
INTRODUCTION
Colorectal cancer (CRC) is the third most common cancer worldwide, conferring significant morbidity, mortality and cost to the public health system [1]. The majority of deaths result from metastasis of the primary cancer to distant organs, predominantly the liver [2]. The standard first-line chemotherapeutic regimen for metastatic CRC is a combination of fluorouracil (5-FU) and folinic acid with oxaliplatin (FOLFOX) or irinotecan (FOLFIRI) with or without targeted agents [3]. Currently, the single molecular targetable treatment available for CRC is epidermal growth factor receptor (EGFR) inhibitors for tumors lacking mutation in the oncogene KRAS [4–6]. However, even among patients with KRAS wild type primary tumors, a recent study showed only 10–20 % of metastatic CRC patients benefitted from an EGFR inhibitor [7]. One contributory reason for this low rate of response may be discordance in KRAS mutation status between primary tumors and corresponding metastases [8, 9]. It is also likely for some cases that effector molecules downstream of the EGFR, including members of the MAPK and AKT signaling pathways [7], may be further altered during the metastatic process or following therapeutic intervention [10, 11]. Clarification of whether it is sufficient to assay the primary tumor or necessary to biopsy metastatic deposits for biomarker assessment is critical for optimizing management of patients with metastatic disease.
Genome-wide assessment of somatic copy number alterations provides opportunities for identifying cancer driver genes in an unbiased manner and may provide novel markers for the early diagnosis and personalized treatment of CRC [12]. For example, DNA copy number amplifications in KRAS, ERBB2, MET and FGFR1 have been found to be predictors of poor prognosis and resistance to anti-EGFR therapy for CRC patients [13, 14]. It has also been recently shown that DNA copy number alterations are major sources of tumor heterogeneity, and one study found a striking discordance in copy number alterations between paired primary tumors and metastatic in samples from 27 patients using a DNA sequencing panel which was limited to only 100 genes [15]. Moreover, an understanding of the clonal composition of primary tumors and the extent to which this contributes to the molecular profile of metastatic disease pre- and post-therapy is vital to fully realize the potential of genomics in precision medicine approaches to improve patient outcomes.
We performed genome-wide chromosomal copy number assessment using single nucleotide polymorphism (SNP) arrays of primary CRC collected before therapy and the paired liver metastasis collected before or after therapeutic intervention. We show that copy number alterations differ significantly between primary tumors and their metastatic deposits.
RESULTS
Clinical and histologic data
Surgical samples of primary colorectal tumors and paired liver metastases from 16 patients (Median age 62.6 years) were analyzed (Table 1). A paired normal control sample was obtained for each patient from macroscopically normal mucosa adjacent to the primary tumor. In all 16 patients, primary CRC tissue was taken prior to chemotherapy. Nine patients had a synchronous liver metastasis at the time of diagnosis and 7 patients developed a liver metastasis metachronously (Figure 1). Metastatic tissue was taken after exposure to chemotherapy in 6 patients, which included 5 metachronous patients and 1 synchronous patient who had neoadjuvant chemotherapy prior to liver surgery (Figure 1).
Table 1: Clinicopathological characteristics of the 16 patients with CRC
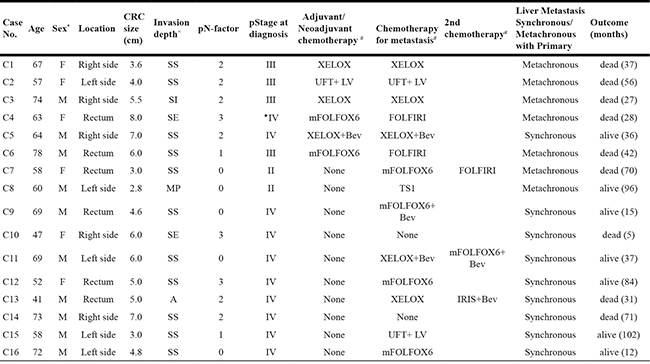
*M–male; F–female ⋆para aortic lymph node metastasis
^SS–Tumor invades through the muscularis propria into pericolorectal tissue; SI–tumor directly invades or is adherent to other organs or structures;
SE–tumor penetrates to the surface of the visceral peritoneum; MP–tumor invades muscularis propria; A–rectal tumor invades through the muscularis propria into pericolorectal tissue
#XELOX–Capecitabine and oxaliplatin; UFT+ LV–uracil-tegafur plus leucovorin; mFOLFOX6–fluorouracil (5-FU) and leucovorin with oxaliplatin;
FOLFIRI–5-FU and leucovorin with irinotecan; Bev–bevacizumab; TS1–Oral fluoropyrimidine anticancer drug; IRIS–irinotecan and TS-1.
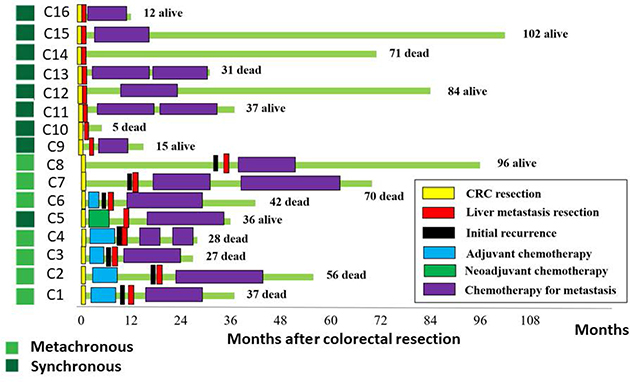
Figure 1: Clinical characteristics of the study cohort. The course of treatment for the 16 CRC patients is indicated schematically. Samples were obtained at surgical resection time points indicated by yellow blocks for primary CRC and red blocks for the liver metastases. The survival status of the patient is shown, through observations made at 1–6 month intervals until death or December 2015.
