracies indicated that the GSR indices could provide sufficient information for the machine learning to recognize and undergo adequate recognition and classification. It also revealed distinct functional regularity patterns of ES, CCC and EC, which can be applied to the molecular classification among ES, CCC and EC. Unsupervised classification by the hierarchical clustering was performed to uncover the relationship between the three diseases (Figure 1B). The clustering data revealed a relatively close relationship between CCC and EC, and the detailed dendrogram of the GO terms were shown in Supplementary Figure 1. The heatmap (Figure 1B) also showed similar patterns between CCC and EC. There were many overlapped deregulated molecular functions and biological processes between CCC and EC, indicating a close etiology of these two types of cancer.
Table 1: Sample number and mean of the gene set regularity indexes for each group
Disease |
Case |
Control |
Case Mean (SD) |
Control Mean (SD) |
P value |
|---|---|---|---|---|---|
ES |
107 |
156 |
0.6299(0.0832) |
0.6715(0.0825) |
<2.2x10-16 |
CCC |
85 |
136 |
0.6304(0.1034) |
0.6532(0.1120) |
<2.2x10-16 |
EC |
90 |
136 |
0.6466(0.0.1051) |
0.6539(0.1116) |
<2.2x10-16 |
Table 2: Accuracies of the binary and multiclass classification and prediction by machine learning
Gene set |
Classification |
Group |
Sensitivity(SD) |
Specificity(SD) |
Accuracy(SD) |
AUC |
|---|---|---|---|---|---|---|
GO term |
Binary |
ES |
1.0000(0.0000) |
1.0000(0.0000) |
1.0000(0.0000) |
1.0000 |
CCC |
1.0000(0.0000) |
1.0000(0.0000) |
1.0000(0.0000) |
1.0000 |
||
EC |
0.9597(0.0303) |
0.9965(0.0109) |
0.9800(0.0163) |
0.9768 |
||
Multiclass |
ES-CCC-EC- control |
NA |
NA |
0.9868(0.0046) |
NA |
Discovering the deregulated functions involving in the malignant transformation of EAOC by mining the DNA microarray gene expression data
We used the set operations to identify commonly deregulated functions from the top 1000 significantly deregulated GO terms among ES, CCC and EC. There were 65 deregulated functions in common (Supplementary Table 2), revealing the possible etiology of malignant transformation of EAOC. Among the 65 deregulated functions, up to 16.9% (11/65) deregulated functions were relating to inflammation/immune, showing the important roles of inflammation/immune playing on the malignant transformation of EAOC. We then focused on the immune/inflammation related functions and extracted them from the functionomes of ES, CCC and EC using the following keys: ‘immune system process’ (GO:0002376), ‘response to stress’ (GO:0006950), ‘cytoplasmic part’ (GO:0044444), and ‘cytokine production’ (GO:0001816) to collect all of their offspring. Table 3 displayed the 114 most significantly deregulated immune/inflammation related GO terms in the three diseases. These immune/inflammation related GO terms were predominately associated with deregulated cytokines production, signaling pathways and activation of immune cells. We carried out the set operations with the 114 GO terms to discover the coexisting immune/inflammation related GO terms among ES, CCC and EC, and displayed the results on the Venn diagram in Figure 2A. The detailed information of the 114 genes were available in Supplementary Table 3. The CCC and EC shared the most number of overlapping deregulated GO terms, accounting for 50% (57/114) of the coexisting deregulated GO terms, indicating the similar immune pathogenesis between these two cancers. There were 9 commonly deregulated GO terms among the ES, CCC and EC as shown on the Figure 2B.
Table 3: The 114 most deregulated immune/inflammation related Gene Ontology terms for the three diseases ranked by the P values
Endometriosis |
Clear cell carcinoma |
Endometrioid carcinoma |
|
|---|---|---|---|
1 |
Golgi cisterna |
Negative regulation of antigen receptor mediated signaling pathway |
Regulation of B cell receptor signaling pathway |
2 |
Golgi stack |
Endoplasmic reticulum quality control compartment |
Wound healing spreading of epidermal cells |
3 |
Positive regulation of interleukin 2 biosynthetic process |
Regulation of B cell receptor signaling pathway |
Regulation of Toll like receptor 4 signaling pathway |
4 |
Interferon gamma mediated signaling pathway |
Regulation of natural killer cell activation |
Regulation of oxidative stress induced neuron death |
5 |
Response to interferon gamma |
Mast cell granule |
Lamellar body |
6 |
Cellular response to interferon gamma |
Positive regulation of endoplasmic reticulum unfolded protein response |
Smooth endoplasmic reticulum |
7 |
Adaptive immune response based on somatic recombination of immune receptors built from immunoglobulin superfamily domains |
Platelet alpha granule lumen |
Negative regulation of antigen receptor mediated signaling pathway |
8 |
Positive regulation of interleukin 8 production |
Lysosomal lumen |
T cell differentiation involved in immune response |
9 |
Intrinsic component of endoplasmic reticulum membrane |
Smooth endoplasmic reticulum |
Angiogenesis involved in wound healing |
10 |
Regulation of interleukin 8 production |
Regulation of Toll like receptor 4 signaling pathway |
Regulation of IRE1 mediated unfolded protein response |
11 |
Lymphocyte mediated immunity |
Platelet alpha granule |
Endoplasmic reticulum quality control compartment |
12 |
Golgi cisterna membrane |
Vacuolar lumen |
Methylosome |
13 |
Recycling endosome |
Positive regulation of macrophage activation |
Interferon gamma production |
14 |
Regulation of T helper cell differentiation |
Recycling endosome |
Regulation of response to interferon gamma |
15 |
Clathrin coated endocytic vesicle |
Secretory granule lumen |
Intrinsic component of Golgi membrane |
16 |
Lytic vacuole membrane |
Regulation of antigen receptor mediated signaling pathway |
Positive regulation of endoplasmic reticulum unfolded protein response |
17 |
Regulation of cellular response to heat |
Humoral immune response |
Autophagosome |
18 |
Late endosome |
ER to Golgi transport vesicle |
Positive regulation of transcription from RNA polymerase II promoter in response to stress |
19 |
Stress activated protein kinase signaling cascade |
Gamma tubulin complex |
Regulation of endoplasmic reticulum unfolded protein response |
20 |
Autophagosome |
IRE1 mediated unfolded protein response |
Axon regeneration |
21 |
Recycling endosome membrane |
Endoplasmic reticulum Golgi intermediate compartment |
Regulation of T cell chemotaxis |
22 |
Leukocyte mediated immunity |
Cellular response to topologically incorrect protein |
Humoral immune response |
23 |
Smooth endoplasmic reticulum |
Mast cell mediated immunity |
Trans Golgi network membrane |
24 |
Positive regulation of t helper cell differentiation |
