
Introduction
While hematological malignancies represent less than 10 % of all cancer cases in the United States [1], studies into their pathogenesis have led to critical insights into the molecular mechanisms of cancer initiation and progression including fundamental paradigm shifts such as the stem cell hypothesis of cancer [2]. Hematological malignancies have also been at the forefront of studies that led to the development of molecularly targeted therapeutics [3]. Thus, identification of a novel driver of oncogenesis in hematological neoplasms is likely to shed new light on mechanisms of oncogenesis relevant to diverse types of cancer. It is in this context that recent identification of mutations in CBL in a small but significant proportion of patients with myeloid malignancies provides an important milestone. In this article, we review the basic functions of Cbl family proteins, survey their mutations in human patients and animal models that manifest as myeloproliferative/myelodysplastic syndromes and propose potential mechanisms of oncogenesis and possible strategies to treat patients with CBL mutations.
Cbl Family Proteins
Members of the Casitas B-lineage lymphoma (Cbl) protein family are evolutionarily-conserved multi-domain regulators of signal transduction (reviewed in [4-6]). In mammals, this family includes Cbl (also known as c-Cbl), Cbl-b and Cbl-c (also known as Cbl-3 or Cbl-SL [7]). Extensive biochemical studies have demonstrated that they act primarily as attenuators of cellular signals by functioning as E3 ubiquitin ligases directed towards protein tyrosine kinase (PTK) pathways [8-10]. The N-terminal regions of all Cbl family members are highly conserved; these include the tyrosine kinase binding (TKB) domain, the RING finger (RF) domain and the short linker region between these two domains. Structural analyses have shown that the TKB domain is composed of a four-helical bundle, a calcium-binding EF hand and a variant SH2 domain [11]; together, these domains constitute a relatively unique platform that mediates specific binding to cognate phosphotyrosine-containing motifs almost exclusively found in activated PTKs. The RING finger domain and the linker region together mediates binding to E2 ubiquitin-conjugating enzymes and both of these motifs are essential for the E3 ubiquitin ligase activity of Cbl proteins [12].
The carboxyl regions of Cbl family members are more divergent; while Cbl-c possesses a relatively short region carboxyl to the RF domain, Cbl and Cbl-b contain multiple protein-protein interactions motifs including: proline-rich regions that bind to SH3 domains of a number of signaling proteins such as Src family kinases and the Grb2 adaptor protein; tyrosine residues that become phosphorylated upon cellular activation and interact with key signaling mediators such as the Vav family guanine nucleotide exchange factors, the p85 regulatory subunit of phosphatidylinositol 3-kinase (PI3K) and Crk family adaptors; and leucine zipper/ubiquitin-associated (LZ/UBA) domain that is proposed to be involved in homo- and hetero-dimerization of Cbl proteins.
Conclusions based on biochemical evidence that Cbl proteins function as attenuators of mammalian PTKs have been validated by animal models that demonstrated enhanced biological responses when Cbl family members are genetically ablated. For example, Cbl-null mice show increased cellularity in the hematopoietic organs [13-15] whereas Cblb-null mice exhibit hyper-responsiveness to immunological insults leading to autoimmunity [16, 17]; this phenotype was further augmented to a fulminant inflammatory disease when Cbl and Cbl-b were concurrently deleted in the T cell compartment [18]. Given that Cbl proteins associate with a variety of growth factor receptors, their negative regulatory roles towards PTKs engendered an expectation that Cbl proteins may function as tumor suppressors and that their mutations and/or deletions could contribute as driving or accessory oncogenic mechanisms. This hypothesis was further supported by the historical background that Cbl was originally identified as a cellular homolog of a murine viral oncogene (v-Cbl); this fibroblast-transforming gene was a fusion of the Cbl TKB domain with the viral gag sequences [19]. Combined with studies on the Cbl linker/RF domain mutants including a pre-B cell line-derived 70Z Cbl as well as various engineered mutants [20], these data altogether indicated that the linker/RF domains were critical for a potential tumor suppressor role of Cbl proteins. Yet, a direct role of Cbl proteins in human cancers remained elusive until recently.
