
INTRODUCTION
Helicobacter pylori colonizes the human gastric mucosa and has currently infected more than half of the entire human population [1]. The bacterium has been estimated to be the cause of approximately 90% of non-cardiac gastric cancers [2]. In addition to H. pylori virulence, host genetic backgrounds and other environmental factors also exhibit complex interactions in gastric cancer development. Gastric cancer develops through a multiple step process known as the gastritis-gastric cancer carcinogenic pathway that is triggered by H. pylori infection. Severe gastritis has been proven to be a precursor condition [1, 3–9]. In addition, H. pylori infection causes various diseases such as gastroduodenal ulcers and MALT lymphoma. Previous genome-wide association studies have revealed the association of SNP rs2294008 in Prostate Stem Cell Antigen (PSCA) with two H. pylori related diseases, gastric cancer and duodenal ulcer [10, 11]. The C allele of rs2294008 increases duodenal ulcer risk and the T allele increases gastric cancer and chronic atrophic gastritis risk [12], while rs2294008 was not associated with H. pylori susceptibility [13], suggesting the important role of this variation in clinical outcomes after H. pylori infection. rs2294008 was associated with PSCA mRNA expression in normal gastric tissues and gastric cancer. Although the gastric cancer-risk T allele is associated with a higher expression of PSCA mRNA, PSCA expression in gastric cancer tissues decreases. Therefore, the role of PSCA in gastric carcinogenesis remains largely unknown [10, 14]. Here, we investigated rs2294008 and PSCA expression using germline DNA and gastric mucosal tissues at different disease stages and revealed novel roles of host genetic factor and bacterial infection in the disease pathogenesis.
RESULTS
Study participants
The flowchart of patient enrollment was shown in Supplementary Figure 1. A total of 280 subjects with H. pylori infection and 28 H. pylori-negative controls were enrolled in this study [15]. Among 280 H. pylori-infected patients, 133 received the second evaluation after successful H. pylori eradication. Genotyping results of 509 H. pylori-negative controls and 2,329 gastric cancer patients analyzed in our previous study [11] were used in this study. The characteristics of the participants are summarized in Table 1 and Supplementary Table 1.
Table 1: Characteristics of study participants
H. pylori-negative Controls |
H. pylori-infection |
P valuea |
H. pylori-negative Controls |
Gastric cancer |
|
|---|---|---|---|---|---|
Organizations |
Toyoshima Endoscopy Clinic |
Aichi Cancer Center |
BioBank Japan |
||
Samples |
Blood DNA + gastric mucosal tissues |
Blood DNA |
|||
n |
28 |
280 |
509 |
2,329 |
|
Age mean |
50.2 +- 13.7 |
49.7 +- 11.9 |
0.93 |
42.0 +- 15.6 |
64.9 +- 9.1 |
Sex female % |
46.4 |
49.6 |
0.75 |
52.7 |
22.2 |
Body mass index kg/m2 |
22.8 +- 2.67 |
22.3 +- 3.07 |
0.29 |
||
Past malignancy except gastric cancer |
1 |
18 |
1.0 |
||
Family history of gastric cancer |
6 |
47 |
0.60 |
||
Drinking |
9 |
71 |
0.43 |
||
Smoking |
5 |
20 |
0.063 |
||
Gastric ulcer |
1 |
28 |
0.44 |
||
Duodenal ulcer |
1 |
35 |
0.27 |
||
Severe gastritis % |
0 |
60.4 |
1.7 × 10-9 |
||
509 H. pylori-negative controls and 2,329 gastric cancer cases were analyzed in our previous study (Tanikawa et al. 2012 Nature Genetics) 11. aP values were calculated using Mann-Whitney U test, chi-square test, or Fisher’s exact test.
The role of the SNP rs2294008 in the gastritis-gastric cancer carcinogenic pathway
To investigate the role of the PSCA variation in gastric cancer and precancerous conditions, we genotyped rs2294008 using DNA from patients at different disease stages (Table 2). As previously reported [10, 12, 13, 16], the T allele of rs2294008 was significantly associated with gastric cancer risk when we used H. pylori-negative individuals as a control (P = 4.5 × 10−6; OR = 1.37). Then, we examined the impact of rs2294008 on the gastritis-gastric cancer carcinogenic pathway. There was no significant difference in the allelic frequency between H. pylori-carriers and H. pylori-negative controls (P = 0.15; OR = 1.14), indicating that rs2294008 is not associated with susceptibility to H. pylori infection. We then examined the impact of rs2294008 on the severity of gastritis as evaluated by histological neutrophil activity according to the updated Sydney system [17]. The individuals with H. pylori infection were divided into two categories, mild and severe gastritis [5], and rs2294008 was significantly associated with severe gastritis when we used mild gastritis or H. pylori-negative controls as references (P = 1.0 × 10−3 and 1.1 × 10−3 with OR of 1.93 and 1.52, respectively). However, rs2294008 was not associated with gastric cancer development from severe gastritis (P = 0.37; OR = 0.90). Similar tendencies were observed when gastritis was assessed by an endoscopic or a serological evaluation (Supplementary Table 2). These results indicated that rs2294008 was associated with the progression to severe gastritis after H. pylori-infection, but not with gastric cancer development from severe gastritis (Figure 1).