Myeloid leukocyte mediated immunity |
Trans Golgi network transport vesicle membrane |
25 |
Humoral immune response mediated by circulating immunoglobulin |
Mast cell activation |
Mature B cell differentiation |
26 |
JNK cascade |
Intrinsic component of mitochondrial outer membrane |
Vesicle coat |
27 |
Positive regulation of cd4 positive alpha beta T cell activation |
Lamellar body |
Complement activation |
28 |
Endosomal part |
Spleen development |
Neutrophil mediated immunity |
29 |
Response to heat |
Regulation of endoplasmic reticulum unfolded protein response |
Microbody lumen |
30 |
Vacuolar membrane |
Regulation of IRE1 mediated unfolded protein response |
Adaptive immune response based on somatic recombination of immune receptors built from immunoglobulin superfamily domains |
31 |
Cellular response to heat |
Autophagosome |
Clathrin coat of trans Golgi network vesicle |
32 |
Regulation of cd4 positive alpha beta T cell activation |
Negative regulation of T cell proliferation |
Humoral immune response mediated by circulating immunoglobulin |
33 |
Endosome |
Toll like receptor 4 signaling pathway |
Cd4 positive alpha beta T cell activation |
34 |
Vacuolar part |
ER associated ubiquitin dependent protein catabolic process |
Trans Golgi network transport vesicle |
35 |
Antigen processing and presentation |
Positive regulation of transcription from RNA polymerase ii promoter in response to stress |
Cellular response to heat |
36 |
Endosome lumen |
Negative regulation of T cell differentiation |
Regulation of acute inflammatory response |
37 |
Microtubule organizing center part |
Cellular senescence |
Thymic T cell selection |
38 |
Regulation of interleukin 2 biosynthetic process |
Natural killer cell mediated immunity |
B cell mediated immunity |
39 |
Thymic T cell selection |
Regulation of p38mapk cascade |
Positive regulation of response to oxidative stress |
40 |
B cell receptor signaling pathway |
Adaptive immune response based on somatic recombination of immune receptors built from immunoglobulin superfamily domains |
Intrinsic component of mitochondrial outer membrane |
41 |
Positive regulation of B cell proliferation |
Wound healing spreading of epidermal cells |
Myeloid leukocyte mediated immunity |
42 |
Golgi membrane |
Trans Golgi network transport vesicle |
Cytokine production involved in immune response |
43 |
Regulation of humoral immune response |
ERAD pathway |
Sarcoplasmic reticulum membrane |
44 |
Regulation of cytokine biosynthetic process |
Positive regulation of natural killer cell mediated immunity |
Regulation of humoral immune response |
45 |
Myeloid leukocyte differentiation |
Axon regeneration |
Positive regulation of interleukin 10 production |
46 |
Lymphocyte costimulation |
Neutrophil mediated immunity |
Regulation of interleukin 13 production |
47 |
Complement activation |
Regulation of DNA repair |
Defense response to gram positive bacterium |
48 |
Perinuclear region of cytoplasm |
Trans Golgi network transport vesicle membrane |
Mast cell granule |
49 |
B cell mediated immunity |
Leukocyte mediated immunity |
Base excision repair |
50 |
Positive regulation of chemokine production |
COPI vesicle coat |
Derlin 1 retrotranslocation complex |
51 |
Regulation of chemokine production |
Base excision repair |
Complement activation alternative pathway |
52 |
Positive regulation of DNA damage response signal transduction by p53 class mediator |
Perk mediated unfolded protein response |
Clathrin vesicle coat |
53 |
Endocytic vesicle |
Cellular response to heat |
Regulation of tumor necrosis factor biosynthetic process |
54 |
Cytosolic part |
Vesicle coat |
Positive regulation of transcription from RNA polymerase ii promoter in response to endoplasmic reticulum stress |
55 |
Vacuole |
Cytoplasmic mRNA processing body |
Gamma tubulin complex |
56 |
Golgi apparatus |
Cellular extravasation |
COPI coated vesicle membrane |
57 |
Positive regulation of cytokine production involved in immune response |
Endosome lumen |
Endosome lumen |
58 |
Inflammasome complex |
Regulation of natural killer cell mediated immunity |
ER to Golgi transport vesicle membrane |
59 |
Cellular response to stress |
ER to Golgi transport vesicle membrane |
Response to pain |
60 |
Regulation of defense response to virus by host |
Blood coagulation fibrin clot formation |
Negative regulation of interferon gamma production |
61 |
Microbody membrane |
Positive regulation of interleukin 6 secretion |
Negative regulation of cytokine biosynthetic process |
62 |
Regulation of interleukin 2 production |
Negative regulation of hemopoiesis |
COPI vesicle coat |
63 |
Golgi apparatus part |
T cell differentiation involved in immune response |
Positive regulation of acute inflammatory response |
64 |
Secretory granule |
Defense response to gram positive bacterium |
Negative regulation of T cell differentiation |
65 |
Cytosolic large ribosomal subunit |
Intrinsic component of mitochondrial membrane |
Error prone translesion synthesis |
66 |
Positive regulation of interleukin 2 production |
Positive regulation of leukocyte migration |
Negative regulation of DNA repair |
67 |
Regulation of cellular response to stress |
Positive regulation of leukocyte chemotaxis |
Negative regulation of response to endoplasmic reticulum stress |
68 |
Innate immune response |
Platelet aggregation |
Positive regulation of vascular endothelial growth factor production |
69 |
Positive regulation of immune response |
Clathrin coat |
Regulation of lymphocyte chemotaxis |
70 |
Adaptive immune response |
Response to misfolded protein |
Regulation of cellular response to hypoxia |
71 |
Intrinsic component of Golgi membrane |
Positive regulation of interleukin 10 production |
Regulation of macrophage activation |
72 |
Large ribosomal subunit |
Clathrin coat of trans Golgi network vesicle |
Defense response to fungus |
73 |
Vacuolar lumen |
Regulation of double strand break repair |
Zymogen granule |
74 |
Macrophage differentiation |
Regulation of leukocyte chemotaxis |
Cell cortex region |
75 |
Activation of immune response |
Regulation of macrophage activation |
Regulation of double strand break repair |
76 |
Regulation of interferon beta production |
Regulation of leukocyte migration |
Lysosomal lumen |
77 |
Leukocyte migration |
Regulation of endoplasmic reticulum stress induced intrinsic apoptotic signaling pathway |
Recycling endosome membrane |
78 |
Positive regulation of response to DNA damage stimulus |
Clathrin vesicle coat |
Toll like receptor 4 signaling pathway |
79 |
Defense response to otherorganism |
Myeloid cell activation involved in immune response |
Retrograde protein transport ER to cytosol |
80 |
Transport vesicle |
Inflammasome complex |
