
INTRODUCTION
Astrocytes play vital roles in maintaining housekeeping functions of the nervous system (CNS), including homeostasis of the extracellular environment, neuronal metabolism, shaping of the brain microarchitecture, and regulation of neurotransmitters, such as glutamate [1]. Besides of neuronal impairment [2–4], astrocyte dysfunction was also contribute to the pathogenesis of neurodegenerative diseases through its effect on neuronal survival [5, 6]. For instance, accumulation of neuron-derived amyloid beta 1-42 in activated astrocytes indicates debris-clearing activity of astrocytes in response to Alzheimer’s disease (AD)-related degeneration of local dendrites and synapses [7]. In Parkinson’s disease (PD), astrocytes confer neuroprotection by providing the antioxidant, glutathione [8, 9] and clearing extracellular synuclein [10]; however, under pathological conditions, astrocytes release toxic molecules [11] and proinflammatory cytokines which promote degeneration of nigral dopaminergic neurons [12].
The prevalence of antidepressant use has gradually increased around the world, especially the adverse of selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs) as first-line treatment options in depression [13]. Antidepressants elevate the transmission of one or more of the monoamines: serotonin, nor-adrenaline, or dopamine [14]. They are classified according to their mechanism of action: tricyclic and tetracyclic antidepressants (TCAs and TeCAs), SSRIs, serotonin antagonist and reuptake inhibitors (SARIs), monoamine oxidase inhibitors (MAOIs), and SNRIs. Antidepressants have been proved to improve cognitive functions [15–17] but there is also a large-scale clinical trial which determined no cognitive improvements in antidepressant users [18]. In addition, Andrews et al. proposed that antidepressants disrupt the serotonin-regulated adaptive process [19, 20] and worsen neurological side-effects, such as dizziness [21], drowsiness, and sedation [22]. Previous studies demonstrated that antidepressants cause cell death of normal cells [23, 24]. Furthermore, a meta-analysis by Moraros et al. revealed that antidepressant medication was associated with a 2-fold increase in odds of several cognitive impairments [25]. We also obtained consistent results of antidepressant medication being associated with an increased risk of dementia in our previous study [26]. According to those studies and integrating the idea of an astroglio-centric perspective of neurodegenerative diseases [6], we proposed to determine the effects of antidepressant on astrocytes in this study.
Both in vitro and in vivo studies proposed that astrocyte apoptosis could be triggered by several pathways, such as Ca2+ overload [27], mitochondrial dysfunction [28], oxidative stress [29], nuclear factor-κB (NF-κB) activation [30], endoplasmic reticulum stress [31], and protease activation [32]. Regulation of calcium is critical for astrocytic signaling [33, 34], while intense elevation of intracellular Ca2+ ([Ca2+]i) may be a possible mechanism linking antidepressants and astrocyte apoptosis. Mounting evidence also suggested calcium deregulation would lead to astrocytic cell death [27, 35–37] via reactive oxygen species (ROS) generation through activation of calpain and xanthine oxidase [30]. Moreover, Liu et al. previously revealed the fluoxetine induced apoptosis of astrocyte-derived glioblastomas via AMPAR-mediated calcium overload [38].
In this study, we evaluated the impacts of antidepressants on astrocyte survival and the underlying mechanisms. After screening 11 different antidepressants, we found that sertraline and paroxetine induced astrocyte apoptosis. Astrocyte apoptosis was mediated by elevation of [Ca2+]i, dysfunction of mitochondria, and activation of caspase, and was accompanied by ROS generation. Our exploration of molecular mechanisms of antidepressant-triggered astrocyte apoptosis in this study revealed that antidepressant medication might be a potential risk factor for neurodegenerative diseases.
RESULTS
Sertraline and paroxetine reduce astrocyte viability
We first investigated the effect of different antidepressants on the viability of an astrocyte cell line. As shown in Figure 1, we treated astrocytes with 0-40 μM of sertraline, paroxetine, citalopram, fluvoxamine, escitalopram, venlafaxine, imipramine, doxepin, mirtazapine, moclobemide, and trazodone for 48 h. The MTT results revealed that 10 μM sertraline or 20 μM paroxetine, two SSRIs, significantly reduced the cell viability of astrocytes. In contrast, we found no cytotoxicity toward astrocytes by the other antidepressants.
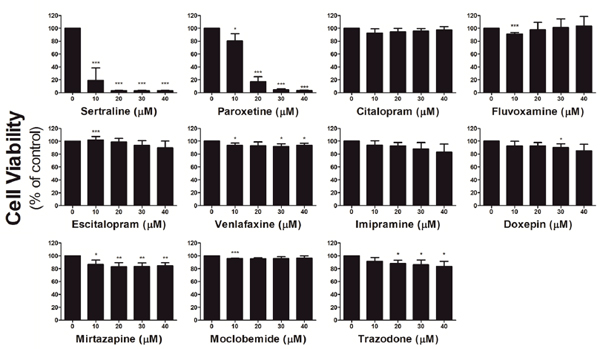
Figure 1: Sertraline and paroxetine reduce astrocyte viability. Astrocyte viability was determined after treatment with indicated concentrations of antidepressants for 48 h by an MTT assay. Data were collected from three independent experiments and statistically analyzed by Student’s t-test, and results are shown as the mean ± SD. *p<0.05, **p<0.01, and ***p<0.001 compared to the control group.
Sertraline and paroxetine induce dose-dependent intracellular calcium elevation in astrocytes
We further studied the underlying mechanism of sertraline- and paroxetine-induced astrocyte cell death. It was reported that sertraline induces calcium levels in human prostate cancer cells. Thus, we used Fluo-4 as a calcium-sensitive probe to determine the influence of antidepressants on calcium flows. Treatment with sertraline and paroxetine increased the intracellular calcium concentration, and the calcium continued to rise in astrocytes at 3 and 6 h; however, only a slight increment in calcium levels was detected after citalopram treatment (Figure 2A). Calcium levels were quantified by flow cytometry (Figure 2B, 2C). Figure 2D and 2E further confirmed the dose-dependent effect of sertraline and paroxetine on the induction of intracellular calcium level.
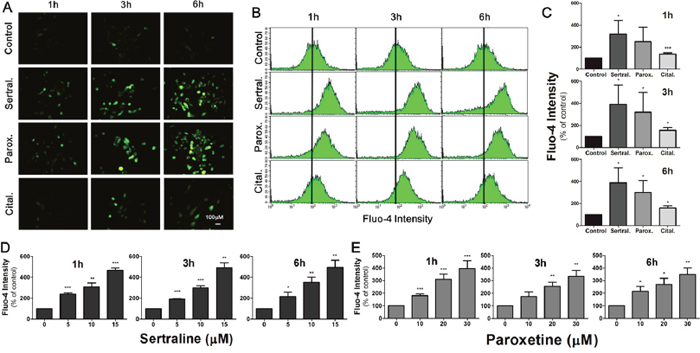
Figure 2: Sertraline and paroxetine induce dose-dependent intracellular calcium elevation in astrocytes. (A) Fluorescence imaging of [Ca2+]i using Fluo-4 was conducted after 1, 3, and 6 h of 10 μM sertraline, 20 μM paroxetine, and 20 μM citalopram treatment. (B, C) Quantitative data were collected by flow cytometry. Significant induction of fluorescence intensity was seen in cells exposed to sertraline and paroxetine compared to the control. (D, E) Sertraline and paroxetine triggered dose-dependent induction of intracellular calcium level. Data were collected from three independent experiments and statistically analyzed by Student’s t-test, and results are shown as the mean ± SD. *p<0.05, ***p<0.001 compared to the control group.