KRAS mutational discordance between primary and paired metastatic tumors was identified in 2 patients (2/16, 13%), whilst BRAF mutation was concordant in the single patient with a BRAF mutant allele (Table 2). These two patients both had Stage III metachronous disease and had received adjuvant chemotherapy prior to sampling of the metastasis. Loss of MLH1 immunostaining, indicating a likely mismatch DNA repair deficiency and microsatellite instability, was detected in 3/16 patients (18.8 %, Table 2) and was concordant between all primary tumors and paired metastases (Supplementary Figure 1).
Table 2: Tumor purity and KRAS/BRAF/mismatch repair status in primary and paired metastasis
Case No. |
Purity PT |
Purity Met |
KRAS PT |
KRAS Met |
BRAF PT |
BRAF Met |
Mismatch Repair PT |
Mismatch Repair Met |
|---|---|---|---|---|---|---|---|---|
C1 |
0.72 |
0.95 |
Mutant |
Mutant |
wt |
wt |
MSS |
MSS |
C2 |
0.87 |
0.88 |
wt |
Mutant |
wt |
wt |
MLH1 Loss |
MLH1 Loss |
C3 |
0.54 |
0.82 |
Mutant |
Mutant |
wt |
wt |
MSS |
MSS |
C4 |
0.63 |
0.78 |
wt |
wt |
wt |
wt |
MSS |
MSS |
C5 |
0.82 |
0.90 |
wt |
wt |
wt |
wt |
MSS |
MSS |
C6 |
0.64 |
0.92 |
Mutant |
wt |
wt |
wt |
MSS |
MSS |
C7 |
0.68 |
0.56 |
wt |
wt |
wt |
wt |
MLH1 Loss |
MLH1 Loss |
C8 |
0.70 |
0.98 |
wt |
wt |
wt |
wt |
MSS |
MSS |
C9 |
0.78 |
0.85 |
wt |
wt |
wt |
wt |
MSS |
MSS |
C10 |
0.90 |
0.75 |
wt |
wt |
Mutant |
Mutant |
MSS |
MSS |
C11 |
0.76 |
0.97 |
wt |
wt |
wt |
wt |
MSS |
MSS |
C12 |
0.92 |
0.96 |
wt |
wt |
wt |
wt |
MSS |
MSS |
C13 |
0.62 |
0.76 |
Mutant |
Mutant |
wt |
wt |
MLH1 Loss |
MLH1 Loss |
C14 |
0.90 |
0.88 |
Mutant |
Mutant |
wt |
wt |
MSS |
MSS |
C15 |
0.77 |
0.91 |
wt |
wt |
wt |
wt |
MSS |
MSS |
C16 |
0.73 |
0.79 |
Mutant |
Mutant |
wt |
wt |
MSS |
MSS |
PT: primary tumors Met: metastasis, wt: Wildtype, MSS: Microsatellite Stable.
Tumor purity was determined from SNP array data using qPure38.
Differences between copy number and LOH profiles for each primary tumor and metastasis pair including the influence of chemotherapy exposure
We calculated the proportion of each genome that was affected by a copy number change from the normal diploid state. Within the 32 samples, the proportion of copy number altered genome ranged from 16.1% to 100%. In 16 samples, over 80% of the genome was copy number altered. Recent studies revealed that whole-genome duplication (WGD) events occur frequently during tumorigenesis and metastasis, resulting in cells bearing complex karyotypes [16, 17]. WGD was identified in both the primary tumor and metastasis in 4 pairs (C2, C12, C14 and C15) whereas in a further 5 pairs only the metastasis had undergone WGD (C3, C8, C11, C13 and C16. Figure 2A). There were no patients in whom only the primary tumor had undergone WGD. WGD was not significantly associated with synchronous versus metachronous disease state, or with treated versus treatment naive metastases.
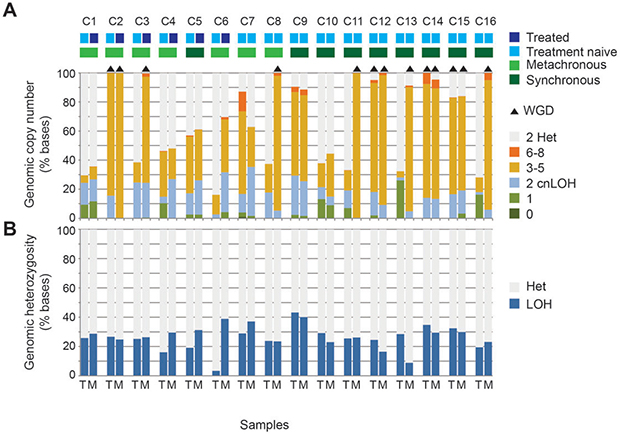
Figure 2: Differences between the copy number profiles of each tumor and metastasis pair including the influence of chemotherapy exposure. The proportion of tumor genomes affected by copy number alteration and loss of heterozygosity (LOH) is shown for paired primary tumors (T) and metastases (M) from the 16 cases. The level of loss or gain of DNA is indicated by different colors (A). Whole genome duplication (WGD) is indicated by black triangles above the bar and is defined as >70% of at least half the chromosomes display copy numbers between 3 and 4 with both parental alleles present. The proportion of tumor genome with LOH is plotted (B).