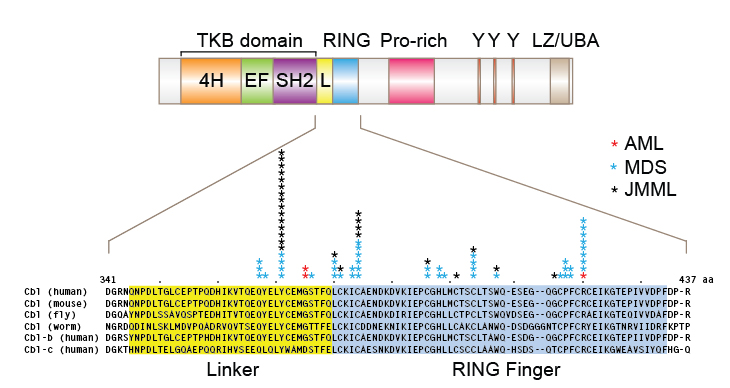
Schematic representation of mammalian Cbl, alignments of amino acid sequences for various Cbl family proteins, and positions of clinically-identified CBL missense mutations. Mutations were compiled from published papers. TKB, tyrosine kinase-binding domain; 4H, four-helix bundle; EF, EF hand; SH2, Src-homology domain 2; L, linker; RING, “really interesting new gene” finger domain; Y, tyrosine residue; LZ/UBA, leucine zipper/ubiquitin associated domain.
Mutant Cbl and Myeloid Malignancies
In 2007, two groups simultaneously identified CBL mutations in acute myeloid leukemia (AML) patient samples [21, 22]. Since then, a number of independent studies confirmed and extended these observations and a consensus has gradually emerged on the nature of CBL mutations and their clinical manifestations [23-32].
First, CBL mutations are most frequently observed in a distinct group of myeloid disorders, namely myelodysplastic syndromes-myeloproliferative neoplasms (MDS/MPN); this subgroup of hematological malignancies includes the chronic myelomonocytic leukemia (CMML), atypical chronic myeloid leukemia (aCML) and juvenile myelomonocytic leukemia (JMML). Notably, these malignancies are often associated with hallmark genetic aberrancies that culminate in activation of the Ras-MAPK signaling pathway [33, 34]. For example, the activating mutations of PTPN11, NRAS and KRAS, and loss of Ras GTPase-activating protein gene NF1 together account for approximately 75% cases of JMML. CBL mutations are now known to account for roughly half of the remaining cases. Among CMML patients, RAS mutations are reported in 20 to 60 % of the cases.
Second, most CBL mutations are missense mutations or small deletions around the linker region and within the RF domain. Where tested, these mutations have been shown to abrogate the E3 ubiquitin ligase activity of Cbl [21, 26, 28]. Complete CBL gene deletion, truncation, or mutations outside of the linker/RING finger regions are rare. These characteristics strongly suggest that the expression of mutant Cbl proteins confers growth and/or survival advantages over cells expressing wild-type Cbl or even those that have lost Cbl expression.
Third, a remarkable feature of patients with Cbl mutations is that the wild-type CBL allele is frequently lost in leukemic clones and is replaced with the mutant allele by acquired uniparental isodisomy (aUPD). Although mutations in CBLB and CBLC have also been reported, they appear to be rare compared to CBL mutations. In this regard, deletion of the wild-type CBL allele and acquisition of a second mutant allele may represent a defining oncogenic event; this hypothesis is most clearly supported by observations in JMML patients, where CBL mutations are often inherited as hemizygous germline mutations [32]. Apparently, a mutant Cbl protein encoded by a hemizygous CBL mutation is not sufficient to counter the function of Cbl family proteins encoded by the remaining wild-type CBL allele plus two wild-type CBLB alleles. This is further backed by experimental data that mutant Cbl proteins confer far more growth advantage on a Cbl-deficient background compared to a Cbl wild-type background [28, 32].