Table 2: Association of rs2294008 with gastritis-gastric cancer sequence
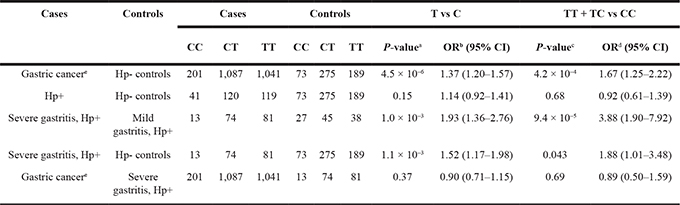
Hp+: H. pylori positive, Hp-: H. pylori negative. aP values are calculated by two-sided Cochran-Armitage test. bORs are calculated by considering the C allele as a reference. cP values are calculated by chi-square test. dORs were calculated by considering the CC genotype as a reference. eData from the previous study (Tanikawa et al. 2012 Nature Genetics) 11.
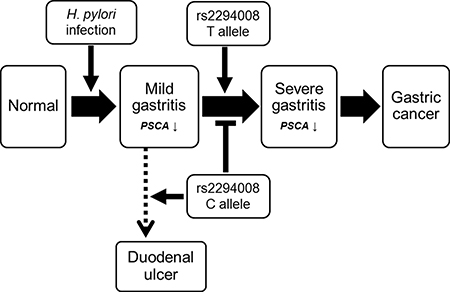
Figure 1: Scheme of SNP rs2294008 contribution to the diseases related with H. pylori infection including gastritis, gastric cancer, and duodenal ulcer.
The proportion of patients with severe gastritis was 32.5% among H. pylori carriers with the CC genotype, but it was as high as 62.2% and 68.1% among those with CT and TT genotypes, respectively. The comparison between the T carriers (TT or TC) and the CC genotype showed more remarkable associations, P = 9.4 × 10−5 and OR = 3.88. These results indicated that individuals with the TT or the TC genotype are susceptible to the progression to severe gastritis after H. pylori-infection and consequently have a high risk for gastric cancer.
The association of the PSCA expression with SNP rs2294008, H. pylori infection, and the severity of gastritis
Next, we evaluated PSCA mRNA in the background gastric mucosa of the incisura angularis at different disease stages. PSCA expression in H. pylori-infected patients (n = 280) was significantly lower than that in H. pylori-negative controls (n = 28) (Figure 2A, P = 5.1 × 10−8), but PSCA expression was not associated with gastritis stage (Figure 2A, P = 0.28). Moreover, PSCA expression increased after H. pylori eradication in 133 paired samples (Figure 2B, P < 1 × 10−11). These results clearly demonstrated the negative regulation of PSCA expression by H. pylori infection. Then, we analyzed the correlation of rs2294008 with PSCA expression. rs2294008 was significantly associated with PSCA expression at all disease stages (Figure 3A–3D, Supplementary Figure 2A–3D, P = 1.8 × 10−4 in H. pylori-negative, 1.3 × 10−12 in H. pylori-infected, 3.6 × 10−8 in mild gastritis, and 8.2 × 10−6 in severe gastritis). Consistent with our results, Sung et al. [14] reported that PSCA expression was associated with rs2294008 in gastric cancer tissues and non-tumor mucosa from gastric cancer patients. These results indicated that the T allele is associated with higher PSCA expression regardless of H. pylori infection status or disease stage.
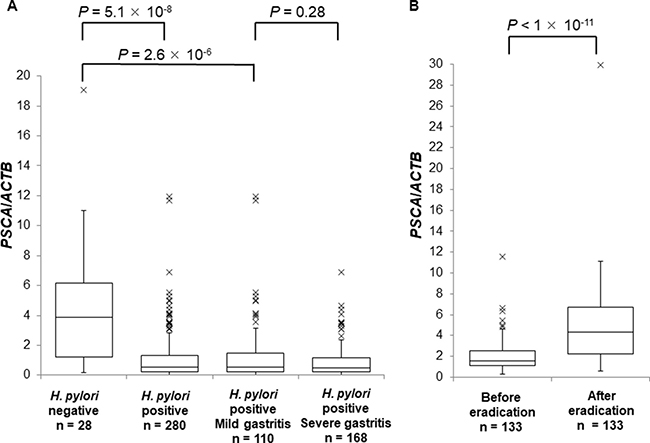
Figure 2: The suppression of PSCA expression by H. pylori infection. Box-plots with medians for PSCA expression normalized to ACTB in biopsy samples of gastric mucosa. (A) PSCA mRNA in H. pylori-negative controls (n = 28), in H. pylori-infected patients (n = 280), in H. pylori-infected mild gastritis patients (n = 110), or in H. pylori-infected severe gastritis patients (n = 168). The P values were calculated by a Mann-Whitney U test. (B) The comparison of the expression levels of PSCA in each individual patient (n = 133) before and after H. pylori eradication. The P value was calculated by a Wilcoxon signed rank test.