Sertraline and paroxetine induce mitochondrial membrane damage with depleted adenosine triphosphate (ATP) production and trigger ROS generation in astrocytes
Mitochondria are important organelles for storing calcium and play a key role in activating apoptosis. To examine the effects of sertraline and paroxetine on mitochondrial function, we used DiOC6 to determine the mitochondrial membrane potential (MMP) and applied mitochondrial functional assay to measure ATP content. Results revealed that induction of MMP at 3 h after treatment with sertraline and paroxetine, which indicated mitochondrial hyperpolarization. Figure 3A shows that treatment with sertraline and paroxetine decreased the MMP at 6 h and 12 h (third and fourth panels). Mitochondrial hypopolarization indicates that mitochondria have been damaged, occurred in most astrocytes of these two treated groups at 24 h (Figure 3A, 3B). Reduction of ATP production in sertraline- and paroxetine- treated astrocytes further confirmed that mitochondrial damage was occurred (Figure 3C, 3D). ROS-induced damage of mitochondria is one of the mechanisms involves in activating apoptosis. We found that both sertraline and paroxetine treatments significantly induced ROS production at 24 h (Figure 3E, 3F).
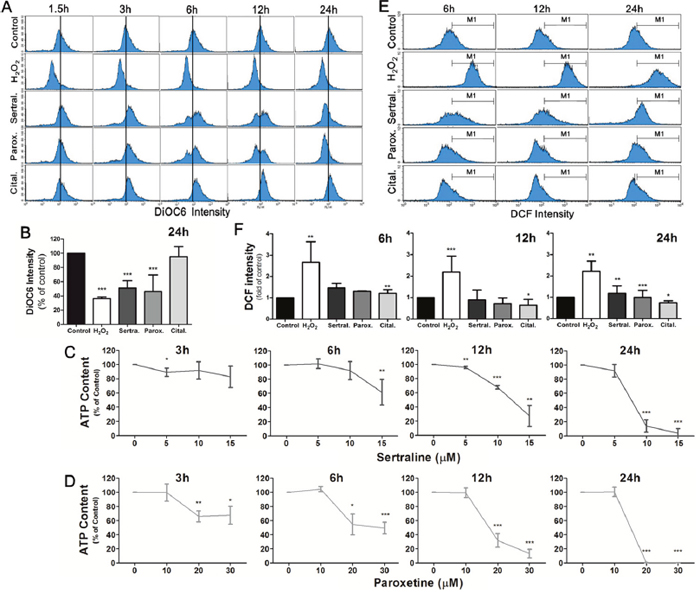
Figure 3: Sertraline and paroxetine induce mitochondrial membrane damage with depleted ATP production and trigger ROS generation in astrocytes. (A, B) Astrocyte cells were treated with sertraline, paroxetine, and citalopram for 1.5, 3, 6, 12, and 24 h, and DiOC6 staining was used to examine the damage of mitochondrial membranes. The distribution of cells according to their mitochondrial membrane potential is shown in each panel of the figure, and means of DiOC6 intensity of different groups are compared in histograms. (C, D) ATP production was measured to monitor the mitochondrial function. (E, F) ROS generation was observed at 24 h after sertraline and paroxetine treatment. The DCF fluorescence intensity was measured by flow cytometric analysis, and the ratio of cells located at the M1 phase was quantified. H2O2 treatment is the positive control for ROS production. Data were collected from three independent experiments and statistically analyzed by Student’s t-test, and results are shown as the mean ± SD. *p<0.05, ***p<0.001 compared to the control group.
Sertraline and paroxetine activate the intrinsic apoptotic pathway in astrocytes
Images in Figure 4A show that the morphology of astrocytes had changed at 6 and 12 h in sertraline- and paroxetine-treated groups, but not in the citalopram-treated group. By a flow cytometric analysis, we also found that treatment with sertraline and paroxetine dramatically increased cell death, as indicated by the accumulation of sub-G1 population (Figure 4B). To define whether sertraline- and paroxetine-induced cell death was through apoptosis, we analyzed expressions of apoptosis-related proteins by a Western blot analysis. We found that treatment with sertraline and paroxetine, but not citalopram, induced cleavage of PARP and caspase-3 (Figure 4C). These data indicated that sertraline and paroxetine induced apoptotic signaling and cell death in astrocytes.
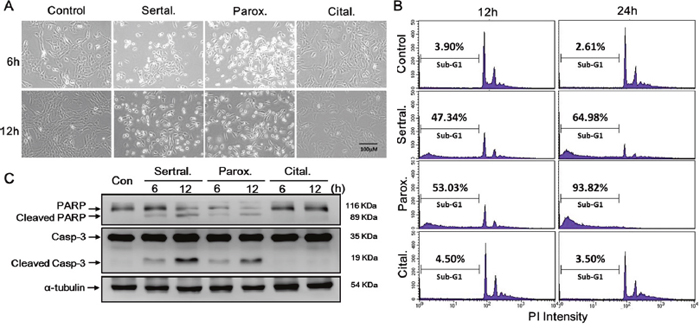
Figure 4: Sertraline and paroxetine activate the intrinsic apoptotic pathway in astrocytes. Cell death was either (A) observed by microscopy or (B) quantified by a flow cytometric analysis. (C) Cleaved caspase-3 and cleaved poly(ADP-ribose) polymerase (PARP) proteins were determined by a Western blot analysis.
Sertraline and paroxetine reduced cell viability in primary astrocytes, neurons, and their primary mixed culture
To further confirm the aforementioned results, we used primary cultured astrocytes and neurons to establish a model to better mimic the nervous system. We further conducted experiments in primary astrocytes, primary neurons, and primary mixed astrocyte-neuron culture. The MTT assay showed that 10-40 μM sertraline had similar cytotoxic effects which significantly reduced primary astrocyte cell viability to <20%, while primary neuron cell viability remained around 50% (Figure 5A, left panel). Paroxetine showed consistent effects on primary cultured cells as did sertraline. Paroxetine at 20 μM significantly decreased the cell viability of primary astrocytes and primary mixed culture to around 20%, while the cell viability of primary neurons remained around 50% under the same conditions (Figure 5A, right panel). We further examined whether sertraline and paroxetine induced primary astrocyte cell death through the same mechanism in an astrocyte cell line. We observed that both sertraline and paroxetine induced intracellular calcium levels at 1, 3, and 6 h according to immunofluorescence staining (Figure 5B). We also found Ca2+ influx in primary cells in response to sertraline and paroxetine treatments by a flow cytometric analysis (Figure 5C, 5D). Sertraline and paroxetine also decreased the MMP in primary cells, which indicated consistent mitochondrial damage as in the astrocyte cell line (Figure 5E).