Positive regulation of macrophage activation |
81 |
Positive regulation of immune effector process |
Granulocyte differentiation |
Innate immune response in mucosa |
82 |
Response to water deprivation |
Non recombinational repair |
Granulocyte differentiation |
83 |
Mitochondrion |
Antimicrobial humoral response |
Regulation of vascular endothelial growth factor production |
84 |
Myeloid cell differentiation |
Regulation of removal of superoxide radicals |
Inflammasome complex |
85 |
Regulation of macrophage activation |
Autophagosome membrane |
Endocytic vesicle lumen |
86 |
Negative regulation of platelet activation |
Negative regulation of wound healing |
Negative regulation of humoral immune response |
87 |
Regulation of response to DNA damage stimulus |
Lymphocyte mediated immunity |
Regulation of cellular extravasation |
88 |
Immune effector process |
Negative regulation of T cell receptor signaling pathway |
Outer mitochondrial membrane protein complex |
89 |
Endocytic vesicle membrane |
Regulation of neutrophil migration |
Regulation of fibrinolysis |
90 |
Negative T cell selection |
Rough endoplasmic reticulum membrane |
B cell proliferation |
91 |
Immune system process |
Pre autophagosomal structure |
Regulation of interleukin 8 secretion |
92 |
Regulation of defense response to virus |
Defense response to fungus |
Hematopoietic stem cell proliferation |
93 |
Negative regulation of innate immune response |
Positive regulation of response to oxidative stress |
PERK mediated unfolded protein response |
94 |
Regulation of immune effector process |
Intrinsic component of Golgi membrane |
Regulation of megakaryocyte differentiation |
95 |
Secretory vesicle |
Error prone translesion synthesis |
Response to misfolded protein |
96 |
Positive regulation of B cell activation |
Negative regulation of lymphocyte differentiation |
Positive regulation of interleukin 8 secretion |
97 |
Early endosome membrane |
Regulation of cellular extravasation |
Mast cell mediated immunity |
98 |
Cytosolic ribosome |
Golgi lumen |
Response to immobilization stress |
99 |
Cellular response to glucose starvation |
Recycling endosome membrane |
Negative regulation of alpha beta T cell activation |
100 |
Centriole |
Regulation of Toll like receptor signaling pathway |
Mature B cell differentiation involved in immune response |
101 |
Regulation of alpha beta T cell differentiation |
Hyperosmotic response |
Mitotic G2 DNA damage checkpoint |
102 |
Regulation of immune response |
COPI coated vesicle membrane |
Negative regulation of alpha beta T cell differentiation |
103 |
Clathrin coated vesicle |
Somatic recombination of immunoglobulin gene segments |
Tricarboxylic acid cycle enzyme complex |
104 |
DNA damage response detection of DNA damage |
Neuron projection regeneration |
Positive regulation of monocyte chemotaxis |
105 |
Regulation of adaptive immune response |
Clathrin coat of endocytic vesicle |
Wash complex |
106 |
Ribosome |
Erythrocyte maturation |
Regulation of removal of superoxide radicals |
107 |
Regulation of B cell proliferation |
Hemoglobin complex |
Protein phosphatase type 1 complex |
108 |
Mitochondrial part |
Response to pain |
Bloc complex |
109 |
Regulation of type I interferon production |
B cell homeostasis |
Negative regulation of platelet activation |
110 |
Activation of JUN kinase activity |
Negative regulation of interferon gamma production |
Regulation of chemokine biosynthetic process |
111 |
Positive regulation of lymphocyte differentiation |
Response to axon injury |
Regulation of regulatory T cell differentiation |
112 |
Regulation of macrophage differentiation |
Negative regulation of interleukin 6 production |
Hemoglobin complex |
113 |
Positive regulation of type 2 immune response |
B cell proliferation |
Negative regulation of endoplasmic reticulum unfolded protein response |
114 |
Regulation of B cell activation |
Negative regulation of alpha beta T cell activation |
Negative regulation of Toll like receptor signaling pathway |

Figure 2: DNA microarray gene expression data mining of deregulated functions involving in the malignant transformation of EAOC. (A) Venn diagram of the deregulated GO term elements from exploratory factor analysis for the three diseases. The figure showed the results of the three diseases with the total factor elements from each of the disease. Their relationship was displayed on the Venn diagram to show the gene set numbers of all possible logical relations among the three diseases. The 9 commonly deregulated GO terms among ES, CCC and EC were listed on the right side of the figure. (B) The nine commonly deregulated GO terms among the ES, CCC, and EC, including ‘inflammasome complex’ was shown.
GO tree analysis of the relationship between deregulated immune/inflammation functions
To concentrate and view the hierarchy of the numerous identified deregulated GO terms, we mapped the immune/inflammation related GO terms to the GO tree based on the parent-child relationship. The related GO terms on the GO tree were then clustered together so the relationship of these GO terms can be visualized and summarized up as Figure 3 shown. The deregulated functions on the GO trees for ES could be summarized to ‘immune response’, ‘inflammation response’, ‘cytokine production’ and ‘inflammasome complex’. The inflammasome complex was highlighted because it was known to be an activator the carcinogenesis in many cancers. The full GO trees of the three diseases are available in Supplementary Figure 2-4. The data-mining approach above revealed the inflammasome complex was one of the most crucial candidate function initiating the malignant transformation of EAOC. In order to discover the genes involving in the inflammasome complex for further investigation and confirmation, we carried out an integrative analysis using the same microarray gene expression datasets to detect the differentially expressed genes (DEGs) of the three diseases. All of the gene expressions of the samples in each dataset were rescaled to the cumulative proportion for the integrative analysis. The full table of the DEGs is available in Supplementary Table 4. We then filtered the genes that were related to inflammasome complex. This filtering obtained a list of 47 genes, as shown in Supplementary Table 5.
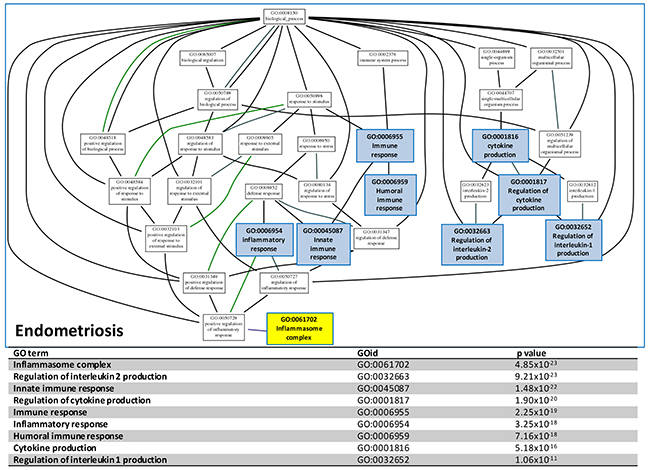
Figure 3: GO tree analysis. The GO tree of deregulated functions of CCC establish with the significant GO terms involving in the inflammation and immune system. After mapping to the GO tree, the similar or related GO terms were clustered together and shown the parent-child relationship. The table listed the immune or inflammation-related GO terms, the GOIDs and their p values in the GO trees.