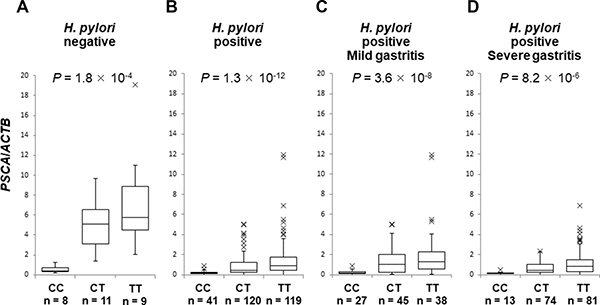
Figure 3: The association of SNP rs2294008 with PSCA expression. Box-plots with medians for PSCA expression normalized to ACTB in biopsy samples of gastric mucosa. The P values were calculated by a Kruskal-Wallis test. (A) PSCA mRNA in H. pylori-negative controls (n = 28), (B) in H. pylori-infected patients (n = 280), (C) in H. pylori-infected mild gastritis patients (n = 110), (D) and in H. pylori-infected severe gastritis patients (n = 168).
In all three genotypes, PSCA expression was decreased in the gastric mucosa by H. pylori infection (Supplementary Figure 3A, CC: P = 0.0023, CT: 4.4 × 10−7, TT: 8.0 × 10−6) and was increased after H. pylori eradication (Supplementary Figure 3B, CC: P = 0.0038, CT: 3.6 × 10−7, TT: 2.7 × 10−11). Interestingly, H. pylori infection caused remarkable reductions in PSCA expression among T allele carriers compared with the CC genotype (Supplementary Figure 3A). We also evaluated PSCA mRNA change by H. pylori eradication in gastric mucosa by using paired samples before and after eradication. As a result, the T allele was associated with increased fold mRNA induction by H. pylori eradication (Figure 4, P = 0.0019). In addition, PSCA expression in severe gastritis patients was lower than in mild gastritis patients only among T carriers, but not among the CC genotype (Supplementary Figure 3C, CC: P = 0.36, CT: 0.048, TT: 0.032). Similar results were observed when the patients were assessed by endoscopic or serological evaluations (Supplementary Figure 4A, 4B). Altogether, H. pylori infection exhibited a stronger impact on PSCA expression among T allele carriers in the development of H. pylori-induced gastritis. Our findings clearly demonstrated that the regulation of PSCA expression by the host genetic factor (i.e., PSCA polymorphism) and the environmental factor (i.e., H. pylori infection) would play important roles in the pathogenesis of H. pylori related diseases.
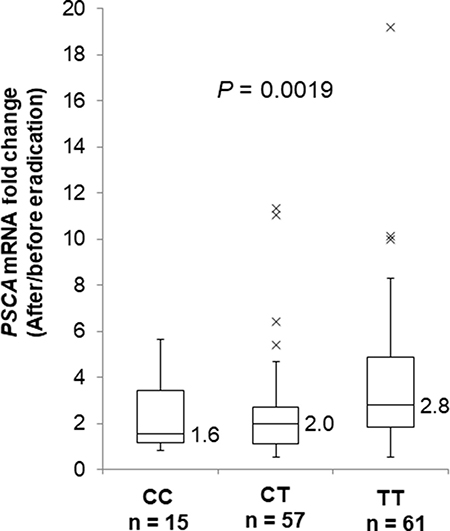
Figure 4: Fold PSCA mRNA induction by H. pylori eradication. Box-plots with medians fold induction of PSCA expression normalized to ACTB. The fold induction was defined as PSCA expression after eradication divided by PSCA expression before eradication using paired samples. PSCA mRNA levels in paired samples before and after H. pylori eradication are quantified by the qRT-PCR analysis (CC; n = 15, CT; n = 57, TT; n = 61). The P values were calculated by a Kruskal-Wallis test.
DISCUSSION
PSCA is a 123-amino acid glycoprotein that is related to the Ly-6 family of cell-surface proteins and has a role in signal transcription and in the regulation of cell proliferation. An increased expression of PSCA was observed in prostatic, bladder, and pancreatic cancers, indicating its oncogenic roles in tumor development of these cancers. However, decreased PSCA expression was documented in gastric cancer, gallbladder cancer, and head-and-neck squamous cell carcinoma, representing the complexity of PSCA function and PSCA regulation in various tissues [10–13, 16, 18–22]. Fu et al. [23] reported that the T allele of rs2294008 was associated with higher PSCA expression in both bladder cancers and in normal tissues. Because PSCA is upregulated in bladder cancer tissue and T allele carriers are associated with higher PSCA expression, the T allele is associated with a higher bladder cancer risk. However, the role of PSCA in gastric cancer development is not well understood, since PSCA expression was suppressed in gastric cancer tissues while the risk T allele is associated with higher PSCA expression.