Then, how do the mutant Cbl proteins function? Considering the high degree of structural similarity between Cbl and Cbl-b (and given the relatively epithelial-restricted expression of Cbl-c), it is conceivable that mutant Cbl proteins function as a dominant-negative mutant towards Cbl-b (or, as seen in some patients, towards both Cbl and Cbl-b if aUPD has not occurred) in hematopoietic cells. Phenotypic differences between mice deficient in Cbl alone or both Cbl and Cbl-b in the hematopoietic compartment are also consistent with the notion that complete loss of Cbl functions is necessary to promote myeloid malignancy. While bone marrows showed an expansion of the hematopoietic stem/progenitor compartments in both genetic backgrounds, rapidly fatal myeloproliferative disorder with peripheral organ involvement was seen only in Cbl, Cblb double-deficient mice but not in Cbl-null mice even at an advanced age [15, 35, 36].
Additionally, mutant Cbl proteins may function as gain-of-function oncogenes. Considering that essentially all leukemia-associated Cbl mutants possess an intact TKB and C-terminal motifs that mediate interactions with a variety of signaling proteins, it is logical to hypothesize that these E3-defective proteins will be recruited to activated PTKs (via the TKB domain) and that this will lead to the formation of a signaling complex that lacks the negative regulatory function of wild-type Cbl. In this scenario, mutant Cbl proteins will associate more persistently with signaling intermediates through phosphotyrosine-containing motifs, proline-rich regions and other potential motifs. Thus, mutant Cbl proteins are likely to serve as supramolecular scaffolds to assemble aberrant signaling complexes that can promote hyperactivation of signaling pathways normally attenuated by E3-competent wild-type Cbl. This idea is compatible with more robust transforming ability of 70Z Cbl compared to v-Cbl-equivalent sequences when expressed in fibroblasts [20] and a stronger biochemical activation of PDGFR signaling cascade by 70Z Cbl and other full-length Cbl mutants compared to v-Cbl [37].
Further support for this idea comes from analyses of a mutant Cbl knock-in mouse model developed by Langdon and colleagues in which a Cbl RING finger mutant (C379A, equivalent to C381 in human) is expressed from the endogenous Cbl promoter [38]. While homozygous mutant mice show early lethality, heterozygous mutant mice with one wild-type Cbl allele do not show hematopoietic abnormalities. However, mice with one C379A mutant allele on a Cbl-null background (with wild-type Cblb) succumb to myeloid malignancies with a median survival time of 47 weeks [36]; although the disease in these mice develops later than in Cbl, Cblb double-deficient mice [35], the overall features of the disease are remarkably similar. Interestingly, Akt was constitutively activated in the C379A mutant hematopoietic cells but not in control or Cbl-null mutant cells [36]. This was accompanied by the enhanced phosphorylation of Y737 (corresponding to Y731 in human) of Cbl, an experimentally-proven binding site for the p85 regulatory subunit of PI3K. These findings in an in vivo model that phenocopies critical features of human diseases support the premise that an E3-deficient oncogenic Cbl mutant is capable of hyperactivating a key pathway through an associated signaling partner whose function would have been attenuated by wild-type Cbl. Thus, it will be highly pertinent in future studies to use this and other mouse models to identify and evaluate the activation status of PTKs that serve as physiological targets of Cbl proteins in hematopoietic stem cells, and to assess which signaling pathways are rendered hyperactive as a result of the expression of a gain-of-function Cbl mutant. Clearly, to distinguish between the dominant-negative and gain-of-function mechanisms of mutant Cbl-dependent oncogenesis, it is imperative to test their activities in cellular environments completely free of endogenous Cbl proteins. Cbl, Cblb double-deficient mice [35] as well as cells derived from these animals should be valuable research tools for this purpose.