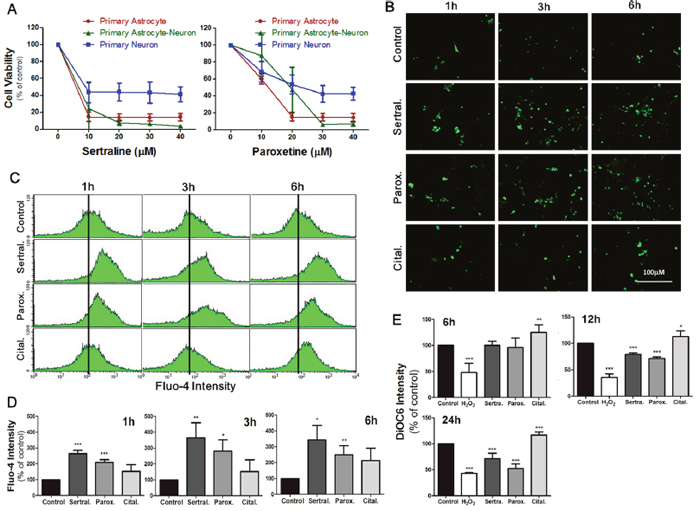
Figure 5: Sertraline and paroxetine reduced cell viability in primary astrocytes, neurons, and their primary mixed culture. (A) Cell viability of different cells with indicated experimental conditions was detected by an MTT assay at 12 h after treatment. Data were collected from three independent experiments, and results are shown as the mean ± SD. (B) Fluorescence imaging of [Ca2+]i of primary astrocytes using Fluo-4 was conducted after 1, 3, and 6 h of 10 μM sertraline, 20 μM paroxetine, and 20 μM citalopram treatment. (C, D) Quantitative data were collected by flow cytometry. (E) Mitochondrial damage of primary astrocytes was observed after 12 and 24 h of sertraline, paroxetine, and citalopram treatment. The DiOC6 fluorescence intensity was measured by a flow cytometric analysis. Data were collected from three independent experiments and statistically analyzed by Student’s t-test, and results are shown as the mean ± SD. *p<0.05, ***p<0.001 compared to the control group.
Sertraline- and paroxetine-induced cell death was not associated with glutamate receptors, P2 receptor or TRPA1 receptor
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor (AMPAR) [39, 40], N-methyl-D-aspartate (NMDA) receptor [41], kainite receptor [42], P2 receptor [43, 44] and transient receptor potential ankyrin 1 (TRPA1) channel [45] are critical for astrocytic calcium influx. It was shown that fluoxetine induces calcium overload and causes apoptosis of glioma cells via activation of the AMPAR [38]. To examine whether sertraline and paroxetine also induce calcium overload via activation of the glutamate receptor, we pretreated astrocytes with several glutamate receptor inhibitors, including NBQX (AMPA/kainate receptor antagonist), NS-102 (kainate receptor antagonist), and MK-801 (NMDA receptor antagonist), followed by sertraline and paroxetine treatment. We found that none of these inhibitors rescued astrocytes from cell death (Figure 6A). We also pretreated astrocytes with the ATP-gated calcium channel inhibitors, PPADs (P2 receptor antagonist) and HC-030031 (TRPA1 channel antagonist), and found no protective effects of these two inhibitors on sertraline- and paroxetine-induced cell death (Figure 6B). These data suggest that sertraline- and paroxetine-induced cell death may occur through a glutamate receptor- and ATP-gated calcium channel-independent pathway.
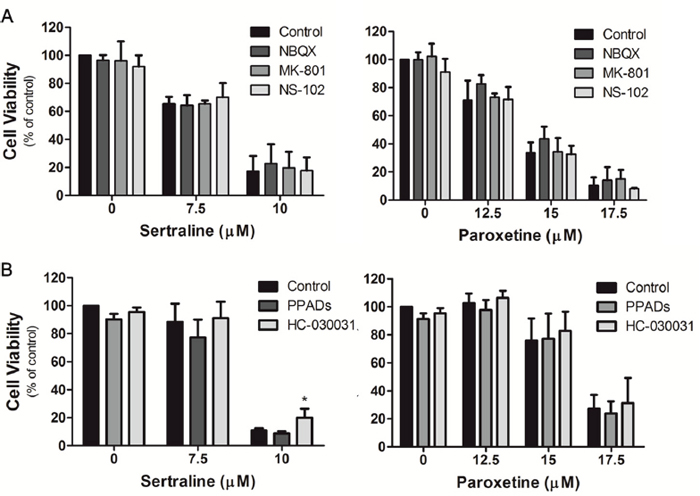
Figure 6: Sertraline- and paroxetine-induced cell death was not associated with glutamate receptors, P2 receptor or TRPA1 receptor. Astrocytes were pretreated with (A) NBQX, MK-801, NS-102, (B) PPADs, and HC-030031, and then exposed to sertraline and paroxetine. Cell viability was analyzed by an MTT assay. Data were collected from three independent experiments and statistically analyzed by Student’s t-test, and results are shown as the mean ± SD. *p<0.05, **p<0.01, ***p<0.001 compared to the control group.
DISCUSSION
We found that sertraline and paroxetine, two SSRIs, reduced CTX TNA2 astrocyte viability but not the other nine commonly used antidepressants, which include three SSRIs, one SNRI, two TCAs, one TeCA, one MAOI, and one SARI. Sertraline and paroxetine initiated a complex response of astrocyte apoptosis. It is shown that calcium overload induced mitochondrial dysfunction, which eventually leads to the activation of an intrinsic apoptotic pathway (Figure 7). Consistent results of sertraline- and paroxetine-induced cell death and mitochondrial dysfunction were also observed in primary astrocytes.
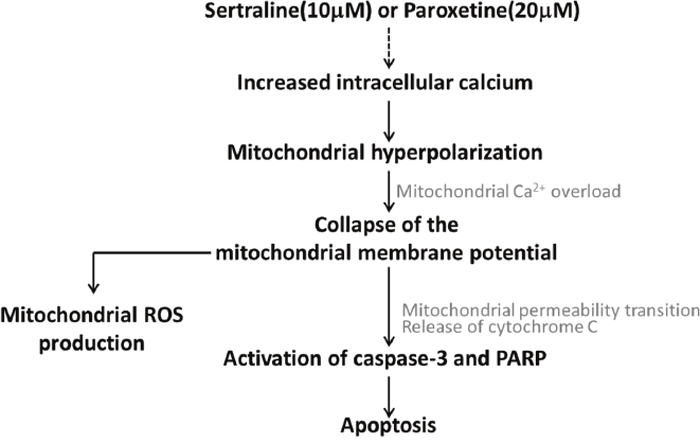
Figure 7: Working model related to sertraline- and paroxetine-induced astrocyte apoptosis. Treatments with sertraline and paroxetine led to induction of intracellular calcium, mitochondrial hyperpolarization followed by mitochondrial damage, and reactive oxygen species (ROS) generation. Ultimately, caspase-3 and poly(ADP-ribose) polymerase (PARP) proteins were activated, and apoptosis occurred.