Copy number alterations may result in shifts in the allelic ratio from the normal heterozygous state through to regions of loss of heterozygosity (LOH) that can occur at any copy number and is measured as the B allele frequency in the array data. For all sample pairs excluding from patient C6, the proportion of the tumor genome affected by LOH was similar between primary and metastatic tumors even when only the metastasis had undergone WGD (Figure 2B).
A comparative analysis was carried out to assess the differences in copy number states between the paired samples. Each genome was segmented into 10Kb regions and the copy number state was compared between corresponding regions in the paired primary and metastasis samples. Initially, in the 5 pairs of samples displaying WGD in only the metastasis, the genomes appeared highly different in metastases compared to matched primary tumors. On further examination, the majority of the differences were due an increase of 2 copies without a change in the LOH status. This large scale moderate copy number increase with no selection for one of the alleles is unlikely to result in driver gene expression changes relative to the background genes, therefore a correction for the WGD was applied. All regions where the copy number change difference was either +1 or +2 copies without LOH were recorded as no difference or concordant between paired samples. With the WGD correction applied, the average percent copy number discordance across all pairs of samples was 22.0% (range 3.3 – 63.6). Interestingly, the treatment naive tumor sample pairs had significantly higher copy number differences compared to sample pairs where the metastasis had been collected post-therapy (Figure 3A, P = 0.014). LOH events unique to primary samples occurred more frequently in those that had not been exposed to therapy, whilst LOH events unique to metastatic tumors was more frequent post-therapy (Figure 3B, P = 0.003). Overall the LOH patterns between the paired tumors were different by an average 12.4% of the genome (ranging from 2.0% to 35.6%, Figure 3B).
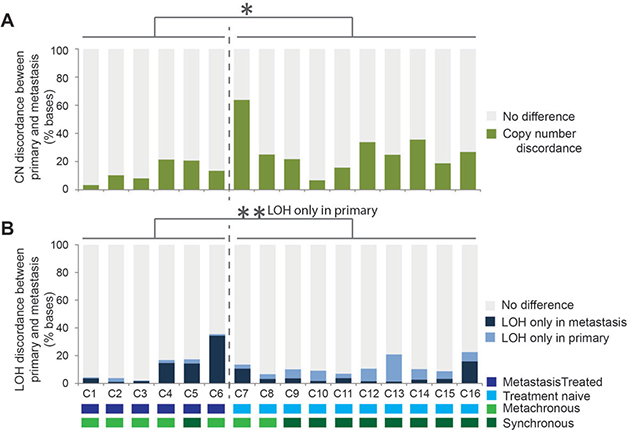
Figure 3: Genomic copy number and loss of heterozygosity (LOH) differences identified between all 16 primary tumor and metastasis pairs. The copy number and LOH was assayed every 10Kb for 287921 sites per metastatic genome and compared with the corresponding region in the paired primary tumor sample with a correction for whole genome duplication applied to all sample pairs. The proportion of regions with copy number that was different between the paired samples is indicated in (A) by the height of the green bars. The proportion of regions for which LOH was observed only in the metastasis sample is indicated by the height of the dark blue bars and those with LOH only in the primary tumor sample by the height of the light blue sections (B). In both charts the grey indicates concordant copy number or LOH states between the paired samples. *P < 0.05, **P < 0.01 (t-test).
Differences in candidate driver genes between primary and metastasis samples
We analyzed the copy number status for 188 candidate CRC genes in the paired primary tumors and metastases (Supplementary Table 1). The copy number status of 123 genes (123/188, 65.4%) was shared between primary tumor and metastasis (Figure 4A). These included genes recurrently mutated in colorectal cancer such as APC, TP53, KRAS, EGFR, SMAD4, FBXW7, NRAS, VEGFA and RNF43. The genes affected most frequently by discordant copy number status between primary and paired metastatic tissues were CSMD3, TRPS1, TGFBR2, CTNNB1, FHIT and MACROD2 (Table 3). TGFBR2, CTNNB1 and FHIT are located on chromosome 3, which was amplified only in metastases (3/16, 18.8 %).
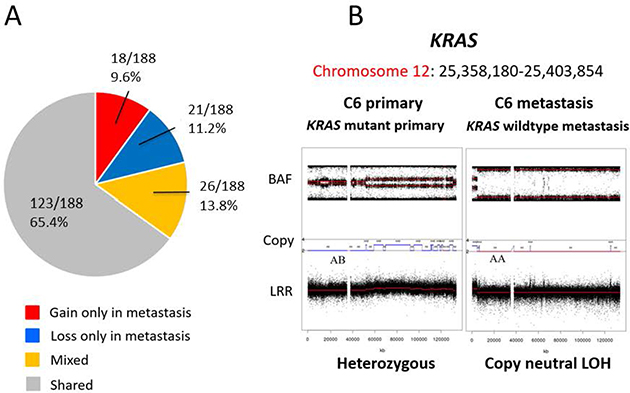
Figure 4: Differences in candidate driver genes between primary and metastasis samples. We analysed the copy number status for 188 candidate CRC genes in the paired primary CRC and metastases (A). The copy number status of 123 genes (123/188 65.4%) was shared between primary and metastasis. KRAS mutation was present in the primary cancers but was not detectable in the metastatic sample following chemotherapy (B). The copy number profile of the genomic region containing KRAS indicates the locus was heterozygous (2 copies; AB) in the primary tumor but showed copy neutral LOH (2 copies; AA) in the paired metastasis (B).