Here, we identified the novel roles of a host genetic factor and bacterial infection in the regulation of PSCA expression in H. pylori induced active gastritis. rs2294008 was associated with disease progression to severe gastritis after H. pylori infection, but not with the development of gastric cancer after severe gastritis. In addition, a subgroup analysis using SNP genotypes revealed that only T allele carriers showed decreased PSCA expression in the progression from mild gastritis to severe gastritis. Here, we found that the CC genotype is associated with a low risk for progression to severe gastritis, which is consistent with our previous report that demonstrated an increased risk for duodenal ulcers in CC genotypes (Figure 1) [11, 24].
The number of active gastritis samples (n = 168) were relatively small compared with those of gastric cancer patients (n = 2,329). The statistical power of our study is estimated to be 62%, when it is assumed that OR of rs2294008 for gastric cancer risk is 1.3. However, information about H. pylori infection status of gastric cancer patients was not available in our study, although nearly 90% of gastric cancer patients were considered to be infected with H. pylori. Therefore, to conclude the role of rs2294008 in the progression from active gastritis to gastric cancer, further analysis using case-control samples with positive H. pylori infection is necessary. Currently we are conducting a follow-up survey of active gastritis patients. We would like to evaluate the role of genetic and clinical parameters as prognostic biomarkers in the future study.
rs2294008 is located 26 bases upstream of the translation initiation codon and the T allele encodes another translation initiation codon for the PSCA gene, resulting in an additional nine amino acids at N-terminal portion of PSCA protein to change protein localization from the cytoplasm to the cell surface [10, 11]. Long form of cell surface PSCA protein encoded by T allele was shown to promote cell proliferation [22, 25], while short cytosolic PSCA proteins associated with the C allele are rapidly degraded by the ubiquitin proteasomal pathway [11]. Therefore, C allele of rs2294008 is associated with low PSCA expression at mRNA and protein level. H. pylori infection has a strong impact among T allele carriers, because H. pylori infection remarkably suppress PSCA expression which is highly expressed in normal gastric mucosal tissues without H. pylori infection. In contrast, subjects with the CC genotype are unlikely to be affected by H. pylori infection because they express PSCA mRNA and protein at very low level regardless of H. pylori infection. Interestingly, individuals with a nonsense SNP (rs138377917[A]) of PSCA that lack functional PSCA protein is associated with a low risk for gastric cancer [26]. These lines of evidence clearly demonstrate that genotypes related with loss of functional PSCA (C allele of rs2294008 and A allele of rs138377917) are associated with a reduced risk for gastric cancer.
There are some limitations in this study because only endoscopy-based Japanese patients were analyzed. In addition, we did not examine CagA of H. pylori, but almost all Japanese H. pylori have CagA proteins and 95% of them are the East-Asia-type [1].
Here we revealed how host genetic variation and bacterial infection regulate PSCA expression and progression to severe gastritis. Our findings elucidated the critical roles of PSCA expression and SNP in the gastritis-gastric cancer progression and their possible implications for risk prediction and personalized disease prevention.
MATERIALS AND METHODS
Study design and participants
This cohort study consisted of subjects who agreed to participate in this study and underwent esophagogastroduodenoscopy (EGD) at Toyoshima Endoscopy Clinic from December 2013 to April 2015. We could obtain a written informed consent from a total of 326 individuals. All EGDs were undertaken by experienced endoscopists. EGDs were performed either for screening, an evaluation of present symptoms, surveillance of previous esophagogastroduodenal disease, positive results of H. pylori infection, an abnormal serum pepsinogen level, or abnormal findings on the barium meal. The inclusion criteria were patients aged 20 years or more without a past history of gastric cancer, esophagogastric junction adenocarcinoma, surgical gastrectomy, or H. pylori eradication. The exclusion criteria were a diagnosis of gastric cancer or adenoma based on an EGD at the time of enrollment, autoimmune gastritis, severe concomitant illness, an unidentified status of H. pylori, or an agreement withdrawal. We diagnosed patients with histologically severe corpus-restricted atrophic gastritis as autoimmune gastritis [27].
We divided the participants into two groups, with or without current H. pylori infection. We defined participants with histological intestinal metaplasia or a serum pepsinogen I/II ratio <3.0 as infected with H. pylori in the past and excluded them from the H. pylori-negative control groups [28]. Participants infected with H. pylori received eradication therapy after informed consent was provided. After the confirmation of successful H. pylori eradication, we performed the second EGD 6 months to 2 years after the index EGD. We collected peripheral blood samples at enrollment. The remaining mucosal biopsy tissues from the gastric incisura angularis at the index and the second EGD were used in this study.