At physiological levels, calcium wave is used for communication among neighboring astrocytes by gap-junctions and works together in the regulation of neuronal and non-neuronal functions of astrocytes, including synaptic neurotransmitter release, synaptic plasticity, and neuronal activity [34]. However, in a pathological state, an intracellular calcium rise is thought to play a primary role in initiating programmed cell death of neurons [46–48] and astrocytes [27, 49, 50]. Chiesa et al. suggested that perturbation of calcium homeostasis is sufficient to activate apoptosis in glial cells according to evidence of chromatin condensation, nucleus fragmentation, and genomic DNA laddering of astrocytes under a condition of calcium deprivation [49]. Astrocyte apoptosis was shown in several neurodegenerative diseases, including AD [51, 52]. Ultimately, astrocyte dysfunction renders them unable to execute housekeeping functions which further lead to neurodegenerative disorders.
Calcium influx is gated by several astroglial ionotropic receptors either directly (through receptor pores) or indirectly (by activating other Ca2+-permeable channels/transporters/exchangers), and they are the AMPAR [53, 54], NMDA receptor [54, 55], P2 receptors [54–56], TRPA1 channels [57], and etc. Lalo et al. demonstrated that AMPA and NMDA glutamate receptors [58] and P2X1/5 purinoceptor [59] are expressed by mouse cortical astrocytes, and their activation can evoke increased astrocytic [Ca2+]i [55]. We observed the intracellular calcium rapidly rise at 1 h after treatment with sertraline and paroxetine. The dose-dependently induction of intracellular calcium levels by sertraline and paroxetine is also further support the idea that sertraline- and paroxetine-induced cell death of astrocyte due to calcium overload. However, the cell death of astrocytes could not be blocked by glutamate receptor inhibitors or ATP-gated ion channel inhibitors. Our results indicated that this calcium overload was not contributed to by activation of glutamate receptor or ATP-activated ion channel, since their blockage could not reverse antidepressant-induced astrocyte cell death. Therefore, we identified that intracellular Ca2+ elevation is invoked as a trigger for the apoptotic effects; however, the mechanism of sertraline- and paroxetine-induced calcium overload remains unclear. Astrocyte calcium signaling is critical for the release of gliotransmitters which act on neurons and vascular smooth muscles, while their dysregulation might be involved in several diseases including AD, HD, and epilepsy [60]. Therefore, whether sertraline and paroxetine disrupt astrocyte calcium signaling and further cause neurodegenerative diseases is an intriguing issue.
In addition, our results showed that sertraline and paroxetine caused mitochondrial hyperpolarization at 3 h. Two peaks respectively represent populations of hyperpolarization and hypopolarization of the MMP. This phenomenon indicated that astrocyte mitochondria were damaged after mitochondrial hyperpolarization, and this may possibly due to calcium overload. Previous studies showed several possible mechanisms of mitochondrial hyperpolarization, which included increased electron transport through mitochondrial respiratory chain (MRC) complexes [61], uncoupling of F0F1 ATP synthase [62], and cytosolic calcium rise [63]. Furthermore, we also showed the possibility of ROS formation may be a consequence of cell damage, not being the dominant pathway in inducing apoptosis.
Our previous article suggested that several antidepressant users were more likely to develop dementia, regardless their depression status [26]. Several studies showed consistent results with our observation and will be further discussed. A meta-analysis conducted by Moraros et al. revealed that antidepressant usage was associated with an increased odds of some types of cognitive impairment or dementia, and consistent results were revealed among two populations who were aged ≥65 (odds ratio (OR)=1.65) or <65 years (OR=3.25) [25]. In addition, Chen et al. demonstrated that sertraline activates the mitogen-activated protein kinase (MAPK) pathway and further triggers apoptosis in hepatic cells [23]. Mitochondrial dysfunction is also a possible mechanism of sertraline-associated hepatotoxicity [64]. Jason et al. indicated that sertraline and paroxetine significantly decreased cell viability and increased apoptosis of human osteoclasts and osteoblasts. Additionally, similar results indicated that citalopram was relatively safe to osteoclasts and osteoblasts, compared to the two other aforementioned SSRIs [24]. Consistent with our results proposing sertraline-induced calcium overload, Huang et al. demonstrated that sertraline induced a calcium rise in human PC3 prostate cancer cells, human OC2 oral cancer cells, and human MG63 osteosarcoma cells via its release from endoplasmic reticulum and multiple calcium influx pathways [65–67]. However, paroxetine evoked Ca2+-independent apoptosis via activation of p38 MAPK-associated caspase-3 and protein kinase C in human osteosarcoma cells [68] and renal tubular cells [69], respectively. Meanwhile, Taler et al. suggested that sertraline caused potent neurotrophic activity in human neuroblastoma cells and conferred a pro-cognitive effect by improving spatial learning memory [70]. The above study showed that lower concentration (below 10 μM) of sertraline and paroxetine enhanced cell viability of neuroblastoma cells [70]; however, in the present study, when the concentration of sertraline and paroxetine was elevated to more than 10 μM, the antidepressants significantly reduced cell viability. In addition, Peng et al. suggested that sertraline is neuroprotective in an HD mouse model from the aspect of improvement of motor performance and amelioration of brain atrophy. Steiner et al. demonstrated that paroxetine was neuroprotective against HIV Tat and gp120 and other mitochondrial toxins and enhanced proliferation of neural progenitor cells in a gp120 neurotoxicity transgenic model [71]. A recent article indicated the anti-inflammatory effect of paroxetine in the impairment of lipopolysaccharide-induced microglial activation [72].
Reis et al. showed that median serum concentrations of sertraline and paroxetine were 67 and 131 nM, respectively; however, no literature shows the information about antidepressant concentrations in the CNS [73]. These antidepressants might occur in low concentrations in the CNS; however, the development of astrocytic toxicity and further astrocyte-mediated neurotoxicity would be long-term processes and might result in brain degenerative changes. The prevalence of antidepressant use in Taiwan was up to 4.63% in 2009 [74]; hence, it is necessary to evaluate the safety of different classes of antidepressants. To the present, this is the first study to illustrate the possible mechanism of cytotoxicity of sertraline and paroxetine on astrocytes via calcium overload. This study is an extension of our previously published article on the potential risk of antidepressant medication [26]. Based on the fact that astrocyte dysfunction threatening the survival of neurons, we conclude that antidepressant-induced astrocyte apoptosis might be involved in the development of neurodegenerative diseases. More studies are warranted to further elucidate other possible effects and mechanisms of antidepressants in astrocyte death and neurodegeneration.
MATERIALS AND METHODS
Cell culture
The rat astrocyte CTX-TNA2 cell line was obtained from American Type Culture Collection and primary cortical neuron cells were obtained from Thermo Fisher Scientific. CTX-TNA2 cells were grown in Dulbecco’s modified Eagle’s medium (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (Biological Industries). Primary astrocyte culture and astrocyte-neuron primary mixed cultures of postnatal day 1-3 Sprague-Dawley rats (BioLASCO Taiwan, Yilan, Taiwan) were prepared as previously described [75].