Expression of inflammasome complex and inflammasome-related genes correlate with poor survival outcome in EAOC patients
To further illustrate the role of inflammasome in EAOC progression, we used Kaplan–Meier plotter (http://www.kmplot.com/ovar) to explore the correlation between EAOC patient survival and the expression levels of inflammasome complex as well as inflammasome-related geneses. Inflammasomes are multimeric protein complexes. Activation of inflammasomes and regulation of related pathway capable of orchestrating host inflammation and immunity [15, 16]. The component of inflammasome in tumorigenesis included inflammasome complex and inflammasome-related pathway [17, 18]. Inflammasome complex included nucleotide-binding domain and leucine-rich repeat receptors (NLRs), absent in melanoma 2(AIM2) and apoptosis-associated speck-like protein containing a CARD (ASC). NLRs and AIM2 recruit pro-caspase and promote its autocatalytic cleavage into active caspase, which leads to a cascade of pro-inflammatory events via the activation of the pro-inflammatory cytokine, which then interacts with their membranes receptors (TLR, TNF, INF, P2RX7) and related pathway amplifying the inflammatory response. We checked the 47 genes in Supplementary Table 5; they included 7 genes of inflammasome complex (NLRP3, AIM2, PYCARD, NAIP, Caspase-4, Caspase-7 and Caspase-8) and 11 genes of the inflammasome-related pathway (TLR1, TLR7, TOLLIP, NFKBIA, TNF, TNFAIP3, INFGR2, P2RX7, IL-1B, IL1RL1 and IL-18). Based on a database created by Gyorffy et al. [19], we correlated the gene expression levels of 18 highly expressed inflammasome markers, including 7 inflammasome complex genes and 11 inflammasome genes related pathway, with EAOC patient survival outcome. We found that high expression levels of the 7 inflammasome complex genes (NLRP3, AIM2, PYCARD, NAIP, Caspase-4, Caspase-7 and Caspase-8) tend to correlate with poor patient survival, and four of them (NLRP3, AIM2, PYCARD, NAIP) were statistically significant (Figure 4). NLRP3 and AIM2 are the initiators of inflammasomes, while PYCARD and NAIP are the core proteins of inflammasomes. These results indicated a potential direct involvement of inflammasome in EAOC progression. In the 11 genes inflammasome-related pathway (TLR1, TLR7, TOLLIP, NFKBIA, TNF, TNFAIP3, INFGR2, P2RX7, IL-1B, IL1RL1 and IL-18), high expression of these genes tended to correlate with poor survival of EAOC patients and 5 of them (TLR1, TLR7, TOLLIP, NFKBIA and TNF) reached statistical significance (Figure 5). The other 9 inflammasome-related genes (Caspase-4, Caspase-7, Caspase-8, TNFAIP3, INFGR2, P2RX7, IL-1B, IL1RL1 and IL-18) were not correlate with survival of the EAOC patients (Supplementary Figure 5-6). The flowchart and selection criteria of the EAOC marker genes were demonstrated as Supplementary Figure 7. These results indicated the involvements of inflammasome complex and inflammasome-related pathways in mediating EAOC disease progression.
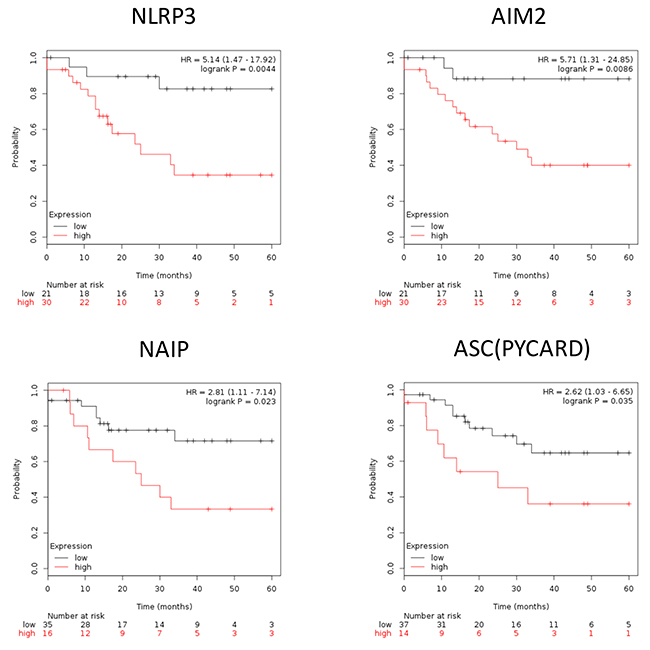
Figure 4: Inflammasome complex correlate with survival outcome in EAOC patients. Kaplan–Meier plotter survival curves showed significant difference of EAOC survival with different expression level of inflammasome complex (NLRP3, AIM2, PYCARD, NAIP). HR = 5.14, 95% CI 1.47 to 17.92, p-value = 0.044; HR = 5.71, 95% CI 1.31 to 24.85, p-value = 0.086; HR = 2.62, 95% CI 1.03 to 6.65, p-value = 0.035; HR = 2.81, 95% CI 1.11 to 7.14, p-value = 0.023, respectively.
Notably, the survival outcome of EAOC patients was highly correlated with NLRP3, AIM2, and TNF. The hazard ratio of NLRP3 / AIM2 / TNF were 5.14(1.47-17.92) / 5.71(1.31-24.85) / 6.08(1.4-26.49), respectively; p = 0.0044 / 0.0086 / 0.0061, respectively) (Figure 4 and 5). These results suggested key roles of the three inflammasome proteins and related pathways in promoting EAOC progression as well as their prognostic value in EAOC.
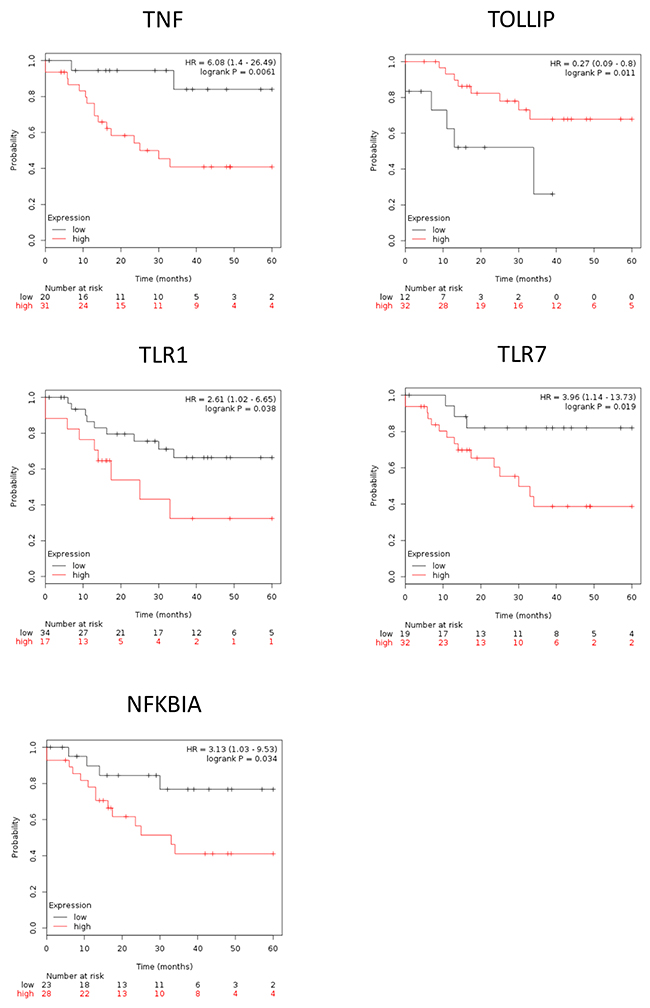
Figure 5: The survival of EAOC patients are correlate with inflammasome-related genes. Kaplan–Meier plotter survival curves showed significant difference of EAOC survival with different expression level of inflammasome-related genes (TNF, FOXO3, TLR7, NFKBIA). HR = 6.08, 95% CI 1.4 to 26.49, p-value = 0.061; HR = 3.15, 95% CI 1.24 to 8.02, p-value = 0.011; HR = 3.96, 95% CI 1.14 to 13.73, p-value = 0.019; HR = 3.13, 95% CI 1.03 to 9.53, p-value = 0.034, respectively.