Considering Cbl’s interaction with various signaling molecules, mutant Cbl is likely to affect a multitude of downstream pathways. We discussed the constitutive activation of the Akt-PI3K pathway above [36]. Because CBL mutations are seen in a sizable fraction of JMML cases, a disease entity strongly linked to hyperactivation of the Ras-Raf-MAPK pathway [34], activity of this pathway needs to be carefully evaluated. Indeed, Erk activation of LSK (Lin-Sca-1+c-Kit+) cells in response to Flt3 ligand stimulation is prolonged in C379A mice [36], suggesting that this is another potential pathway affected by CBL mutation. Molecular basis of Cbl-mediated regulation of the MAPK pathway has been proposed previously [6]. Furthermore, mutant Cbl proteins may also influence cytoskeletal reorganization and cell motility through their interaction with the Rac1 and Cdc42 pathways [39, 40]. Comprehensive structure-function analyses using ectopic expression of mutant Cbl proteins in hematopoietic stem cells and, in the long run, from its endogenous promoter through knock-in approaches should help directly test these potential mechanisms in leukemogenesis and disease progression. Finally, aberrations of other pathways such as RUNX1, JAK2, and FLT3 are found in human patients with CBL mutations [28, 41, 42]. Therefore, modeling these additional oncogenic events in cellular and animal models should help provide a fuller picture of mutant Cbl-driven leukemogenic process.
Importantly, many of the signaling pathways that appear to be linked to mutant Cbl-driven oncogenesis are also known to be hyperactive in other cancer, and are currently being pursued actively as potential therapeutic targets. Therefore, a better understanding of the spectrum of signaling alterations provoked by mutant Cbl proteins is likely to reveal logical therapeutic strategies for patients with CBL mutations. Because mutant Cbl proteins may unleash pathways that are distinct from those engaged by its normal counterpart, unbiased genomic and proteomic approaches may help identify potential therapeutic targets. Finally, it may be possible to develop therapeutics that directly target mutant Cbl. Existing biochemical data indicate that mutant Cbl proteins need to interact with their PTK targets through the TKB domain to exert their transforming activity. Therefore, it is conceivable that interruption of this interaction can block mutant Cbl-driven oncogenesis. While not as widely pursued as inhibitors that target specific catalytically active sites in enzymes (such as protein tyrosine kinases), successful development of small molecule inhibitors of protein-protein interaction has begun to emerge, validating it as a practical approach [43]. The interaction interfaces that mediate the Cbl TKB domain binding to cognate phospho-peptide motifs on target PTKs have been structurally characterized and should be useful for a peptidomimetic approach to design small molecule inhibitors. This approach can be complemented with more unbiased chemical library screens. Our laboratories have established high throughput assays that are suitable for screening small molecules as well as for characterization of rationally-designed inhibitors of Cbl interaction with PTKs [44].
In conclusion, recent identification of mutations of Cbl in MDS/MPN and development of models that recapitulate features of these diseases in mice have opened exciting new avenues to translate what we have learned over the last two decades about the multi-faceted roles of Cbl family proteins as negative regulators of PTK signaling. These new findings also pose new questions about the functional roles of Cbl proteins, mechanisms by which these proteins regulate hematopoietic (and potentially other) stem cell programs, and how these new findings can be channeled into new diagnostic and therapeutic opportunities.
Acknowledgements
Work in the authors’ laboratories was supported by grants from the US National Institutes of Health (CA87986, CA99163, CA105489 and CA116552 to H.B.; CA96844 to V.B.; 127239 to A.N.; CA134660 to M.S.), the US Department of Defense Breast Cancer Research Program (W81XWH-11-1-0166 to H.B; W81XWH-07-1-0351 and W81XWH-11-1-0171 to V.B; W81XWH-10-1-0740 to M.N.), University of Nebraska Medical Center (UNMC)-Eppley Cancer Center (Pilot Grants to M.N. and V.B.), Nebraska Department of Health and Human Services (LB-506 grant to S.M.R.) and Nebraska Center for Nanomedicine-Center for Biomedical Research Excellence (NCN-COBRE; seed grant to S.M.R.).
References
1. Cancer Facts & Figures 2010. 2010; Available from: http://www.cancer.org/acs/groups/content/@epidemiologysurveilance/documents/document/acspc-026238.pdf
2. Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994; 367: 645-648.
3. Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, Sawyers CL. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001; 344: 1031-1037.
4. Thien CB, Langdon WY. Cbl: many adaptations to regulate protein tyrosine kinases. Nat Rev Mol Cell Biol. 2001; 2: 294-307.