H. pylori negative controls (n = 509) and gastric cancer patients (n = 2,329) were recruited at Aichi Cancer Center and BioBank Japan, respectively [11].
This research project was approved by the institutional review board at each institute. All participants provided written informed consent as approved by the institutional review board.
Assessment of H. pylori status
At enrollment, we determined current H. pylori infection by either a positive result of the 13C-urea breath test (UBIT, Otsuka, Tokushima, Japan), the stool antigen test, or a combination of the serum immunoglobulin G antibody test (E-plate, Eiken, Tokyo, Japan) and culture or pathology (hematoxylin and eosin staining). The 13C-urea breath test requires a 2-week cessation of maintenance therapy with acid suppressors. At least 8 weeks after the completion of eradication therapy, successful eradication was confirmed using a negative 13C-urea breath test or stool antigen test [7].
Diagnosis of gastritis
We divided gastritis into two categories, mild and severe gastritis, by three methods: histology, endoscopy, and serology.
The histological estimation was conducted according to the updated Sydney system [17]. To grade the neutrophil activity, we used two biopsy specimens from the greater curvature of the corpus and the antrum obtained by the EGD. We topographically classified the gastritis into four categories (i.e., no gastritis, antrum-predominant gastritis, pangastritis, and corpus-predominant gastritis). No gastritis and antrum-predominant gastritis were defined as mild gastritis and pangastritis and corpus-predominant gastritis were defined as severe gastritis. The histological diagnosis was performed by an expert gastrointestinal pathologist independently of the endoscopists. Chronic active gastritis identified by the distribution of neutrophil activity represents persistent active inflammation that is associated with tissue damage. Chronic active gastritis has been proven to be strongly associated with gastric cancer (corpus-predominant gastritis; relative risk [RR]: 34.5 [95% CI: 7.1−166.7], pangastritis; 15.6 [6.5−36.8], antrum-predominant gastritis as the reference group) [5, 29].
Endoscopic gastritis was assessed by atrophic-border identification by the different color and height of the mucosa according to Kimura-Takemoto classification [30]. In closed-type atrophic gastritis, the atrophic-border lies between the antrum and the lesser curvature of the gastric body and is defined as mild gastritis. In open-type gastritis, the border lies beyond the cardia and reaches the greater curvature of the gastric body and is defined as severe gastritis. The endoscopic-atrophic-border represents the histological border of atrophy and the range of atrophy spreads with the progression of gastritis [31]. Endoscopic gastritis has been demonstrated as a risk factor for gastric cancer (severe gastritis; RR: 4.9 [95% CI: 2.8–19.2], none or mild gastritis as the reference group) [5].
We used serum levels of pepsinogen I and II for the serological evaluation and defined a pepsinogen I/II ratio <3.0 as severe gastritis [8, 32]. Serum levels of pepsinogen I and II were measured using a commercially available kit (Pepsinogen CLEIA; Fuji Rebio Ltd., Tokyo, Japan). We excluded patients receiving proton pump inhibitors from the serological evaluation. The pepsinogen I/II ratio has been shown to be associated with the risk for gastric cancer (OR: 2.78−10.92) [8, 33].
H. pylori eradication
The H. pylori eradication first-line regimen included clarithromycin 400 mg, amoxicillin 1500 mg, and a daily proton pump inhibitor for 7 days. In patients with infection of clarithromycin-resistant H. pylori or those with allergies to clarithromycin, the second-line regimen included metronidazole 500 mg, amoxicillin 1500 mg, and a daily proton pump inhibitor for 7 days.
SNP Genotyping
DNA was isolated from peripheral blood leukocytes by using QIAamp DNA mini Kits (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. The samples were genotyped by the Invader assay system (Third Wave Technologies Madison, WI) using the purified DNA form peripheral blood. Aliquot of DNA from patients with active or mild gastritis was randomly assigned to a 96 well plate and subjected to SNP analysis. The genotype results of healthy controls and gastric cancer patients analyzed in the previous study was used in this study [11].
qRT-PCR analysis
The RNA was extracted from the fresh mucosa of the gastric angulus as background by EGD biopsy. Total RNA were isolated from human tissues using AllPrep DNA/RNA/miRNA Universal Kits (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. RNA was treated with DNase during purification. Quality and quantity of RNA were evaluated by using NanoDrop (Thermo Scientific). Total RNA with > 1.9 of OD 260/280 ratio were used for further analysis. Complementary DNAs were synthesized from 500 ng of total RNA using Super Script III reverse transcriptase (Invitrogen). Quantitative real-time PCR (qRT-PCR) was conducted using SYBR Green Master Mix with a Light Cycler 480 (Roche, Basel, Switzerland). The expression of the beta-actin (ACTB) gene was used for normalization. A serial dilution of plasmid containing PCR fragment and distilled water (no template control) were used to calculate copy number of ACTB and PSCA. Primers used for qRT-PCR were as follows; PSCA_F:ctgctgtgctactcctgcaa, PSCA_R:ttgctgatgacggtcagg, ACTB_F:ccctggagaagagctacgag, ACTB_R:tgaaggtagtttcgt ggatgc.