Table 3: Candidate gene list summarized to highlight differences in the copy number
Gene |
Chromosome |
Amplification private to metastasis |
Loss private to metastasis |
|---|---|---|---|
CSMD3 |
8 |
3/16 (18.8 %) |
1/16 (6.3 %) |
TRPS1 |
8 |
3/16 (18.8 %) |
1/16 (6.3 %) |
TGFBR2 |
3 |
3/16 (18.8 %) |
0/16 (0 %) |
CTNNB1 |
3 |
3/16 (18.8 %) |
0/16 (0 %) |
FHIT |
3 |
3/16 (18.8 %) |
0/16 (0 %) |
MACROD2 |
20 |
0/16 (0 %) |
3/16 (18.8 %) |
RBFOX1 |
16 |
0/16 (0 %) |
2/16 (12.5 %) |
PTK2 |
8 |
2/16 (12.5 %) |
2/16 (12.5 %) |
VPS13B |
8 |
2/16 (12.5 %) |
1/16 (6.3 %) |
IRS2 |
13 |
2/16 (12.5 %) |
1/16 (6.3 %) |
CBLB |
3 |
2/16 (12.5 %) |
0/16 (0 %) |
KALRN |
3 |
2/16 (12.5 %) |
0/16 (0 %) |
WHSC1L1 |
8 |
2/16 (12.5 %) |
0/16 (0 %) |
FGFR1 |
8 |
2/16 (12.5 %) |
0/16 (0 %) |
MYC |
8 |
2/16 (12.5 %) |
0/16 (0 %) |
Colorectal cancer candidate genes are reported where copy number differences were observed between the paired samples for at least 2 cases.
An example of mutational discordance is shown in Figure 4B where KRAS mutation was detected in the primary tumor, which would have rendered the patient ineligible for anti-EGFR therapy. However, the paired liver metastasis was wild type for KRAS. The KRAS locus was heterozygous in the primary tumor but showed copy neutral LOH in the paired metastasis, suggesting deletion of the mutant allele in the metastatic tumor.
Copy number status for genes with clinically available targeted agents
The copy number status of 24 genes known to be clinically actionable in colorectal cancer [18, 19] were assessed for amplification occurring specifically in the metastasis but not the primary tumor, as these alterations may have been clinically useful in informing therapy selection. A subset of these can be targeted by therapies that are already approved or in late-phase development (Phase II or III). These include members of the RTK and RAS pathways, such as KRAS, BRAF, NRAS, and ERBB2, which have known associated with resistance to anti-EGFR targeted therapies [13]. In our study, amplification of ERBB2, FGFR1, PIK3CA or CDK8 loci was observed in metastatic but not the paired primary tumors of patients C6, C8, C13 and C16, respectively (Figure 5). ERBB2 encodes HER2, which is the target of trastuzumab. ERBB2 was amplified only in the metastasis of patient C6, therefore trastuzumab may have been beneficial this patient following relapse from standard chemotherapy. Similarly, FGFR1, a target of regorafenib, was specifically amplified only in the metastasis of patient C8 (Figure 5).
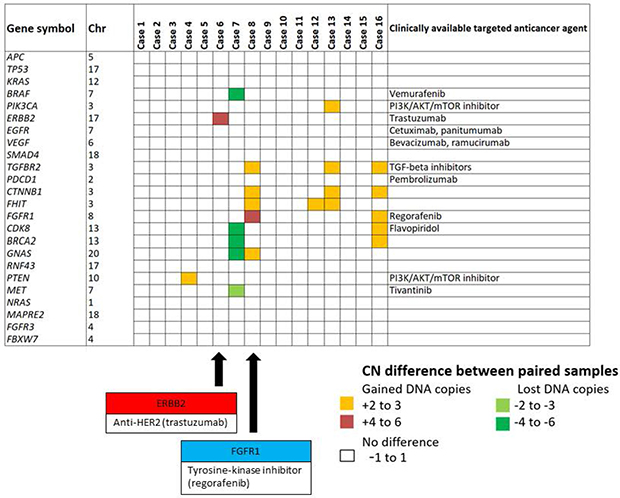
Figure 5: Copy number status private to the metastasis sample affecting clinically actionable targets. The degree of copy number difference was calculated by subtraction of the primary tumor copy number state from that of the liver metastasis. The copy number status of 24 clinically actionable genes was assessed for alterations occurring specifically in metastatic but not primary tissue. ERBB2, a known target receptor of trastuzumab (anti-HER2), was specifically amplified only in the metastatic tissue of C6. Similarly, FGFR1, a known target receptor of regorafenib was specifically amplified only in the metastatic tissue of C8. Black arrow indicates specific gene amplification of C6 (ERBB2) and C8 (FGFR1) respectively.
DISCUSSION
The significant morbidity of colorectal cancer is largely due to metastatic dissemination of the primary tumor. These distant metastatic deposits may have a very different molecular landscape to the primary disease as they may have arisen from minor clones of the primary disease or have undergone molecular changes through selective pressures during the metastatic process. However, actionable biomarkers used for informing therapeutic regimens for metastatic disease are usually based on the molecular status of the primary tumor sample alone. Here, we performed SNP arrays to investigate copy number profiles between paired primary and metastatic colorectal cancer samples to better understand the global differences that may occur between these two disease states.