5. Duan L, Reddi AL, Ghosh A, Dimri M, Band H. The Cbl family and other ubiquitin ligases: destructive forces in control of antigen receptor signaling. Immunity. 2004; 21: 7-17.
6. Schmidt MHH, Dikic I. The Cbl interactome and its functions. Nat Rev Mol Cell Biol. 2005; 6: 907-918.
7. Lupher ML, Rao N, Eck MJ, Band H. The Cbl protooncoprotein: a negative regulator of immune receptor signal transduction. Immunol Today. 1999; 20: 375-382.
8. Joazeiro CA, Wing SS, Huang H, Leverson JD, Hunter T, Liu YC. The tyrosine kinase negative regulator c-Cbl as a RING-type, E2-dependent ubiquitin-protein ligase. Science. 1999; 286: 309-312.
9. Levkowitz G, Waterman H, Ettenberg SA, Katz M, Tsygankov AY, Alroy I, Lavi S, Iwai K, Reiss Y, Ciechanover A, Lipkowitz S, Yarden Y. Ubiquitin ligase activity and tyrosine phosphorylation underlie suppression of growth factor signaling by c-Cbl/Sli-1. Mol Cell. 1999; 4: 1029-1040.
10. Yokouchi M, Kondo T, Houghton A, Bartkiewicz M, Horne WC, Zhang H, Yoshimura A, Baron R. Ligand-induced ubiquitination of the epidermal growth factor receptor involves the interaction of the c-Cbl RING finger and UbcH7. J Biol Chem. 1999; 274: 31707-31712.
11. Meng W, Sawasdikosol S, Burakoff SJ, Eck MJ. Structure of the amino-terminal domain of Cbl complexed to its binding site on ZAP-70 kinase. Nature. 1999; 398: 84-90.
12. Zheng N, Wang P, Jeffrey PD, Pavletich NP. Structure of a c-Cbl-UbcH7 complex: RING domain function in ubiquitin-protein ligases. Cell. 2000; 102: 533-539.
13. Murphy MA, Schnall RG, Venter DJ, Barnett L, Bertoncello I, Thien CB, Langdon WY, Bowtell DD. Tissue hyperplasia and enhanced T-cell signalling via ZAP-70 in c-Cbl-deficient mice. Mol Cell Biol. 1998; 18: 4872-4882.
14. Naramura M, Kole HK, Hu RJ, Gu H. Altered thymic positive selection and intracellular signals in Cbl-deficient mice. Proc Natl Acad Sci USA. 1998; 95: 15547-15552.
15. Rathinam C, Thien CBF, Langdon WY, Gu H, Flavell RA. The E3 ubiquitin ligase c-Cbl restricts development and functions of hematopoietic stem cells. Genes Dev. 2008; 22: 992-997.
16. Bachmaier K, Krawczyk C, Kozieradzki I, Kong YY, Sasaki T, Oliveira-dos-Santos A, Mariathasan S, Bouchard D, Wakeham A, Itie A, Le J, Ohashi PS, Sarosi I, Nishina H, Lipkowitz S, Penninger JM. Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor Cbl-b. Nature. 2000; 403: 211-216.
17. Chiang YJ, Kole HK, Brown K, Naramura M, Fukuhara S, Hu RJ, Jang IK, Gutkind JS, Shevach E, Gu H. Cbl-b regulates the CD28 dependence of T-cell activation. Nature. 2000; 403: 216-220.
18. Naramura M, Jang I, Kole H, Huang F, Haines D, Gu H. c-Cbl and Cbl-b regulate T cell responsiveness by promoting ligand-induced TCR down-modulation. Nat Immunol. 2002; 3: 1192-1199.
19. Blake TJ, Heath KG, Langdon WY. The truncation that generated the v-cbl oncogene reveals an ability for nuclear transport, DNA binding and acute transformation. EMBO J. 1993; 12: 2017-2026.
20. Andoniou CE, Thien CB, Langdon WY. Tumour induction by activated abl involves tyrosine phosphorylation of the product of the cbl oncogene. EMBO J. 1994; 13: 4515-4523.