Cell survival analysis
MTT assay was applied to assess cell viability. Briefly, 0.25 mg/mL MTT (Sigma) was added to cells at 37 °C for 1 h, and the absorbance at 595 nm of MTT-formazan was detected spectrophotometrically using an ELISA reader (μQuant, Bio-Tek) after dissolution of the crystals in isopropanol. The percentage of cell viability was calculated by the formula: [Experimental group / Control group] x 100%.
Cell cycle analysis by flow cytometry
To analyze the cell cycle of astrocyte cells, trypsin-EDTA was applied to dissociate cells from the culture dish, and cells were spun down at 8944 ×g for 5 min at 4 °C. The supernatant was removed, and the cell pellet was suspended in 70% v/v ethanol at -20 °C overnight. After the ethanol was removed by centrifugation, 0.5 mL of 0.5% Triton X-100 with RNase A (7 μg/ml) was used to suspend cell pellets, which were then incubated at 37 °C for 30 min. Ultimately, 50 μg/ml propidium iodide (PI, Sigma) was added to the tube, and the fluorescent intensity at 637 nm was detected.
Measurement of ROS generation by intact cells
Intracellular production of ROS by CTX-TNA2 cells was detected by oxidation of the probes DCFH-DA to DCF. DCFH-DA can readily enter cells due to its non-polar property. It is trapped within cells once it is hydrolyzed to the non-fluorescent polar derivative, DCFH. It turns into the highly fluorescent DCF if it undergoes oxidization. Before different treatments, cells were incubated in the dark for 1 h at 37 °C with 50 μM DCFH-DA. Cells were harvested at 6, 12, and 24 h after treatment and were suspended in plain medium. CTX-TNA2 cells of each sample were analyzed, and the intracellular fluorescence was detected using a FACScan (Becton Dickinson, Sunnyvale, CA) flow cytometer with excitation at 488 nm and emission at 530 nm. The rise in peroxide levels was quantitated by measuring the percentage of cells at the M1 interval.
Measurement of the mitochondrial membrane potential (MMP)
Cells were treated with the indicated concentration of sertraline and paroxetine for 1.5, 3, 6, 12, and 24 h. Before being harvested, cells were incubated with 40 nM DiOC6(3) (Sigma) for 30 min at 37 °C. After that, cells were washed and suspended in phosphate-buffered saline (PBS). DiOC6(3) fluorescence intensities of FL-1 (530±15 nm) were measured with a flow cytometer CellQuest program (FACScan, Becton Dickinson).
Cellular adenosine triphosphate (ATP) level measurement
ATP content, as a measurement of mitochondrial function, was detected and quantified by CellTiter-Glo Luminescent Cell Viability Assay (Promega Corporation, Madison, WI). The luminescent signals released from the assay were measured with a Spark™ 10M multimode microplate reader (Tecan Trading AG, Männedorf, Switzerland). The cellular ATP content was calculated by comparing the luminescence of the treated cells with that of the control.
Intracellular calcium ([Ca2+]i) measurement
After treatment with antidepressants, cells were loaded with 5 μM Fluo-4, AM (Molecular Probes) for 30 min, and then real-time [Ca2+]i images were immediately captured by fluorescence microscopy (Olympus IX81 microscope, NY, USA). To quantitate [Ca2+]i, cells were suspended in PBS, and the fluorescence intensities were measured with a flow cytometer (FACScan, Becton Dickinson) equipped with an excitation wavelength of 494 nm and an emission wavelength of 516 nm.
Western blot analysis
Cell lysates were prepared by suspending cells in RIPA lysis buffer (50 mM Tris-HCl at pH 7.4, 1% Nonidet P-40, 150 mM sodium chloride, 1 mM sodium fluoride, 1 mM sodium orthovanadate, and 1 mM phenylmethylsulfonyl fluoride). An equal amount of protein was separated on 10% sodium dodecylsulfate-polyacrylamide gels and transferred to polyvinylidene difluoride membranes (Millipore). Membranes were probed with specific antibodies: anti-α-tubulin (Neomarker), anti-caspase-3 (Cell Signaling), anti-PARP (Imgenes) antibodies, and then probed with horseradish peroxidase (HRP)-labeled secondary antibodies. After enhanced chemiluminescent substrate (ECL) was added to the membrane, images were captured by the BioSpectrum Imaging System (AnAnalytik Jena Company, Ultra-Violet Products Ltd).
Statistical analysis
Data are expressed as the mean ± standard deviation (SD), and each value in this study was repeated from three independent experiments. Statistical significance was determined using Student’s t-test. p values of <0.05, <0.01, and <0.001 were considered statistically significant differences.
Abbreviations
AD: Alzheimer’s disease; HD: Huntington’s disease; PD: Parkinson’s disease; CNS: central nervous system (CNS); SSRIs: selective serotonin reuptake inhibitors; SNRIs: serotonin-norepinephrine reuptake inhibitors TCAs: tricyclic antidepressants; TeCA: tetracyclic antidepressant; SARIs: serotonin antagonist and reuptake inhibitors; MAOIs: monoamine oxidase inhibitors; ATP: adenosine triphosphate; IP3: inositol-1:4:5-trisphosphate; AMPA: α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; NMDA: N-methyl-D-aspartate; NF-κB: nuclear factor-κB; ROS: reactive oxygen species; PTP: permeability transition pore; MMP: mitochondrial membrane potential; PARP: poly(ADP-ribose) polymerase; MRC: mitochondrial respiratory chain; OR: odds ratio; MAPK: mitogen-activated protein kinase; PI: propidium iodide; PBS: phosphate-buffered saline; HRP: horseradish peroxidase; ECL: enhanced chemiluminescent substrate; SD: standard deviation.
Author contributions
Chee-Kin Then takes responsibilities for the research, data, and their analyses and interpretation. Chee-Kin Then, Kao-Hui Liu, Kuo-Hsuan Chung, Jia-Yi Wang, Ming-Hsuan Liao and Shing-Chuan Shen takes responsibilities for methodology and project administration. Shing-Chuan Shen takes additional responsibilities for the accuracy of data analyses. Kuo-Hsuan Chung, Jia-Yi Wang and Shing-Chuan Shen also critically revised the manuscript for important intellectual content. Shing-Chuan Shen has a full access to all data in this study, have the right to publish any or all of the data, and take responsibility for the final manuscript.
ACKNOWLEDGMENTS
We thank the Department of Biochemistry and Molecular Cell Biology of Taipei Medical University (TMU) provided the Spark™ 10M multimode microplate reader (Tecan Trading AG, Männedorf, Switzerland).
CONFLICTS OF INTEREST
We have confirmed that there are no known conflicts of interest associated with this publication.
GRANT SUPPORT
This work was supported by the Ministry of Science and Technology (MOST), Taiwan [grant numbers MOST105-2314-B-038-013-MY3(1-3), MOST103-2320-B-038-024, MOST104-2923-B-038-001- MY3(3–3)]; the National Science Council (NSC), Taiwan [grant number NSC 102-2314-B-038-042]; and Taipei Medical University (TMU) [grant number TMU104-F-004].