Based on the survival analysis (Figure 4 and 5), we used the 9 inflammasome markers and STRING database (https://string-db.org) to establish a functional interaction network (Figure 6A). As members of inflammasome complex and inflammasome pathway related genes, the 9 proteins showed intensive interactions and regulatory crosstalk. This interactive network supported the involvement and key role of inflammation in EAOC malignant progression. Collectively, we demonstrated that the NLRP3, AIM2, PYCARD, NAIP, TLR1, TLR7, TOLLIP, NFKBIA and TNF would be the potential markers of prognosis in EAOC (Figure 6B).
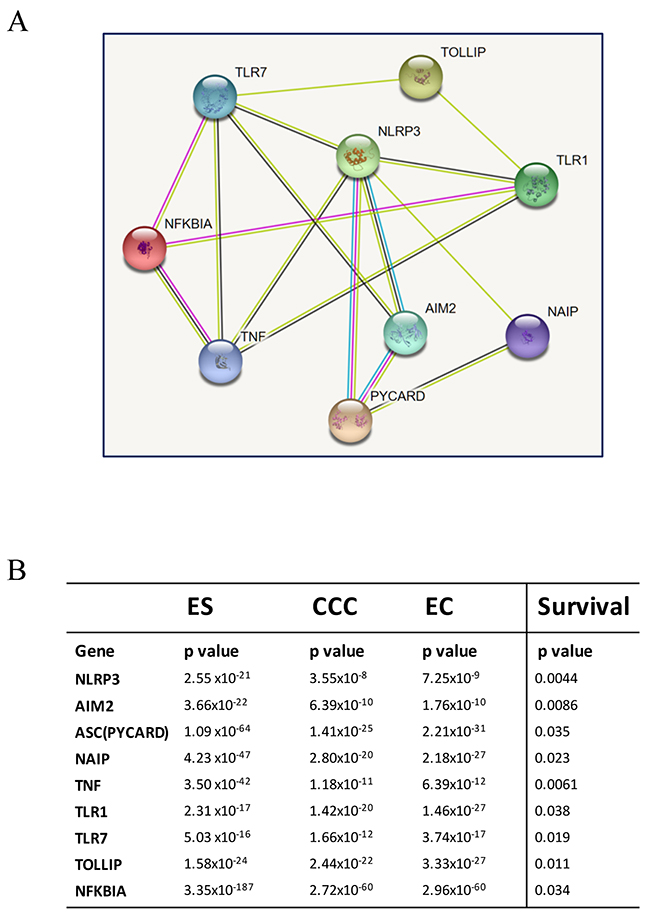
Figure 6: Interaction analysis of identified genes. (A) The identified potential involving genes were subjected to a protein-protein interaction (PPI) analysis by establishing an interactive network from the STRING database (https://string-db.org). As members of inflammasome complex and inflammasome-related genes, their proteins showed intensive interactions. The average node degree is 3.56, and the PPI enrichment p-value is 3.33x10-15, significantly more interactions than expected. (B) The p values of each gene in the three diseases were showed in the chart. The progressive changes of p values from ES to CCC and EC demonstrated that the NLRP3, AIM2, PYCARD, NAIP, TLR7, NFKBIA, TNF, FOXO3 would be the potential markers of prognosis in EAOC.
Immunohistochemistrical analysis for AIM2 expression among the three diseases
To evaluate the clinical significance of the identified inflammasome-related genes in ovarian cancer transformation, we collected a cohort of clinical samples (ES, n = 13; CCC, n = 15; EC, n = 15) and immunostained them with anti-AIM2 antibody. We found increased AIM2 protein level in CCC and EC samples in comparison to ES samples (Figure 7A). Quantification of AIM2 levels in all samples showed a higher mean value of AIM2 protein expression in both cancer types than in ES (Figure 7B). We then calculated the case numbers of AIM2-high and AIM2-low, as well as Ki-67-high and Ki-67-low in ES, CCC, and EC samples, and correlated the status of the two markers in each type of samples. As shown in Figure 7C–7E, we generally observed a positive correlation between the expression levels of Ki-67 and AIM2. In the 13 ES samples, all of them exhibited only low levels of Ki-67 and AIM2 (Figure 7C), while in the CCC samples, 12 out of 15 expressed high levels of Ki-67 and AIM2 (Figure 7E). In the group of EC, 5 samples expressed high Ki-67 and AIM2 level, and 6 expressed low levels of the two proteins (Figure 7D). Calculation of the percentage of AIM2-high case showed a progressive increase from ES to EC and to CCC (Figure 7C–7E). These results provid clinical evidence supporting the involvement of AIM2 in the malignant transformation of ES to CCC/EC.
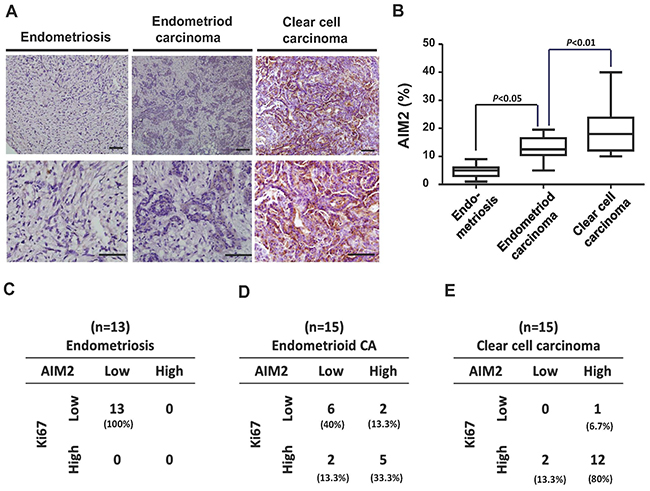
Figure 7: Immunohistochemistrical analysis of clinical samples from patients with ES, EC, and CCC. (A) Clinical samples from patients with ES (n = 13), EC (n = 15), and CCC (n = 15) were immunostained with anti-AIM2 antibody. (B) The expression levels of AIM2 in all clinical samples were quantified and presented in the chart. The mean values of AIM2 expression in EC and CCC were higher than that in ES. (C-E) Samples were stained with Ki-67 and AIM2. The case numbers of ES, EC, and CCC with high and low expression levels of Ki-67 and AIM2 were calculated and displayed in the chart. The percentages of each combination were also calculated. The AIM2 levels was positively correlated with Ki-67 levels.