When comparing the primary tumors to paired metastatic samples, almost a quarter (22.0%) of the genomes contained copy number differences. Whole genome duplication occurred in 4 primary tumors and their paired metastases. In another 5 paired samples, WGD was identified in the metastasis but not in the primary tumor. There were no paired samples where only the primary tumor had undergone WGD. A recent report revealed that the majority (62.5%) of esophageal adenocarcinoma undergo WGD and that tumors with WGD had different patterns of genomic alterations with more frequent oncogenic amplifications and less frequent inactivation of tumor suppressors [20]. Dewhurst et al. showed that in colorectal cancer, WGD allows for a higher rate of chromosomal instability, resulting in poor relapse-free survival [21]. These studies are consistent with our finding of greater rates of WGD in metastases compared to primary tumours. Further studies are now required to determine whether WGD is associated with distinctive clinical or molecular features.
In colorectal cancer patients with liver metastases, it has recently been reported that intra-tumoral heterogeneity is more common in patients previously exposed to chemotherapy compared with patients who are chemotherapy-naive [22]. A similar finding is seen in patients with urothelial carcinoma metastases [23]. Understanding how selective pressure from chemotherapy is involved in evolution from primary tumor to metastasis is a central biological question with clinical implications. In our study, LOH events unique to the primary tumor were commonly observed in therapy naive cases, which likely reflects the reduced clonal complexity of the secondary tumor due to the metastasis and expansion of a single primary tumor clone. By contrast, we predominantly observed an acquisition of LOH events in metastases following chemotherapy, suggesting the therapy has driven the increase in genomic complexity.
For colorectal cancer, the mutation status of KRAS and BRAF in is critical for directing choice of therapy. Here we have demonstrated a case where the primary tumor was KRAS mutant but was wildtype in the metastatic sample following chemotherapy. This suggests that the mutant allele was either lost from the metastatic tumor or the metastatic deposit arose from a KRAS wildtype clone in the primary tumor and highlights the importance of assessing clinically actionable markers in the metastatic rather than primary tumor sample. In this case, targeting the metastatic disease with an anti-EGFR agent may have improved the outcome for this patient.
Our data provide a strong rationale for investigating copy number variations throughout the genome to identify the full complement of potentially actionable genomic alterations that could be developed as clinical biomarkers or therapeutic targets. Amplifications may give rise to overexpression of oncoproteins that are targets of currently available drugs. We hypothesized that genes in regions of altered copy number would include known targets for currently available drugs that may be of benefit in the management of individual patients. We did find a number of examples including amplification private to the metastasis at loci for genes including ERBB2, FGFR1, CDK8 and PIK3CA, which may have increased sensitivity to trastuzumab [24], regorafenib [25], flavopiridol [26], or PI3K/AKT/mTOR inhibitor, respectively (Figure 5). These metastasis-specific gene amplifications are at present insufficient to guide patient management. However, this demonstrates there are potentially actionable changes that occur exclusively in the metastatic tumor but not in the primary tumor.
Studies in lung, gastric, urothelial, thyroid and breast cancer have demonstrated somatic copy number discordance between primary tumors and their metastases [27–31]. Consistent with our results, these studies reported a high rate of concordance when examining known driver mutations or copy number alterations. In CRC, a recent report from Mamlouk is in line with these findings, however, they additionally validated cases of discordance in MMP17, TCF7L2, GNAS, CARD11 and TP53 at the level of gene copy number [15].
Here we provide proof of concept that metastatic samples can be substantially different from their matched primary tumors, even prior to chemotherapeutic intervention. Understanding intra-tumoral heterogeneity is clinically important because it could underlie failure of targeted systemic therapy. Further studies using more comprehensive approaches such as whole-genome sequencing will be needed to characterize all relevant classes of mutations, including single nucleotide variations, indels, copy number and structural rearrangements and non-protein coding mutations with regulatory significance between paired primary and metastatic cancers.
In conclusion, our results demonstrate that the molecular discordance between primary and metastatic tissue may be of clinical relevance in the era of genomically directed precision colorectal cancer medicine. Our data suggest that clinically actionable molecular targets for metastatic chemotherapy may be missed when relying only on biopsies of the primary tumor at the time of diagnosis. These differences could be driven by clonal evolution of the primary tumor due to the metastatic process or as a result of therapy. Serial metastatic or liquid biopsies obtained during the course of clinical care may improve outcomes by more accurately capturing the rapidly changing molecular landscape of a given patient’s disease to better rationalize strategies for personalized therapy.
MATERIALS AND METHODS
Human tissue samples
Samples from 16 patients who had undergone radical surgery for colorectal carcinoma between 2003 and 2014 at Hokkaido University Hospital were analyzed in this study. A total of 48 tissue samples from these 16 patients were obtained in a fresh state including from normal colorectal mucosa, primary tumors and the associated colorectal liver metastases. All tumors were snap frozen in liquid nitrogen within 20 minutes of extirpation. Normal tissue was sampled at least 10cm from the tumor margin. Clinicopathological information for the study patients is summarized in Table 1. Clinical staging and grading of liver metastases were based on the definition set by the Japanese Society for Cancer of Colon and Rectum Guidelines [32]. Patients were observed at 1–6 month intervals until death or December 2015. All patients provided written, informed consent. Approval was obtained from the Hokkaido University Human Research Ethics Committee (HREC) (14-005) and the QIMR Berghofer HREC (P2139). Nucleic acids were extracted using the Qiagen Allprep Kit in accordance with the manufacturer’s instruction. DNA was quantified using Qubit HS DNA Assay (Invitrogen).
Clinical gene hotspot mutation testing
The presence of the BRAF V600E and KRAS codon 12 and 13 mutations were detected using allelic discrimination or high resolution melta analysis, respectively, as described previously [33–35]. Putative mutations were confirmed by Sanger sequencing.