REFERENCES
1. Araque A, Navarrete M. Glial cells in neuronal network function. Philos Trans R Soc Lond B Biol Sci. 2010; 365: 2375-81. https://doi.org/10.1098/rstb.2009.0313.
2. Mattson MP. Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol. 2000; 1: 120-9. https://doi.org/10.1038/35040009.
3. Choonara YE, Pillay V, du Toit LC, Modi G, Naidoo D, Ndesendo VM, Sibambo SR. Trends in the molecular pathogenesis and clinical therapeutics of common neurodegenerative disorders. Int J Mol Sci. 2009; 10: 2510-57. https://doi.org/10.3390/ijms10062510.
4. Martin LJ. Neuronal cell death in nervous system development, disease, and injury (review). Int J Mol Med. 2001; 7: 455-78.
5. Maragakis NJ, Rothstein JD. Mechanisms of disease: astrocytes in neurodegenerative disease. Nat Clin Pract Neurol. 2006; 2: 679-89. https://doi.org/10.1038/ncpneuro0355.
6. Takuma K, Baba A, Matsuda T. Astrocyte apoptosis: implications for neuroprotection. Prog Neurobiol. 2004; 72: 111-27. https://doi.org/10.1016/j.pneurobio.2004.02.001.
7. Nagele RG, D’Andrea MR, Lee H, Venkataraman V, Wang HY. Astrocytes accumulate A beta 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res. 2003; 971: 197-209. https://doi.org/10.1016/S0006-8993(03)02361-8.
8. Hirrlinger J, Schulz JB, Dringen R. Glutathione release from cultured brain cells: multidrug resistance protein 1 mediates the release of GSH from rat astroglial cells. J Neurosci Res. 2002; 69: 318-26. https://doi.org/10.1002/jnr.10308.
9. Zeevalk GD, Rampour R, Bernard LP. Glutathione and Parkinson’s disease: is this the elephant in the room? Biomed Pharmacother. 2008; 62: 236-49. https://doi.org/10.1016/j.biopha.2008.01.017.
10. Lee HJ, Suk JE, Patrick C, Bae EJ, Cho JH, Rho S, Hwang D, Masliah E, Lee SJ. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem. 2010; 285: 9262-72. https://doi.org/10.1074/jbc.M109.081125.
11. McNaught KS, Carrupt PA, Altomare C, Cellamare S, Carotti A, Testa B, Jenner P, Marsden CD. Isoquinoline derivatives as endogenous neurotoxins in the aetiology of Parkinson’s disease. Biochem Pharmacol. 1998; 56: 921-33. https://doi.org/10.1016/S0006-2952(98)00142-7.
12. Rappold PM, Tieu K. Astrocytes and therapeutics for Parkinson’s disease. Neurotherapeutics. 2010; 7: 413-23. https://doi.org/10.1016/j.nurt.2010.07.001.
13. Gorman JM, Kent JM. SSRIs and SNRIs: broad spectrum of efficacy beyond major depression. J Clin Psychiatry. 1999; 60: 33-8; discussion 9.
14. Bleakley S. Review of the choice and use of antidepressant drugs. Prog Neurol Psychiatry. 2013.
15. Hunsberger J, Austin DR, Henter ID, Chen G. The neurotrophic and neuroprotective effects of psychotropic agents. Dialogues Clin Neurosci. 2009; 11: 333-48.
16. Yang D, Chen M, Russo-Neustadt A. Antidepressants are neuroprotective against nutrient deprivation stress in rat hippocampal neurons. Eur J Neurosci. 2012; 36: 2573-87. https://doi.org/10.1111/j.1460-9568.2012.08187.x.
17. Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci. 1995; 15: 7539-47.
18. Shilyansky C, Williams LM, Gyurak A, Harris A, Usherwood T, Etkin A. Effect of antidepressant treatment on cognitive impairments associated with depression: a randomised longitudinal study. Lancet Psychiatry. 2016; 3: 425-35. https://doi.org/10.1016/S2215-0366(16)00012-2.
19. Andrews PW, Thomson JA Jr, Amstadter A, Neale MC. Primum non nocere: an evolutionary analysis of whether antidepressants do more harm than good. Front Psychol. 2012; 3: 117. https://doi.org/10.3389/fpsyg.2012.00117.
20. Schmitt JA, Ramaekers JG, Kruizinga MJ, van Boxtel MP, Vuurman EF, Riedel WJ. Additional dopamine reuptake inhibition attenuates vigilance impairment induced by serotonin reuptake inhibition in man. J Psychopharmacol. 2002; 16: 207-14. https://doi.org/10.1177/026988110201600303.
21. Rosenberg PB, Drye LT, Martin BK, Frangakis C, Mintzer JE, Weintraub D, Porsteinsson AP, Schneider LS, Rabins PV, Munro CA, Meinert CL, Lyketsos CG, DIADS-2 Research Group. Sertraline for the treatment of depression in Alzheimer disease. Am J Geriatr Psychiatry. 2010; 18: 136-45. https://doi.org/10.1097/JGP.0b013e3181c796eb.
22. Banerjee S, Hellier J, Dewey M, Romeo R, Ballard C, Baldwin R, Bentham P, Fox C, Holmes C, Katona C, Knapp M, Lawton C, Lindesay J, et al. Sertraline or mirtazapine for depression in dementia (HTA-SADD): a randomised, multicentre, double-blind, placebo-controlled trial. Lancet. 2011; 378: 403-11. https://doi.org/10.1016/S0140-6736(11)60830-1.
23. Chen S, Xuan J, Wan L, Lin H, Couch L, Mei N, Dobrovolsky VN, Guo L. Sertraline, an antidepressant, induces apoptosis in hepatic cells through the mitogen-activated protein kinase pathway. Toxicol Sci. 2014; 137: 404-15. https://doi.org/10.1093/toxsci/kft254.
24. Hodge JM, Wang Y, Berk M, Collier FM, Fernandes TJ, Constable MJ, Pasco JA, Dodd S, Nicholson GC, Kennedy RL, Williams LJ. Selective serotonin reuptake inhibitors inhibit human osteoclast and osteoblast formation and function. Biol Psychiatry. 2013; 74: 32-9. https://doi.org/10.1016/j.biopsych.2012.11.003.
25. Moraros J, Nwankwo C, Patten SB, Mousseau DD. The association of antidepressant drug usage with cognitive impairment or dementia, including Alzheimer disease: a systematic review and meta-analysis. Depress Anxiety. 2017; 34: 217-26. https://doi.org/10.1002/da.22584.
26. Then CK, Chi NF, Chung KH, Kuo L, Liu KH, Hu CJ, Shen SC, Lin YK. Risk analysis of use of different classes of antidepressants on subsequent dementia: a nationwide cohort study in Taiwan. PLoS One. 2017. doi: ARTN e0175187 10.1371/journal.pone.0175187.
27. Guo GW, Liang YX. Aluminum-induced apoptosis in cultured astrocytes and its effect on calcium homeostasis. Brain Res. 2001; 888: 221-6.