Working model of inflammasome in endometriosis associated ovarian carcinoma
Based on our data-driven analysis and lab validation, we proposed a working model of the association between inflammasome in endometriosis and the progression of ovarian cancer. In the microenvironment of ovarian endometrioma, inflammasome is driven directly by specific DAMPs or by the two-signal model as in the case of NLRP3 in the microenvironment of ovarian endometrioma, The recognition of DAMPs by extracellular TLRs leads to the activation of NF-κB (first signal), which in turn promotes the transcription of pro- inflammatory cytokines or some NLRs (e.g.NLRP3). NLRs assemble into the inflammasome complex which via the CARD domain can recruit pro-caspase and promote its autocatalytic cleavage (second signal). Active caspase can lead to a cascade of pro-inflammatory events via the activation of pro- inflammatory cytokines, which then interact with their own membrane receptors amplifying the inflammatory response. On the other hand, active caspase can lead to cell pyroptosis with the consequence of the release of inflammatory cytokines. Inflammatory cytokines activated oncogene over-expression then induced EAOC carcinogenesis (Figure 8).
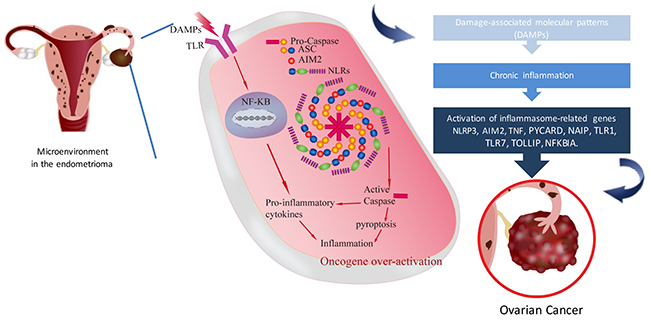
Figure 8: Working model of the inflammasome in endometriosis associated ovarian cancer. This model presents the microenvironment in endometrioma of the ovary. Retrograded menstruation accumulated in ovary provoked DAMPs and caused chronic inflammation. Inflammasome related genes (NLRP3, AIM2, PYCARD, NAIP, TNF, FOXO3, TLR7, NFKBIA) were activated subsequently. Activated caspase can lead to cell pyroptosis with the consequence of the release of inflammatory cytokines. Finally, inflammatory cytokines induced oncogene over-expression then produced EAOC carcinogenesis.
DISCUSSION
Complex diseases usually involve in a spectrum of variable deregulated functions. So we investigated the pathogenesis of EAOC with the functionome consisted of 5917 GO defined functions computed from large-scale microarray gene expression profiles. We demonstrated the informativeness of the GSR indices was sufficient for machine learning to accurately recognize and classify these complex diseases based on the functional regularity patterns. The patterns were similar between CCC and EC as showed on the heatmap (Figure 2), revealing the possibility of homogeneous etiology between these two cancers. We further investigated the common deregulated functions among ES, CCC, and EC to discover the candidate elements involving in the malignant transformation from ES to CCC or EC. Our study revealed the consistent findings: the ‘activation of immune response’ in ES; the ‘humoral immune response’ deregulated GO terms for CCC and EC. Moreover, the deregulated GO term ‘inflammatory response’ (GO:0006954) coexisted in ES, CCC, and EC. We further checked the immune/inflammation related GO terms in the functionomes of the three diseases. The set analysis using the top significant 114 immunes/inflammation related GO terms for the three diseases showed nine common deregulated GO terms, and the existence of inflammasome complex in this list is noticeable because it has been demonstrated to be a critical promoter of carcinogenesis in various cancers. Then we checked the DEGs detected from the same DNA microarray datasets, the inflammasome related genes, including NLRP3, AIM2, PYCARD, NAIP, TLR1, TLR7, TOLLIP, NFKBIA and TNF were demonstrated to be differentially expressed in the three diseases and also significantly correlated with poor progression-free survival. Finally, high expressions level of AIM2 were confirmed in EC and CCC, in comparison to ES, by immunohistochemical analysis, and is correlated with high level of Ki-67. Our results support that a close relationship between endometriosis and clear cell carcinoma/endometrioid carcinoma, and support the hypothesis of endometriosis associated ovarian carcinoma. Dysregulated inflammasome could be a fundamental role in modulating the malignant transformation of EAOC, which also broadens the scope of the inflammation/immunity as a molecular biomarker in monitoring the malignant transformation of endometriosis and also could be the treatment target of endometriosis associated ovarian cancer. To the best of our knowledge, these findings are vital to clarify the role of the inflammasome in EAOC carcinogenesis.
The inflammatory microenvironment has been revealed to play crucial roles in all stages of tumor development [20]. Pathogen or damage signals that trigger inflammation have been reported to drive tumorigenesis in many forms of cancer [11]; immune cells that trigger inflammation were also associated with tumor development [21]. The immune microenvironment is critical for the carcinogenesis of EAOC. The cell proliferation resulted from aberrations humoral immunity and complement pathway activation was postulated to play a major role in the pathogenesis of EAOC [22]. Cancer-immune phenotypes in humans can be divided into three main categories: the immune-desert phenotype, the immune–excluded phenotype and the inflamed phenotype. Each is related to specific underlying biological mechanisms that may prevent the host’s immune response from eliminating cancer. Inflamed tumors are infiltrated by a variety subtypes of immune cells including immune-inhibitory regulatory T cells, myeloid-derived suppressor cells, and cancer-associated fibroblasts [23]. The presence of intratumoral T cells independently associated with delayed recurrence or prolonged survival in multivariate analysis of advanced ovarian carcinoma and was related to increased expression of interferon-γ, interleukin-2, and lymphocyte-attracting chemokines within the tumor [24]. Anti-inflammatory effects in autoimmune diseases and neurodegeneration also appeared to suppress the inflammatory activity of TLR4-NF-κB/ NLRP3 inflammasome pathway and provided novel mechanistic insights for the potential therapeutic for cervical cancer [25]. The inflammasome of NOD-like receptor family pyrin domain-containing 3 (NLRP3) is a complex protein involved in the induction of innate inflammatory/immune responses. The complex consists of the NLRP3 protein, which serve as a sensor for the activation of the inflammasome, and an apoptosis-associated speck-like protein containing a CARD complex (ASC), which recruits pro-caspase through its CARD domain. Pro-caspase is then interchange to active caspase, which, in turn, cleaves pro-inflammatory cytokines (pro-IL-1β and pro-IL-18) to their active forms. IL-1β and IL-18 to promote inflammation by recruiting additional inflammatory/immune cells. Then oncogene could be activated. Thus, NLRP3 signaling persistent sterile inflammation could be the initial stage of carcinogenesis.