Immunohistochemical evaluation
Formalin-fixed paraffin-embedded tissue blocks were prepared from surgical specimens, and sections were stained with hematoxylin and eosin for histopathological examination by two gastrointestinal pathologists. Immunohistochemical staining for MLH1 was performed as described previously [36]. In brief, 4μM sections were cut, dewaxed and rehydrated. High pH antigen retrieval solution (pH 9.0; Dako, Glostrup, Denmark) was used for MLH1 at 112°C for 7 min. All sections were stained using the MLH1 antibody (clone G168-15, 1:100; BD Pharmingen). Loss of MLH1 expression was recorded when nuclear staining was observed in normal tissue but not in adjacent malignant cells.
SNP arrays and copy number analysis
DNA from all samples was assayed with the Omni 2.5–8, V1.0 and V1.1 IlluminaBeadChips as per manufacturer’s instructions (Illumina). SNP arrays were scanned on an iScan (Illumina), data was processed using the Genotyping module (v.1.9.4) in GenomeStudio v.2011.1 (Illumina) to calculate B-allele frequencies (BAF) and logR ratios. GAP [37] software was used to segment the SNP array data and determine the level of copy number which was classified into one of 5 categories: homozygous deletion (copy number: 0), loss (copy number:1), copy neutral LOH (copy number:2), gain (copy number: 3–5) and amplification (copy number: 6–8). Loss of heterozygosity segments were generated using the segmented B-allele frequency data. Tumor cellularity was determined from SNP array data using qPure [38] to ensure a minimum tumor cellularity of 50%. Whole genome duplication (WGD) whereby >70% of at least half the chromosomes display copy numbers between 3 and 4 with both parental alleles present that typically totaled >45% of all genomic copy number (Supplementary Table 2). All array data has been deposited into the NCBI Gene Expression Omnibus (GEO) repository under accession GSE100787.
Identification of discordant copy number and/or LOH between the sample pairs
A pair wise comparison analysis of genomic copy number and heterozygosity states over 10Kb windows for each primary and liver metastasis pair was carried out. The copy number and heterozygosity for each 10Kb was determined by the largest segment in that region identified using the GAP tool. Discordant copy number regions were identified by subtraction of the primary tumor copy number from that of the liver metastasis. To account for WGD inflating the number of discordant regions, all regions where the copy number change difference was either +1 or +2 copies without LOH were recorded as no difference or concordant between paired samples. Discordant regions of loss of heterozygosity were sought separately and classified into: unchanged; loss of heterozygosity in the liver metastasis; or retained heterozygosity in the liver metastasis. The genomic proportion of these discordant regions, that were made up of single or multiple adjacent 10Kb regions, was recorded.
Genome-wide profiling of potential related genes
We sought to investigate the copy number state of 188 genes (listed in Supplementary Table 1) which were previously identified as significantly mutated genes in CRC [12, 15], and reported by The Cancer Genome Atlas (TGGA) [19] as well as the Catalogue of Somatic Mutations in Cancer (COSMIC) [39]. Copy number and LOH status for each gene was determined by the largest segment produced using GAP, within or spanning the gene footprint (from transcription start site to the last base of the longest transcript). Differences in copy number, heterozygosity status and segment boundaries between tumor pairs were summed across the patients.
Statistical analysis
Two-tailed student’s t-tests were used to compare differences between groups. All differences were considered significant at a p-value of less than 0.05.
Abbreviations
CRC: colorectal cancer; SNP: Single-Nucleotide-Polymorphism; LOH: loss of heterozygosity; MSI: microsatellite instability; MSS: microsatellite stable; BAF: B-allele frequency; WGD: whole-genome duplication.
Author contributions
F.K, A.P, H.N, M.B, C.K, N.W, B.L, M.F, A.T and V.W conceived and designed the experiments. F.K, K.N, D.M, C.B, T.D, L.F, S.P, S.H and C.L performed the experiments. T.K, N.K, H.Y, G.H and A.P analyzed the data. F.K, A.P, N.W and K.N contributed materials/analysis tools. F.K, A.P, N.W and V.W wrote the manuscript.
ACKNOWLEDGMENTS AND FUNDING
We are grateful for fellowship support from the Uehara Memorial Foundation Fellowship (grant 201430015, FK), the Gastroenterological Society of Australia Senior Research Fellowship (VW), the Keith Boden Fellowship (KN), the Australian Postgraduate Award (CL and the Royal College of Pathologists of Australasia (CL). This work was funded by the National Health and Medical Research Council of Australia (1063105, 1112113), the Study Education and Research Trust Fund of Pathology Queensland, the Royal Brisbane and Women’s Hospital Research Foundation, the Ian Potter Foundation (20162154) and Perpetual Trustees (IPAP201601894).
CONFLICTS OF INTEREST
The authors have nothing to declare.
REFERENCES
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA: Cancer J Clin. 2016; 66:7–30.
2. Foster JH. Treatment of metastatic disease of the liver: a skeptic’s view. Semin Liver Dis. 1984; 4:170–179.
3. O’Neil BH, Goldberg RM. Innovations in chemotherapy for metastatic colorectal cancer: an update of recent clinical trials. Oncologist. 2008; 13:1074–1083.
4. Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, Patterson SD, Chang DD. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008; 26:1626–1634.
5. Lievre A, Bachet JB, Boige V, Cayre A, Le Corre D, Buc E, Ychou M, Bouche O, Landi B, Louvet C, Andre T, Bibeau F, Diebold MD, et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol. 2008; 26:374–379.