21. Sargin B, Choudhary C, Crosetto N, Schmidt MHH, Grundler R, Rensinghoff M, Thiessen C, Tickenbrock L, Schwäble J, Brandts C, August B, Koschmieder S, Bandi SR, Duyster J, Berdel WE, Müller-Tidow C, et al. Flt3-dependent transformation by inactivating c-Cbl mutations in AML. Blood. 2007; 110: 1004-1012.
22. Caligiuri MA, Briesewitz R, Yu J, Wang L, Wei M, Arnoczky KJ, Marburger TB, Wen J, Perrotti D, Bloomfield CD, Whitman SP. Novel c-CBL and CBL-b ubiquitin ligase mutations in human acute myeloid leukemia. Blood. 2007; 110: 1022-1024.
23. Abbas S, Rotmans G, Löwenberg B, Valk PJM. Exon 8 splice site mutations in the gene encoding the E3-ligase CBL are associated with core binding factor acute myeloid leukemias. Haematologica. 2008; 93: 1595-1597.
24. Dunbar AJ, Gondek LP, O’Keefe CL, Makishima H, Rataul MS, Szpurka H, Sekeres MA, Wang XF, McDevitt MA, Maciejewski JP. 250K single nucleotide polymorphism array karyotyping identifies acquired uniparental disomy and homozygous mutations, including novel missense substitutions of c-Cbl, in myeloid malignancies. Cancer Res. 2008; 68: 10349-10357.
25. Reindl C, Quentmeier H, Petropoulos K, Greif PA, Benthaus T, Argiropoulos B, Mellert G, Vempati S, Duyster J, Buske C, Bohlander SK, Humphries KR, Hiddemann W, Spiekermann K. CBL exon 8/9 mutants activate the FLT3 pathway and cluster in core binding factor/11q deletion acute myeloid leukemia/myelodysplastic syndrome subtypes. Clin Cancer Res. 2009; 15: 2238-2247.
26. Grand FH, Hidalgo-Curtis CE, Ernst T, Zoi K, Zoi C, McGuire C, Kreil S, Jones A, Score J, Metzgeroth G, Oscier D, Hall A, Brandts C, Serve H, Reiter A, Chase AJ, et al. Frequent CBL mutations associated with 11q acquired uniparental disomy in myeloproliferative neoplasms. Blood. 2009; 113: 6182-6192.
27. Loh ML, Sakai DS, Flotho C, Kang M, Fliegauf M, Archambeault S, Mullighan CG, Chen L, Bergstraesser E, Bueso-Ramos CE, Emanuel PD, Hasle H, Issa J, van den Heuvel-Eibrink MM, Locatelli F, Stary J, et al. Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood. 2009; 114: 1859-1863.
28. Sanada M, Suzuki T, Shih L, Otsu M, Kato M, Yamazaki S, Tamura A, Honda H, Sakata-Yanagimoto M, Kumano K, Oda H, Yamagata T, Takita J, Gotoh N, Nakazaki K, Kawamata N, et al. Gain-of-function of mutated C-CBL tumour suppressor in myeloid neoplasms. Nature. 2009; 460: 904-908.
29. Makishima H, Cazzolli H, Szpurka H, Dunbar A, Tiu R, Huh J, Muramatsu H, O’Keefe C, Hsi E, Paquette RL, Kojima S, List AF, Sekeres MA, McDevitt MA, Maciejewski JP. Mutations of e3 ubiquitin ligase cbl family members constitute a novel common pathogenic lesion in myeloid malignancies. J Clin Oncol. 2009; 27: 6109-6116.
30. Muramatsu H, Makishima H, Jankowska AM, Cazzolli H, O’Keefe C, Yoshida N, Xu Y, Nishio N, Hama A, Yagasaki H, Takahashi Y, Kato K, Manabe A, Kojima S, Maciejewski JP. Mutations of an E3 ubiquitin ligase c-Cbl but not TET2 mutations are pathogenic in juvenile myelomonocytic leukemia. Blood. 2010; 115: 1969-1975.