AIM (absent in melanoma 2) can induce inflammasome upon intracellularly delivery of double-stranded DNA (dsDNA) to protect cells against pathogens like virus and bacteria. AIM2 is a cytosolic dsDNA sensor and directly interacts with dsDNA, mainly from virus or bacteria, through its C-terminal HIN-200 domain, leading to a serial activation of inflammation proteins and form AIM2 inflammasome. Activation of the AIM2 inflammasome and other canonical inflammasomes results in a type of inflammatory cell death called pyroptosis. Chronic inflammation of the benign prostate hyperplasia was reported closely related to prostate cancer. Recent studies showed that AIM2 inflammasome plays a critial roles in the tumor progression of prostate cancer. Activation of AIM2 was served as a biomarker to identify the molecular mechanisms through prostatic infections and/or sterile inflammation contribute to the carcinogenesis of prostatic cancer [26]. In our comparative bioinformatic analysis between endometriosis and ovarian carcinoma, we found AIM2 diversely expressed in the two groups of data, suggesting a role of AIM2 in promoting the progression of ovarian carcinoma. Notably, the analysis of immunohistochemistry staining further confirmed a correlation between high AIM2 expression and high Ki-67 activity in clinical EAOC samples, supporting that AIM2 and inflammasome play a key regulatory role in EAOC transformation and disease progression. Therefore, based on our findings, inflammation mechanism is suggested as the key regulatory step mediated the malignant transformation of endometriosis. Further in vivo study to investigate whether NLRP3/AIM2 contributes to EAOC carcinogenesis and the role of the inflammasome in EAOC is imperative.
This investigation has limitations, though. First, the GO term gene set database does not define the comphehensive human functions yet. Therefore, undefined immulogical functions involving in the malignant transformation may be missed in the current analysis. Second, the GSR model may produce false positive results because of similar gene elements in different gene sets. For example, the 47th desregulated functions for EC ‘Defense response to gram positive bacterium (GO:0050830)’ in the Table 3 may potentially be a false positivity; because to our knowledge, there is no evidence showing the involvement of gram positive vacterial infection in the etiology of EC. It may raise from the duplicated gene elements in the gene set definitions. Third, the case number for the immunohistochemistrical analysis is relatively small. More cases are necessary to clarify the pathogenesis of EAOC in the future.
In conclusion, we established a bioinformatic platform of gene-set integrative molecular functionome to dissect the molecular pathogenetic pathways of EAOC and demonstrated dysregulated inflammasomes play a fundamental role in modulating the malignant transformation and cancer progression in EAOC. Our results support the hypothesis that endometriosis shares similar genetic signatures with EAOC that validated by data-driven analysis and tissue array, which also broaden the scope of the inflammation/immunity as a molecular biomarker in monitoring the malignant transformation of endometriosis and also could be the treatment target of endometriosis associated ovarian cancer.
MATERIALS AND METHODS
Computing the GSR indices
The regulation of the GO terms were quantified by the GSR model, which converted gene expression profiles to quantified functions by the modifying the Differential Rank Conservation (DIRAC) [27] algorithm. This model quantifies the ordering change of the gene elements in a gene set between the gene expression orderings in ES, CCC or EC and the most common gene expression ordering in the normal control population in this study. Microarray gene expression profiles were downloaded from the Gene Expression Omnibus (GEO) database as.SOFT format, and then the gene expression levels were extracted according to the corresponding gene elements in the GO term gene set and converted to the ordinal data based on their expression levels. The GSR index is the ratio of gene expression ordering in a gene set between each case or normal control sample and the most common gene expression ordering among the normal tissue samples. Computing the GSR indices was executed in R environment. The detail of the GSR model and the computing procedures are described in our previous study [21].
Gene set definition, microarray datasets and data processing
The versions of the GO gene set definitions were c5.all.v6.0.symbols.gmt (2017), downloaded from the MSigDB and contained 5917 GO gene sets. The selection criteria for the downloaded microarray gene expression datasets were: 1. The datasets should provide definite information on the diagnosis for each sample; 2. Because this study utilized the common genes among the selected datasets; a dataset was discarded if it resulted in the number of common genes less than 8000 when it was integrated.
Statistical analysis
The differences of the GSR indices between the three diseases and the control groups were tested by Mann Whitney U test and corrected by multiple hypotheses using false discovery rate (Benjamini-Hochberg procedure). The significance level was set at <0.01. Progression-free survival (PFS) data of endometrioid ovarian cancer were available for 51 patients obtained from kmplot.com. The Kaplan–Meier survival curves for endometrioid ovarian cancer can be reached at http://www.kmplot.com/ovar. Hazard ratio (HR; and 95% confidence intervals) and logrank P are calculated and displayed with website.
Classification and prediction by machine learning
GSR indices computed through the GO term gene sets were classified and predicted by the support vector machine (SVM) with kernlab [28], an R package for kernel-based machine learning methods and was used to classify patterns of the GSR indices with the setting of kernel = ‘vanilladot’ (linear kernel function). The performance of classification and prediction by SVM were measured by 5-fold cross-validation: samples were randomly sampled and divided into 5 parts, 4 parts were used for training sets and the remainder one part for prediction. The performance of binary classification was assessed with sensitivity, specificity, accuracy and AUC. Sensitivity, specificity, accuracy and AUC were computed using the results of successive 10 classifications. AUC was computed by an R package pROC [29]. The accuracy of multiclass classification was computed from the fraction of correct predictions within total prediction number.
Hierarchical clustering, dendrogram and heatmaps
The GSR indices in each gene set were averaged then underwent hierarchical clustering with the function ‘heatmap.2’ in R package ‘gplots’ (version 3.0.1) as default. This function executed hierarchical clustering, and drew dendrogram and heatmaps.
All possible logical relations among the deregulated functions of the three diseases were evaluated and displayed by Venn diagram using the R package ‘VennDiagram’ (version 1.6.17).
Reconstruction of GO trees and detection of differentially expressed genes
The GO tree were reconstructed by the ‘RamiGO’ [30], an R package providing functions to interact with the AmiGO 2 web server (http://amigo2.berkeleybop.org/amigo) and retrieves GO trees. To discover the DEGs for each of ES, CCC and EC, we carried out an integrative analysis with the same DNA microarray datasets. The gene expression levels of all samples in each dataset were transformed and rescaled to cumulative proportion values from 0 (lowest expression) to 1 (highest expression) with an R package “YuGene” (version 1.1.5) before integration. The DEGs were discovered using linear model computed with empirical Bayes analysis by the functions “lmFit” and “eBayes” provided by the R package “limna” (version 3.26.9).
Clinical samples
The present study included 30 archived ovarian cancers (clear cell, N = 15 and endometrioid, N = 15), 13 ovarian endometrioma. In the cases of ovarian cancer, tissues were collected from women underwent surgery as their treatment guideline, and tissue specimens of endometrioma were collected from women who had ovarian endometrioma undergone a surgery of ovarian cystectomy. The patients were diagnosed and treated and had their tissues placed in a bank at the Tri-Service General Hospital, Taipei, Taiwan. All invasive cancers were confirmed by histopathology. The Institutional Review Board of the Tri-Service General Hospital, National Defense Medical Center approved the study. Informed consent was acquired from all patients and control subjects.