28. Takuma K, Phuagphong P, Lee E, Mori K, Baba A, Matsuda T. Anti-apoptotic effect of cGMP in cultured astrocytes: inhibition by cGMP-dependent protein kinase of mitochondrial permeable transition pore. J Biol Chem. 2001; 276: 48093-9. https://doi.org/10.1074/jbc.M108622200.
29. Chen SH, Lin JK, Liu SH, Liang YC, Lin-Shiau SY. Apoptosis of cultured astrocytes induced by the copper and neocuproine complex through oxidative stress and JNK activation. Toxicol Sci. 2008; 102: 138-49. https://doi.org/10.1093/toxsci/kfm292.
30. Takuma K, Lee E, Kidawara M, Mori K, Kimura Y, Baba A, Matsuda T. Apoptosis in Ca2 + reperfusion injury of cultured astrocytes: roles of reactive oxygen species and NF-kappaB activation. Eur J Neurosci. 1999; 11: 4204-12.
31. Zhang C, Wang C, Ren J, Guo X, Yun K. Morphine protects spinal cord astrocytes from glutamate-induced apoptosis via reducing endoplasmic reticulum stress. Int J Mol Sci. 2016. https://doi.org/10.3390/ijms17101523.
32. Keane RW, Srinivasan A, Foster LM, Testa MP, Ord T, Nonner D, Wang HG, Reed JC, Bredesen DE, Kayalar C. Activation of CPP32 during apoptosis of neurons and astrocytes. J Neurosci Res. 1997; 48: 168-80.
33. Agulhon C, Petravicz J, McMullen AB, Sweger EJ, Minton SK, Taves SR, Casper KB, Fiacco TA, McCarthy KD. What is the role of astrocyte calcium in neurophysiology? Neuron. 2008; 59: 932-46. https://doi.org/10.1016/j.neuron.2008.09.004.
34. Bazargani N, Attwell D. Astrocyte calcium signaling: the third wave. Nat Neurosci. 2016; 19: 182-9. https://doi.org/10.1038/nn.4201.
35. Matsuda T, Takuma K, Nishiguchi E, Hashimoto H, Azuma J, Baba A. Involvement of Na+-Ca2+ exchanger in reperfusion-induced delayed cell death of cultured rat astrocytes. Eur J Neurosci. 1996; 8: 951-8.
36. Swarnkar S, Goswami P, Kamat PK, Gupta S, Patro IK, Singh S, Nath C. Rotenone-induced apoptosis and role of calcium: a study on Neuro-2a cells. Arch Toxicol. 2012; 86: 1387-97. https://doi.org/10.1007/s00204-012-0853-z.
37. Bondarenko A, Chesler M. Calcium dependence of rapid astrocyte death induced by transient hypoxia, acidosis, and extracellular ion shifts. Glia. 2001; 34: 143-9.
38. Liu KH, Yang ST, Lin YK, Lin JW, Lee YH, Wang JY, Hu CJ, Lin EY, Chen SM, Then CK, Shen SC. Fluoxetine, an antidepressant, suppresses glioblastoma by evoking AMPAR-mediated calcium-dependent apoptosis. Oncotarget. 2015; 6: 5088-101. https://doi.org/10.18632/oncotarget.3243.
39. Glaum SR, Holzwarth JA, Miller RJ. Glutamate receptors activate Ca2+ mobilization and Ca2+ influx into astrocytes. Proc Natl Acad Sci U S A. 1990; 87: 3454-8.
40. Condorelli DF, Dell’Albani P, Corsaro M, Barresi V, Giuffrida Stella AM. AMPA-selective glutamate receptor subunits in astroglial cultures. J Neurosci Res. 1993; 36: 344-56. https://doi.org/10.1002/jnr.490360312.
41. Jimenez-Blasco D, Santofimia-Castano P, Gonzalez A, Almeida A, Bolanos JP. Astrocyte NMDA receptors’ activity sustains neuronal survival through a Cdk5-Nrf2 pathway. Cell Death Differ. 2015; 22: 1877-89. https://doi.org/10.1038/cdd.2015.49.
42. Garcia-Barcina JM, Matute C. Expression of kainate-selective glutamate receptor subunits in glial cells of the adult bovine white matter. Eur J Neurosci. 1996; 8: 2379-87.
43. Abbracchio MP, Ceruti S. Roles of P2 receptors in glial cells: focus on astrocytes. Purinergic Signal. 2006; 2: 595-604. https://doi.org/10.1007/s11302-006-9016-0.
44. Fumagalli M, Brambilla R, D’Ambrosi N, Volonte C, Matteoli M, Verderio C, Abbracchio MP. Nucleotide-mediated calcium signaling in rat cortical astrocytes: role of P2X and P2Y receptors. Glia. 2003; 43: 218-03. https://doi.org/10.1002/glia.10248.
45. Shigetomi E, Jackson-Weaver O, Huckstepp RT, O’Dell TJ, Khakh BS. TRPA1 channels are regulators of astrocyte basal calcium levels and long-term potentiation via constitutive D-serine release. J Neurosci. 2013; 33: 10143-53. https://doi.org/10.1523/JNEUROSCI.5779-12.2013.
46. Tecoma ES, Monyer H, Goldberg MP, Choi DW. Traumatic neuronal injury in vitro is attenuated by NMDA antagonists. Neuron. 1989; 2: 1541-5.
47. Dunlap K, Luebke JI, Turner TJ. Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 1995; 18: 89-98.
48. Joseph R, Li W, Han E. Neuronal death, cytoplasmic calcium and internucleosomal DNA fragmentation: evidence for DNA fragments being released from cells. Brain Res Mol Brain Res. 1993; 17: 70-6.
49. Chiesa R, Angeretti N, Del Bo R, Lucca E, Munna E, Forloni G. Extracellular calcium deprivation in astrocytes: regulation of mRNA expression and apoptosis. J Neurochem. 1998; 70: 1474-83.
50. Rakkar K, Bayraktutan U. Increases in intracellular calcium perturb blood-brain barrier via protein kinase C-alpha and apoptosis. Biochim Biophys Acta. 2016; 1862: 56-71. https://doi.org/10.1016/j.bbadis.2015.10.016.
51. Nishimura T, Akiyama H, Yonehara S, Kondo H, Ikeda K, Kato M, Iseki E, Kosaka K. Fas antigen expression in brains of patients with Alzheimer-type dementia. Brain Res. 1995; 695: 137-45.
52. Smale G, Nichols NR, Brady DR, Finch CE, Horton WE Jr. Evidence for apoptotic cell death in Alzheimer’s disease. Exp Neurol. 1995; 133: 225-30. https://doi.org/10.1006/exnr.1995.1025.
53. Lind BL, Brazhe AR, Jessen SB, Tan FC, Lauritzen MJ. Rapid stimulus-evoked astrocyte Ca2+ elevations and hemodynamic responses in mouse somatosensory cortex in vivo. Proc Natl Acad Sci U S A. 2013; 110: E4678-87. https://doi.org/10.1073/pnas.1310065110.