31. Fernandes MS, Reddy MM, Croteau NJ, Walz C, Weisbach H, Podar K, Band H, Carroll M, Reiter A, Larson RA, Salgia R, Griffin JD, Sattler M. Novel oncogenic mutations of CBL in human acute myeloid leukemia that activate growth and survival pathways depend on increased metabolism. J Biol Chem. 2010; 285: 32596-605
32. Niemeyer CM, Kang MW, Shin DH, Furlan I, Erlacher M, Bunin NJ, Bunda S, Finklestein JZ, Sakamoto KM, Gorr TA, Mehta P, Schmid I, Kropshofer G, Corbacioglu S, Lang PJ, Klein C, et al. Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nat Genet. 2010; 42: 794-800.
33. Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007; 7: 295-308.
34. Emanuel PD. Juvenile myelomonocytic leukemia and chronic myelomonocytic leukemia. Leukemia. 2008; 22: 1335-1342.
35. Naramura M, Nandwani N, Gu H, Band V, Band H. Rapidly fatal myeloproliferative disorders in mice with deletion of Casitas B-cell lymphoma (Cbl) and Cbl-b in hematopoietic stem cells. Proc Natl Acad Sci USA. 2010; 107: 16274-16279.
36. Rathinam C, Thien CBF, Flavell RA, Langdon WY. Myeloid leukemia development in c-Cbl RING finger mutant mice is dependent on FLT3 signaling. Cancer Cell. 2010; 18: 341-352.
37. Bonita DP, Miyake S, Lupher ML, Langdon WY, Band H. Phosphotyrosine binding domain-dependent upregulation of the platelet-derived growth factor receptor alpha signaling cascade by transforming mutants of Cbl: implications for Cbl’s function and oncogenicity. Mol Cell Biol. 1997; 17: 4597-4610.
38. Thien CBF, Blystad FD, Zhan Y, Lew AM, Voigt V, Andoniou CE, Langdon WY. Loss of c-Cbl RING finger function results in high-intensity TCR signaling and thymic deletion. EMBO J. 2005; 24: 3807-3819.
39. Wu WJ, Tu S, Cerione RA. Activated Cdc42 sequesters c-Cbl and prevents EGF receptor degradation. Cell. 2003; 114: 715-725.
40. Duan L, Raja SM, Chen G, Virmani S, Williams SH, Clubb RJ, Mukhopadhyay C, Rainey MA, Ying G, Dimri M, Chen J, Reddi AL, Naramura M, Band V, Band H. Negative regulation of EGFR-Vav2 signaling axis by Cbl ubiquitin ligase controls EGF receptor-mediated epithelial cell adherens junction dynamics and cell migration. J Biol Chem. 2011; 286: 620-633.
41. Pérez B, Kosmider O, Cassinat B, Renneville A, Lachenaud J, Kaltenbach S, Bertrand Y, Baruchel A, Chomienne C, Fontenay M, Preudhomme C, Cavé H. Genetic typing of CBL, ASXL1, RUNX1, TET2 and JAK2 in juvenile myelomonocytic leukaemia reveals a genetic profile distinct from chronic myelomonocytic leukaemia. Br J Haematol. 2010; 151:460-8.
42. Makishima H, Jankowska AM, McDevitt MA, O’Keefe C, Dujardin S, Cazzolli H, Przychodzen B, Prince C, Nicoll J, Siddaiah H, Shaik M, Szpurka H, Hsi E, Advani A, Paquette R, Maciejewski JP. CBL, CBLB, TET2, ASXL1, and IDH1/2 mutations and additional chromosomal aberrations constitute molecular events in chronic myelogenous leukemia. Blood. 2011 Feb 23. [Epub ahead of print]
43. Azmi AS, Mohammad RM. Non-peptidic small molecule inhibitors against Bcl-2 for cancer therapy. J Cell Physiol. 2009; 218: 13-21.
44. Kumar EA, Charvet CD, Lokesh GL, Natarajan A. High-throughput fluorescence polarization assay to identify inhibitors of casitas B-lineage lymphoma(tyrosine kinase binding)-protein tyrosine kinase interactions. Anal Biochem. 2010 Dec 1. [Epub ahead of print]