Statistical analysis
The comparison of the demographic characteristics between the groups was performed using the Mann-Whitney U-test in continuous variables and the chi-square test or Fisher’s exact test in categorical variables. The association of rs2294008 with various diseases related with H. pylori infection was tested by the two-sided Cochran-Armitage trend test among the three genotypes and by the chi-square test between the CC genotype and T carriers. The odds ratios were calculated by considering the C allele or the CC genotype as reference. The comparison of the gene expression between the two groups was analyzed using the Mann-Whitney U-test and among the three groups using the Kruskal-Wallis test. The comparison of gene expression before and after H. pylori eradication was analyzed using the Wilcoxon signed rank test. Significance was indicated by a P value less than 0.05. Calculations were carried out using statistical software Ekuseru-Toukei 2015 (Social Survey Research Information Co., Ltd., Tokyo, Japan).
Abbreviations
PSCA: Prostate Stem Cell Antigen; SNP: single nucleotide polymorphism; H. pylori: Helicobacter pylori; EGD: esophagogastroduodenoscopy; RR: relative risk; CI: confidence interval; OR: odds ratio; qRT-PCR: quantitative real-time PCR; ACTB: beta-actin.
Author contributions
O Toyoshima, C Tanikawa, R Yamamoto, Y Seto, and K Matsuda designed the study. O Toyoshima, H Yamashita., K Sakitani, S Yoshida, and K Koike recruited the subject, obtained informed consent, and collected clinical data and the samples. C Tanikawa, R Yamamoto, M Kubo, K Matsuo, H Ito, and K Matsuda performed genotyping and qRT-PCR analysis. H Watanabe carried out pathological diagnosis. O Toyoshima and K Matsuda analyzed and summarized all the results, and wrote the manuscript. K Matsuda obtained funding for the study.
ACKNOWLEDGMENTS
We thank Satoyo Oda, Akane Sei, and Satomi Takahashi for technical assistance and Yoshihiro Isomura, Satoki Shichijo, Shuhei Matsumoto, Takamitsu Kanazawa, and Toru Arano for collecting the samples.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This work was supported by Ministry of Education, Culture, Sports, Science and Technology of Japan [25134707 and 16H01566 to Ko.M., 15K14377 to C.T.]. This study was also supported by funding from the Tailor-Made Medical Treatment with the BBJ Project from Japan Agency for Medical Research and Development, AMED (from April 2015), and the Ministry of Education, Culture, Sports, Science, and Technology of Japan (from April 2003 to March 2015).
REFERENCES
1. Yamaoka Y. Mechanisms of disease: Helicobacter pylori virulence factors. Nat Rev Gastroenterol Hepatol. 2010; 7:629–41. https://doi.org/10.1038/nrgastro.2010.154.
2. Plummer M, Franceschi S, Vignat J, Forman D, de Martel C. Global burden of gastric cancer attributable to Helicobacter pylori. Int J Cancer. 2015; 136:487–90. https://doi.org/10.1002/ijc.28999.
3. Correa P. Human gastric carcinogenesis: a multistep and multifactorial process--First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 1992; 52:6735–40.
4. Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, Pukkala E, Skytthe A, Hemminki K. Environmental and heritable factors in the causation of cancer--analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000; 343:78–85. https://doi.org/10.1056/nejm200007133430201.
5. Uemura N, Okamoto S, Yamamoto S, Matsumura N, Yamaguchi S, Yamakido M, Taniyama K, Sasaki N, Schlemper RJ. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med. 2001; 345:784–9. https://doi.org/10.1056/NEJMoa001999.
6. Ekstrom AM, Held M, Hansson LE, Engstrand L, Nyren O. Helicobacter pylori in gastric cancer established by CagA immunoblot as a marker of past infection. Gastroenterology. 2001; 121:784–91.
7. Malfertheiner P, Megraud F, O’Morain CA, Atherton J, Axon AT, Bazzoli F, Gensini GF, Gisbert JP, Graham DY, Rokkas T, El-Omar EM, Kuipers EJ, European Helicobacter Study G. Management of Helicobacter pylori infection--the Maastricht IV/ Florence Consensus Report. Gut. 2012; 61:646–64. https://doi.org/10.1136/gutjnl-2012-302084.