6. Whitehall V, Tran K, Umapathy A, Grieu F, Hewitt C, Evans TJ, Ismail T, Li WQ, Collins P, Ravetto P, Leggett B, Salto-Tellez M, Soong R, et al. A multicenter blinded study to evaluate KRAS mutation testing methodologies in the clinical setting. J Mol Diagn. 2009; 11:543–552.
7. Van Cutsem E, Kohne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, D’Haens G, Pinter T, Lim R, Bodoky G, Roh JK, Folprecht G, Ruff P, et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med. 2009; 360:1408–1417.
8. Richman SD, Chambers P, Seymour MT, Daly C, Grant S, Hemmings G, Quirke P. Intra-tumoral heterogeneity of KRAS and BRAF mutation status in patients with advanced colorectal cancer (aCRC) and cost-effectiveness of multiple sample testing. Anal Cell Pathol (Amst). 2011; 34:61–66.
9. Oliveira C, Velho S, Moutinho C, Ferreira A, Preto A, Domingo E, Capelinha AF, Duval A, Hamelin R, Machado JC, Schwartz S Jr, Carneiro F, Seruca R. KRAS and BRAF oncogenic mutations in MSS colorectal carcinoma progression. Oncogene. 2007; 26:158–163.
10. Baba H, Baba Y, Uemoto S, Yoshida K, Saiura A, Watanabe M, Maehara Y, Oki E, Ikeda Y, Matsuda H, Yamamoto M, Shimada M, Taketomi A, et al. Changes in expression levels of ERCC1, DPYD, and VEGFA mRNA after first-line chemotherapy of metastatic colorectal cancer: results of a multicenter study. Oncotarget. 2015; 6:34004–34013. http://doi.org/10.18632/oncotarget.5227.
11. Tan IB, Malik S, Ramnarayanan K, McPherson JR, Ho DL, Suzuki Y, Ng SB, Yan S, Lim KH, Koh D, Hoe CM, Chan CY, Ten R, et al. High-depth sequencing of over 750 genes supports linear progression of primary tumors and metastases in most patients with liver-limited metastatic colorectal cancer. Genome Biol. 2015; 16:32.
12. Wang H, Liang L, Fang JY, Xu J. Somatic gene copy number alterations in colorectal cancer: new quest for cancer drivers and biomarkers. Oncogene. 2016; 35:2011–2019.
13. Bertotti A, Papp E, Jones S, Adleff V, Anagnostou V, Lupo B, Sausen M, Phallen J, Hruban CA, Tokheim C, Niknafs N, Nesselbush M, Lytle K, et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature. 2015; 526:263–267.
14. Mohan S, Heitzer E, Ulz P, Lafer I, Lax S, Auer M, Pichler M, Gerger A, Eisner F, Hoefler G, Bauernhofer T, Geigl JB, Speicher MR. Changes in colorectal carcinoma genomes under anti-EGFR therapy identified by whole-genome plasma DNA sequencing. PLoS Genet. 2014; 10:e1004271.
15. Mamlouk S, Childs LH, Aust D, Heim D, Melching F, Oliveira C, Wolf T, Durek P, Schumacher D, Blaker H, von Winterfeld M, Gastl B, Mohr K, et al. DNA copy number changes define spatial patterns of heterogeneity in colorectal cancer. Nat Commun. 2017; 8:14093.
16. Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, Beroukhim R, Pellman D, Levine DA, et al. Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol. 2012; 30:413–421.
17. Jemaa M, Manic G, Lledo G, Lissa D, Reynes C, Morin N, Chibon F, Sistigu A, Castedo M, Vitale I, Kroemer G, Abrieu A. Whole-genome duplication increases tumor cell sensitivity to MPS1 inhibition. Oncotarget. 2016; 7:885–901. http://doi.org/10.18632/oncotarget.6432.
18. Nagahashi M, Wakai T, Shimada Y, Ichikawa H, Kameyama H, Kobayashi T, Sakata J, Yagi R, Sato N, Kitagawa Y, Uetake H, Yoshida K, Oki E, et al. Genomic landscape of colorectal cancer in Japan: clinical implications of comprehensive genomic sequencing for precision medicine. Genome Med. 2016; 8:136.
19. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487:330–337.
20. Stachler MD, Taylor-Weiner A, Peng S, McKenna A, Agoston AT, Odze RD, Davison JM, Nason KS, Loda M, Leshchiner I, Stewart C, Stojanov P, Seepo S, et al. Paired exome analysis of Barrett’s esophagus and adenocarcinoma. Nat Genet. 2015; 47:1047–1055.
21. Dewhurst SM, McGranahan N, Burrell RA, Rowan AJ, Gronroos E, Endesfelder D, Joshi T, Mouradov D, Gibbs P, Ward RL, Hawkins NJ, Szallasi Z, Sieber OM, et al. Tolerance of whole-genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Discov. 2014; 4:175–185.
22. Sveen A, Loes IM, Alagaratnam S, Nilsen G, Holand M, Lingjaerde OC, Sorbye H, Berg KC, Horn A, Angelsen JH, Knappskog S, Lonning PE, Lothe RA. Intra-patient Inter-metastatic Genetic Heterogeneity in Colorectal Cancer as a Key Determinant of Survival after Curative Liver Resection. PLoS Genet. 2016; 12:e1006225.