54. Hamilton N, Vayro S, Kirchhoff F, Verkhratsky A, Robbins J, Gorecki DC, Butt AM. Mechanisms of ATP- and glutamate-mediated calcium signaling in white matter astrocytes. Glia. 2008; 56: 734-49. https://doi.org/10.1002/glia.20649.
55. Palygin O, Lalo U, Verkhratsky A, Pankratov Y. Ionotropic NMDA and P2X1/5 receptors mediate synaptically induced Ca2+ signalling in cortical astrocytes. Cell Calcium. 2010; 48: 225-31. https://doi.org/10.1016/j.ceca.2010.09.004.
56. Newman EA. Calcium increases in retinal glial cells evoked by light-induced neuronal activity. J Neurosci. 2005; 25: 5502-10. https://doi.org/10.1523/JNEUROSCI.1354-05.2005.
57. Shigetomi E, Tong X, Kwan KY, Corey DP, Khakh BS. TRPA1 channels regulate astrocyte resting calcium and inhibitory synapse efficacy through GAT-3. Nat Neurosci. 2011; 15: 70-80. https://doi.org/10.1038/nn.3000.
58. Lalo U, Pankratov Y, Kirchhoff F, North RA, Verkhratsky A. NMDA receptors mediate neuron-to-glia signaling in mouse cortical astrocytes. J Neurosci. 2006; 26: 2673-83. https://doi.org/10.1523/JNEUROSCI.4689-05.2006.
59. Lalo U, Pankratov Y, Wichert SP, Rossner MJ, North RA, Kirchhoff F, Verkhratsky A. P2X1 and P2X5 subunits form the functional P2X receptor in mouse cortical astrocytes. J Neurosci. 2008; 28: 5473-80. https://doi.org/10.1523/JNEUROSCI.1149-08.2008.
60. Bazargani N, Attwell D. Astrocyte calcium signaling: the third wave. Nat Neurosci. 2016; 19: 182-9. https://doi.org/10.1038/nn.4201.
61. Joshi B, Li L, Taffe BG, Zhu ZY, Wahl S, Tian H, Ben-Josef E, Taylor JD, Porter AT, Tang DG. Apoptosis induction by a novel anti-prostate cancer compound, BMD188 (a fatty acid-containing hydroxamic acid), requires the mitochondrial respiratory chain. Cancer Res. 1999; 59: 4343-55.
62. Giovannini C, Matarrese P, Scazzocchio B, Sanchez M, Masella R, Malorni W. Mitochondria hyperpolarization is an early event in oxidized low-density lipoprotein-induced apoptosis in Caco-2 intestinal cells. FEBS Lett. 2002; 523: 200-6. https://doi.org/10.1016/S0014-5793(02)02972-1.
63. Perry SW, Norman JP, Barbieri J, Brown EB, Gelbard HA. Mitochondrial membrane potential probes and the proton gradient: a practical usage guide. Biotechniques. 2011; 50: 98-115. https://doi.org/10.2144/000113610.
64. Li Y, Couch L, Higuchi M, Fang JL, Guo L. Mitochondrial dysfunction induced by sertraline, an antidepressant agent. Toxicol Sci. 2012; 127: 582-91. https://doi.org/10.1093/toxsci/kfs100.
65. Huang JK, Chang HT, Chou CT, Shu SS, Kuo CC, Tsai JY, Liao WC, Wang JL, Lin KL, Lu YC, Chen IS, Liu SI, Ho CM, et al. The mechanism of sertraline-induced [Ca2+](i) rise in human PC3 prostate cancer cells. Basic Clin Pharmacol Toxicol. 2011; 109: 103-10. https://doi.org/10.1111/j.1742-7843.2011.00690.x.
66. Chien JM, Chou CT, Pan CC, Kuo CC, Tsai JY, Liao WC, Kuo DH, Shieh P, Ho CM, Chu ST, Su HH, Chi CC, Jan CR. The mechanism of sertraline-induced [Ca2+](i) rise in human OC2 oral cancer cells. Hum Exp Toxicol. 2011; 30: 1635-43. https://doi.org/10.1177/0960327110396523.
67. Lin KL, Chi CC, Lu T, Tseng LL, Wang JL, Lu YC, Jan CR. Effect of sertraline on [Ca2+](i) and viability of human MG63 osteosarcoma cells. Drug Chem Toxicol. 2013; 36: 231-40. https://doi.org/10.3109/01480545.2012.710625.
68. Chou CT, He SP, Jan CR. Paroxetine-induced apoptosis in human osteosarcoma cells: activation of p38 MAP kinase and caspase-3 pathways without involvement of [Ca2+](i) elevation. Toxicol Appl Pharmacol. 2007; 218: 265-73. https://doi.org/10.1016/j.taap.2006.11.012.
69. Chen WC, Huang CC, Huang CJ, Chien JM, Lin KL, Lu YC, Chen IS, Liu SI, Hsu SS, Chang HT, Chou CT, Jan CR. Mechanism of paroxetine-induced cell death in renal tubular cells. Basic Clin Pharmacol Toxicol. 2008; 103: 407-13. https://doi.org/10.1111/j.1742-7843.2008.00319.x.
70. Taler M, Miron O, Gil-Ad I, Weizman A. Neuroprotective and procognitive effects of sertraline: in vitro and in vivo studies. Neurosci Lett. 2013; 550: 93-7. https://doi.org/10.1016/j.neulet.2013.06.033.
71. Steiner JP, Bachani M, Wolfson-Stofko B, Lee MH, Wang T, Li G, Li W, Strayer D, Haughey NJ, Nath A. Interaction of paroxetine with mitochondrial proteins mediates neuroprotection. Neurotherapeutics. 2015; 12: 200-16. https://doi.org/10.1007/s13311-014-0315-9.
72. Liu RP, Zou M, Wang JY, Zhu JJ, Lai JM, Zhou LL, Chen SF, Zhang X, Zhu JH. Paroxetine ameliorates lipopolysaccharide-induced microglia activation via differential regulation of MAPK signaling. J Neuroinflammation. 2014; 11: 47. https://doi.org/10.1186/1742-2094-11-47.
73. Reis M, Aamo T, Spigset O, Ahlner J. Serum concentrations of antidepressant drugs in a naturalistic setting: compilation based on a large therapeutic drug monitoring database. Ther Drug Monit. 2009; 31: 42-56. https://doi.org/10.1097/FTD.0b013e31819114ea.
74. Wu CS, Shau WY, Chan HY, Lee YC, Lai YJ, Lai MS. Utilization of antidepressants in Taiwan: a nationwide population-based survey from 2000 to 2009. Pharmacoepidemiol Drug Saf. 2012; 21: 980-8. https://doi.org/10.1002/pds.3255.
75. Lin MS, Sun YY, Chiu WT, Hung CC, Chang CY, Shie FS, Tsai SH, Lin JW, Hung KS, Lee YH. Curcumin attenuates the expression and secretion of RANTES after spinal cord injury in vivo and lipopolysaccharide-induced astrocyte reactivation in vitro. J Neurotrauma. 2011; 28: 1259-69. https://doi.org/10.1089/neu.2011.1768.