8. Dinis-Ribeiro M, Areia M, de Vries AC, Marcos-Pinto R, Monteiro-Soares M, O’Connor A, Pereira C, Pimentel-Nunes P, Correia R, Ensari A, Dumonceau JM, Machado JC, Macedo G, et al. Management of precancerous conditions and lesions in the stomach (MAPS): guideline from the European Society of Gastrointestinal Endoscopy (ESGE), European Helicobacter Study Group (EHSG), European Society of Pathology (ESP), and the Sociedade Portuguesa de Endoscopia Digestiva (SPED). Endoscopy. 2012; 44:74–94. https://doi.org/10.1055/s-0031-1291491.
9. Hatakeyama M. Helicobacter pylori CagA and gastric cancer: a paradigm for hit-and-run carcinogenesis. Cell Host Microbe. 2014; 15:306–16. https://doi.org/10.1016/j.chom.2014.02.008.
10. Sakamoto H, Yoshimura K, Saeki N, Katai H, Shimoda T, Matsuno Y, Saito D, Sugimura H, Tanioka F, Kato S, Matsukura N, Matsuda N, Nakamura T, et al. Genetic variation in PSCA is associated with susceptibility to diffuse-type gastric cancer. Nat Genet. 2008; 40:730–40. https://doi.org/10.1038/ng.152.
11. Tanikawa C, Urabe Y, Matsuo K, Kubo M, Takahashi A, Ito H, Tajima K, Kamatani N, Nakamura Y, Matsuda K. A genome-wide association study identifies two susceptibility loci for duodenal ulcer in the Japanese population. Nat Genet. 2012; 44:430–4, S1-2. https://doi.org/10.1038/ng.1109.
12. Lochhead P, Frank B, Hold GL, Rabkin CS, Ng MT, Vaughan TL, Risch HA, Gammon MD, Lissowska J, Weck MN, Raum E, Muller H, Illig T, et al. Genetic variation in the prostate stem cell antigen gene and upper gastrointestinal cancer in white individuals. Gastroenterology. 2011; 140:435–41. https://doi.org/10.1053/j.gastro.2010.11.001.
13. Matsuo K, Tajima K, Suzuki T, Kawase T, Watanabe M, Shitara K, Misawa K, Ito S, Sawaki A, Muro K, Nakamura T, Yamao K, Yamamura Y, et al. Association of prostate stem cell antigen gene polymorphisms with the risk of stomach cancer in Japanese. Int J Cancer. 2009; 125:1961–4. https://doi.org/10.1002/ijc.24519.
14. Sung H, Yang HH, Hu N, Su H, Taylor PR, Hyland PL. Functional annotation of high-quality SNP biomarkers of gastric cancer susceptibility: the Yin Yang of PSCA rs2294008. Gut. 2016; 65:361–4. https://doi.org/10.1136/gutjnl-2015-310059.
15. Mori J, Tanikawa C, Ohnishi N, Funauchi Y, Toyoshima O, Ueda K, Matsuda K. EPSIN 3, A Novel p53 Target, Regulates the Apoptotic Pathway and Gastric Carcinogenesis(). Neoplasia (New York, NY). 2017; 19:185–95. https://doi.org/10.1016/j.neo.2016.12.010.
16. Lu Y, Chen J, Ding Y, Jin G, Wu J, Huang H, Deng B, Hua Z, Zhou Y, Shu Y, Liu P, Hu Z, Shen J, et al. Genetic variation of PSCA gene is associated with the risk of both diffuse- and intestinal-type gastric cancer in a Chinese population. Int J Cancer. 2010; 127:2183–9. https://doi.org/10.1002/ijc.25228.
17. Dixon MF, Genta RM, Yardley JH, Correa P. Classification and grading of gastritis. The updated Sydney System. International Workshop on the Histopathology of Gastritis, Houston 1994. Am J Surg Pathol. 1996; 20:1161–81.
18. Reiter RE, Gu Z, Watabe T, Thomas G, Szigeti K, Davis E, Wahl M, Nisitani S, Yamashiro J, Le Beau MM, Loda M, Witte ON. Prostate stem cell antigen: a cell surface marker overexpressed in prostate cancer. Proc Natl Acad Sci U S A. 1998; 95:1735–40.
19. Argani P, Rosty C, Reiter RE, Wilentz RE, Murugesan SR, Leach SD, Ryu B, Skinner HG, Goggins M, Jaffee EM, Yeo CJ, Cameron JL, Kern SE, et al. Discovery of new markers of cancer through serial analysis of gene expression: prostate stem cell antigen is overexpressed in pancreatic adenocarcinoma. Cancer Res. 2001; 61:4320–4.
20. de Nooij-van Dalen AG, van Dongen GA, Smeets SJ, Nieuwenhuis EJ, Stigter-van Walsum M, Snow GB, Brakenhoff RH. Characterization of the human Ly-6 antigens, the newly annotated member Ly-6K included, as molecular markers for head-and-neck squamous cell carcinoma. Int J Cancer. 2003; 103:768–74. https://doi.org/10.1002/ijc.10903.