23. Faltas BM, Prandi D, Tagawa ST, Molina AM, Nanus DM, Sternberg C, Rosenberg J, Mosquera JM, Robinson B, Elemento O, Sboner A, Beltran H, Demichelis F, et al. Clonal evolution of chemotherapy-resistant urothelial carcinoma. Nat Genet. 2016; 48:1490–1499.
24. Sartore-Bianchi A, Trusolino L, Martino C, Bencardino K, Lonardi S, Bergamo F, Zagonel V, Leone F, Depetris I, Martinelli E, Troiani T, Ciardiello F, Racca P, et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): a proof-of-concept, multicentre, open-label, phase 2 trial. Lancet Oncol 2016; 17:738–746.
25. Grothey A, Van Cutsem E, Sobrero A, Siena S, Falcone A, Ychou M, Humblet Y, Bouche O, Mineur L, Barone C, Adenis A, Tabernero J, Yoshino T, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013; 381:303–312.
26. Dickson MA, Shah MA, Rathkopf D, Tse A, Carvajal RD, Wu N, Lefkowitz RA, Gonen M, Cane LM, Dials HJ, Schwartz GK. A phase I clinical trial of FOLFIRI in combination with the pan-cyclin-dependent kinase (CDK) inhibitor flavopiridol. Cancer Chemother Pharmacol. 2010; 66:1113–1121.
27. Bambury RM, Bhatt AS, Riester M, Pedamallu CS, Duke F, Bellmunt J, Stack EC, Werner L, Park R, Iyer G, Loda M, Kantoff PW, Michor F, et al. DNA copy number analysis of metastatic urothelial carcinoma with comparison to primary tumors. BMC Cancer. 2015; 15:242.
28. Liu H, Li F, Zhu Y, Li T, Huang H, Lin T, Hu Y, Qi X, Yu J, Li G. Whole-exome sequencing to identify somatic mutations in peritoneal metastatic gastric adenocarcinoma: A preliminary study. Oncotarget. 2016; 7:43894–43906. http://doi.org/10.18632/oncotarget.9707.
29. Vignot S, Frampton GM, Soria JC, Yelensky R, Commo F, Brambilla C, Palmer G, Moro-Sibilot D, Ross JS, Cronin MT, Andre F, Stephens PJ, Lazar V, et al. Next-generation sequencing reveals high concordance of recurrent somatic alterations between primary tumor and metastases from patients with non-small-cell lung cancer. J Clin Oncol. 2013; 31:2167–2172.
30. Le Pennec S, Konopka T, Gacquer D, Fimereli D, Tarabichi M, Tomas G, Savagner F, Decaussin-Petrucci M, Tresallet C, Andry G, Larsimont D, Detours V, Maenhaut C. Intratumor heterogeneity and clonal evolution in an aggressive papillary thyroid cancer and matched metastases. Endocr Relat Cancer. 2015; 22:205–216.
31. Tang MH, Dahlgren M, Brueffer C, Tjitrowirjo T, Winter C, Chen Y, Olsson E, Wang K, Torngren T, Sjostrom M, Grabau D, Bendahl PO, Ryden L, et al. Remarkable similarities of chromosomal rearrangements between primary human breast cancers and matched distant metastases as revealed by whole-genome sequencing. Oncotarget. 2015; 6:37169–37184. http://doi.org/10.18632/oncotarget.5951.
32. Watanabe T, Itabashi M, Shimada Y, Tanaka S, Ito Y, Ajioka Y, Hamaguchi T, Hyodo I, Igarashi M, Ishida H, Ishihara S, Ishiguro M, Kanemitsu Y, et al. Japanese Society for Cancer of the Colon and Rectum (JSCCR) Guidelines 2014 for treatment of colorectal cancer. Int J Clin Oncol. 2015; 20:207–239.
33. Bond CE, Umapathy A, Buttenshaw RL, Wockner L, Leggett BA, Whitehall VL. Chromosomal instability in BRAF mutant, microsatellite stable colorectal cancers. PloS One. 2012; 7:e47483.
34. Whitehall VL, Rickman C, Bond CE, Ramsnes I, Greco SA, Umapathy A, McKeone D, Faleiro RJ, Buttenshaw RL, Worthley DL, Nayler S, Zhao ZZ, Montgomery GW, et al. Oncogenic PIK3CA mutations in colorectal cancers and polyps. Int J Cancer. 2012; 131:813–820.
35. Nakamura H, Mochizuki A, Shinmyo T, Ando K, Kurimoto N, Yokote K, Takagi M. Immunohistochemical detection of mutated epidermal growth factor receptors in pulmonary adenocarcinoma. Anticancer Res. 2010; 30:5233–5237.
36. Bettington M, Walker N, Rosty C, Brown I, Clouston A, McKeone D, Pearson SA, Klein K, Leggett B, Whitehall V. Serrated tubulovillous adenoma of the large intestine. Histopathology. 2016; 68:578–587.
37. Popova T, Manie E, Stoppa-Lyonnet D, Rigaill G, Barillot E, Stern MH. Genome Alteration Print (GAP): a tool to visualize and mine complex cancer genomic profiles obtained by SNP arrays. Genome Biol. 2009; 10:R128.
38. Song S, Nones K, Miller D, Harliwong I, Kassahn KS, Pinese M, Pajic M, Gill AJ, Johns AL, Anderson M, Holmes O, Leonard C, Taylor D, et al. qpure: A tool to estimate tumor cellularity from genome-wide single-nucleotide polymorphism profiles. PloS One. 2012; 7:e45835.
39. Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, Teague JW, Campbell PJ, Stratton MR, et al. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011; 39:D945–950.