Author contributions
Conception and design of the experiments: Chia-Ming Chang, Cheng-Chang Chang, Peng-Hwei Wang
The performance of the experiments: Yi-Ping Yang
Analyses of the data: Chia-Ming Chang, Kai-Hsi Lu, Chi-Mou Juang
Contribution of reagents/materials/analysis tools: Yi-Ping Yang
Drafting and editing of the manuscript: Chia-Ming Chang, Mong-Lien Wang, Cheng-Chang Chang
Recruitment and collection of clinical samples: Cheng-Chang Chang, Ren-Jun Hsu, and Mu-Hsien Yu.
ACKNOWLEDGMENTS
We thank Hui-Yin Su for figures editing and Huan-Shuo Chang for assistant preparation of experimental materials.
CONFLICTS OF INTEREST
All authors declared no conflicts of interest.
FUNDING
This work was supported in part by the following grants: TSGH-C105-010 and TSGH-C106-080 from the Tri-Service General Hospital; MOST 106-2314-B-016-042 from Ministry of Science and Technology, R.O.C.; the Teh-Tzer Study Group for Human Medical Research. This work was also supported by grants from the Ministry of Science and Technology (MOST 103-2314-B-010-043-MY3, and MOST 106-2314-B-075-061-MY3) and the Taipei Veterans General Hospital (Grant V104C-095, V105C-096, V106C-129; V106D23-001-MY2-1; and V106A-012), Taipei, Taiwan. We thank Hui-Yin Su for figure editing.
REFERENCES
1. Vaughan S, Coward JI, Bast RC Jr, Berchuck A, Berek JS, Brenton JD, Coukos G, Crum CC, Drapkin R, Etemadmoghadam D, Friedlander M, Gabra H, Kaye SB, et al. Rethinking ovarian cancer: recommendations for improving outcomes. Nat Rev Cancer. 2011; 11: 719-25.
2. Brinton LA, Sakoda LC, Sherman ME, Frederiksen K, Kjaer SK, Graubard BI, Olsen JH, Mellemkjaer L. Relationship of benign gynecologic diseases to subsequent risk of ovarian and uterine tumors. Cancer Epidemiol Biomarkers Prev. 2005; 14: 2929-35.
3. Rossing MA, Cushing-Haugen KL, Wicklund KG, Doherty JA, Weiss NS. Risk of epithelial ovarian cancer in relation to benign ovarian conditions and ovarian surgery. Cancer Causes Control. 2008; 19: 1357-64.
4. Van Gorp T, Amant F, Neven P, Vergote I, Moerman P. Endometriosis and the development of malignant tumours of the pelvis. A review of literature. Best Pract Res Clin Obstet Gynaecol. 2004; 18: 349-71.
5. Fukunaga M, Nomura K, Ishikawa E, Ushigome S. Ovarian atypical endometriosis: its close association with malignant epithelial tumours. Histopathology. 1997; 30: 249-55.
6. LaGrenade A, Silverberg SG. Ovarian tumors associated with atypical endometriosis. Hum Pathol. 1988; 19: 1080-4.
7. Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011; 474: 609-15.
8. Zhang D, Chen P, Zheng CH, Xia J. Identification of ovarian cancer subtype-specific network modules and candidate drivers through an integrative genomics approach. Oncotarget. 2016; 7: 4298-309. https://doi.org/10.18632/oncotarget.6774.
9. Sampson JA. Metastatic or Embolic Endometriosis, due to the Menstrual Dissemination of Endometrial Tissue into the Venous Circulation. Am J Pathol. 1927; 3: 93-110.43.
10. Herington JL, Bruner-Tran KL, Lucas JA, Osteen KG. Immune interactions in endometriosis. Expert Rev Clin Immunol. 2011; 7: 611-26.
11. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010; 140: 883-99.
12. Chang CM, Chuang CM, Wang ML, Yang YP, Chuang JH, Yang MJ, Yen MS, Chiou SH, Chang CC. Gene Set-Based Integrative Analysis Revealing Two Distinct Functional Regulation Patterns in Four Common Subtypes of Epithelial Ovarian Cancer. Int J Mol Sci. 2016; 17.
13. Chang CM, Chuang CM, Wang ML, Yang MJ, Chang CC, Yen MS, Chiou SH. Gene Set-Based Functionome Analysis of Pathogenesis in Epithelial Ovarian Serous Carcinoma and the Molecular Features in Different FIGO Stages. Int J Mol Sci. 2016; 17.
14. Kolb R, Liu GH, Janowski AM, Sutterwala FS, Zhang W. Inflammasomes in cancer: a double-edged sword. Protein Cell. 2014; 5: 12-20.
15. Latz E, Xiao TS, Stutz A. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013; 13: 397-411.
16. Man SM, Kanneganti TD. Regulation of inflammasome activation. Immunol Rev. 2015; 265: 6-21.
17. Lin C, Zhang J. Inflammasomes in Inflammation-Induced Cancer. Front Immunol. 2017; 8: 271.
18. Karki R, Man SM, Kanneganti TD. Inflammasomes and Cancer. Cancer Immunol Res. 2017; 5: 94-9.
19. Gyorffy B, Lanczky A, Szallasi Z. Implementing an online tool for genome-wide validation of survival-associated biomarkers in ovarian-cancer using microarray data from 1287 patients. Endocr Relat Cancer. 2012; 19: 197-208.
20. Guo B, Fu S, Zhang J, Liu B, Li Z. Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci Rep. 2016; 6: 36107.
21. de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer. 2006; 6: 24-37.
22. Edwards RP, Huang X, Vlad AM. Chronic inflammation in endometriosis and endometriosis-associated ovarian cancer: New roles for the "old" complement pathway. Oncoimmunology. 2015; 4: e1002732.
23. Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017; 541: 321-30.
24. Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, Makrigiannakis A, Gray H, Schlienger K, Liebman MN, Rubin SC, Coukos G. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003; 348: 203-13.
25. He A, Shao J, Zhang Y, Lu H, Wu Z, Xu Y. CD200Fc reduces LPS-induced IL-1beta activation in human cervical cancer cells by modulating TLR4-NF-kappaB and NLRP3 inflammasome pathway. Oncotarget. 2017; 8: 33214-24. https://doi.org/10.18632/oncotarget.16596.
26. Ponomareva L, Liu H, Duan X, Dickerson E, Shen H, Panchanathan R, Choubey D. AIM2, an IFN-inducible cytosolic DNA sensor, in the development of benign prostate hyperplasia and prostate cancer. Mol Cancer Res. 2013; 11: 1193-202.
27. Eddy JA, Hood L, Price ND, Geman D. Identifying tightly regulated and variably expressed networks by Differential Rank Conservation (DIRAC). PLoS Comput Biol. 2010; 6: e1000792.
28. Karatzoglou A, Smola A, Hornik K, Zeileis A. kernlab - An S4 Package for Kernel Methods in R. Journal of Statistical Software. 2004; 11: 1-20.
29. Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, Muller M. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics. 2011; 12: 77.
30. Schroder MS, Gusenleitner D, Quackenbush J, Culhane AC, Haibe-Kains B. RamiGO: an R/Bioconductor package providing an AmiGO visualize interface. Bioinformatics. 2013; 29: 666-8.