21. Wu X, Ye Y, Kiemeney LA, Sulem P, Rafnar T, Matullo G, Seminara D, Yoshida T, Saeki N, Andrew AS, Dinney CP, Czerniak B, Zhang ZF, et al. Genetic variation in the prostate stem cell antigen gene PSCA confers susceptibility to urinary bladder cancer. Nat Genet. 2009; 41:991–5. https://doi.org/10.1038/ng.421.
22. Marra E, Uva P, Viti V, Simonelli V, Dogliotti E, De Rinaldis E, Lahm A, La Monica N, Nicosia A, Ciliberto G, Palombo F. Growth delay of human bladder cancer cells by Prostate Stem Cell Antigen downregulation is associated with activation of immune signaling pathways. BMC Cancer. 2010; 10:129. https://doi.org/10.1186/1471-2407-10-129.
23. Fu YP, Kohaar I, Rothman N, Earl J, Figueroa JD, Ye Y, Malats N, Tang W, Liu L, Garcia-Closas M, Muchmore B, Chatterjee N, Tarway M, et al. Common genetic variants in the PSCA gene influence gene expression and bladder cancer risk. Proc Natl Acad Sci U S A. 2012; 109:4974–9. https://doi.org/10.1073/pnas.1202189109.
24. Hansson LE, Nyren O, Hsing AW, Bergstrom R, Josefsson S, Chow WH, Fraumeni JF Jr, Adami HO. The risk of stomach cancer in patients with gastric or duodenal ulcer disease. N Engl J Med. 1996; 335:242–9. https://doi.org/10.1056/nejm199607253350404.
25. Gu Z, Yamashiro J, Kono E, Reiter RE. Anti-prostate stem cell antigen monoclonal antibody 1G8 induces cell death in vitro and inhibits tumor growth in vivo via a Fc-independent mechanism. Cancer Res. 2005; 65:9495–500. https://doi.org/10.1158/0008-5472.CAN-05-2086.
26. Helgason H, Rafnar T, Olafsdottir HS, Jonasson JG, Sigurdsson A, Stacey SN, Jonasdottir A, Tryggvadottir L, Alexiusdottir K, Haraldsson A, le Roux L, Gudmundsson J, Johannsdottir H, et al. Loss-of-function variants in ATM confer risk of gastric cancer. Nat Genet. 2015; 47:906–10. https://doi.org/10.1038/ng.3342.
27. Coati I, Fassan M, Farinati F, Graham DY, Genta RM, Rugge M. Autoimmune gastritis: Pathologist’s viewpoint. World J Gastroenterol. 2015; 21:12179–89. https://doi.org/10.3748/wjg.v21.i42.12179.
28. Graham DY. Helicobacter pylori update: gastric cancer, reliable therapy, and possible benefits. Gastroenterology. 2015; 148:719–31.e3. https://doi.org/10.1053/j.gastro.2015.01.040.
29. Shiotani A, Iishi H, Uedo N, Kumamoto M, Nakae Y, Ishiguro S, Tatsuta M, Graham DY. Histologic and serum risk markers for noncardia early gastric cancer. Int J Cancer. 2005; 115:463–9. https://doi.org/10.1002/ijc.20852.
30. Kimura K, Takemoto T. An endoscopic recognition of the atrophic border and its significance in chronic gastritis. Endoscopy. 1969;3:87–97. Endoscopy. 1969; 3:87–97.
31. Satoh K, Kimura K, Taniguchi Y, Yoshida Y, Kihira K, Takimoto T, Kawata H, Saifuku K, Ido K, Takemoto T, Ota Y, Tada M, Karita M, et al. Distribution of inflammation and atrophy in the stomach of Helicobacter pylori-positive and -negative patients with chronic gastritis. Am J Gastroenterol. 1996; 91:963–9.
32. Nardone G, Rocco A, Staibano S, Mezza E, Autiero G, Compare D, De Rosa G, Budillon G. Diagnostic accuracy of the serum profile of gastric mucosa in relation to histological and morphometric diagnosis of atrophy. Aliment Pharmacol Ther. 2005; 22:1139–46. https://doi.org/10.1111/j.1365-2036.2005.02734.x.
33. Dinis-Ribeiro M, da Costa-Pereiray A, Lopes C, Barbosa J, Guilherme M, Moreira-Dias L, Lomba-Viana H, Silva R, Abreu N, Lomba-Viana R. Validity of Serum Pepsinogen I/II Ratio for the Diagnosis of Gastric Epithelial Dysplasia and Intestinal Metaplasia during the Follow-Up of Patients at Risk for Intestinal-Type Gastric Adenocarcinoma. Neoplasia (New York, NY). 2004; 6:449–56.