
INTRODUCTION
Breast cancer is the second most common cancer in female patients, causing extensive mortality, psychological stress and health care burden worldwide [1–3]. Numerous strategies have been developed for the treatment of breast cancer over the past two decades, including chemotherapy, radiotherapy, hormonal therapy and immunological therapy [4]. However, disease progression, relapse and treatment resistance often resulted in treatment failures [5, 6], which raise the question of whether these conventional treatments have targeted the right tumor cells. Recently, it suggests that tumors contain a bulk of heterogeneous cells that derived from a small subset of cell population, which shows the characteristics of stem cell, termed cancer stem cell (CSC) or tumor-initiating cell (TIC) [7, 8]. CSCs contribute to tumor initiation, progression, metastasis, recurrence and treatment resistance [9]. Since the first identification of CSCs in leukemia, CSCs have been identified in almost all cancer types, including breast cancer, pancreatic cancer, lung cancer, gastric cancer and colorectal cancer [10–14]. Conventional cancer treatments are successful at killing the differentiated tumor cells but fail to eliminate CSCs [9]. Therefore, therapeutic strategies that target breast CSCs could potentially improve breast cancer treatment outcomes.
The Wnt pathway is an ancient and evolutionary conserved self-renewal pathway, which regulates stem cells to determine cellular fate during embryonic development and keep tissue homeostasis in adults [15]. The canonical Wnt pathway is mediated by β-catenin, a key intracellular mediator of the pathway, whose degradation is controlled by a proteasomal complex consisting of adenomatous polyposis coli (APC), glycogen synthase kinase 3β (GSK-3β) and AXIN. In the absence of Wnt ligands, cytosolic β-catenin levels are kept low by proteasomal degradation. In the presence of Wnt ligands, ligand-receptor binding induces the stabilization of β-catenin and promotes β-catenin nuclear accumulation. In the nucleus, β-catenin interacts with transcription factors of the LEF/TCF family and induces the expression of Wnt target genes such as SURVIVIN, CYCLIN-D1, MMP-2, C-MYC and AXIN-2 [16]. Deregulation of Wnt pathway has been associated with various human cancers such as breast cancer, gastric cancer, and colorectal cancer [17]. Most importantly, previous studies have shown that CSCs require high Wnt signaling activity to maintain their self-renewal and tumorigenic properties, indicating that Wnt signaling pathway is a potential target for breast CSCs [18].
Celecoxib is one of the most commonly used non-steroidal anti-inflammatory drugs (NSAIDs) for the treatment of fever, pain, stiffness, and swelling. Numerous experimental and epidemiological studies have demonstrated that NSAIDs have chemo-preventive activity against cancers, including breast cancer and colorectal cancer [19–22]. Many case-control studies have also shown a significant decrease in the risk of breast cancer among women with regular NSAIDs use [23, 24]. However, the mechanisms by which NSAIDs exert its cancer prevention effects have yet been completely understood. The anti-inflammatory action of NSAIDs is mediated via their inhibitory effect on cyclooxygenase-2 (COX-2) activity and the synthesis of prostaglandin E2 (PGE2), while both COX-2 and PGE2 are strong inducers for inflammation. Earlier studies have shown that PGE2 was able to enhance the expansion of stem cells in the hematopoietic system and CSCs in colorectal tumors through the activation of Wnt pathway [25–30]. In the present study, we showed for the first time that celecoxib targets breast CSCs by inhibiting the synthesis of PGE2 and down-regulating the Wnt pathway activity.
RESULTS
Celecoxib suppresses breast cancer cell proliferation, CSC growth and self-renewal
The cytotoxic effects of celecoxib on monolayer culture of breast cancer cell lines MCF-7 and MDA-MB-231 were evaluated by CCK-8 detection kit. MCF-7 cells are estrogen-positive and poorly invasiveness, while MDA-MB-231 cells are triple negative and highly invasiveness. Celecoxib inhibited cell proliferation of both MCF-7 and MDA-MB-231 cells in a concentration dependent manner (Figure 1A). Celecoxib inhibited MCF-7 cells more efficiently than that of MDA-MB-231 cells as the IC50 value was much lower in the former than that in the latter (Table 1).
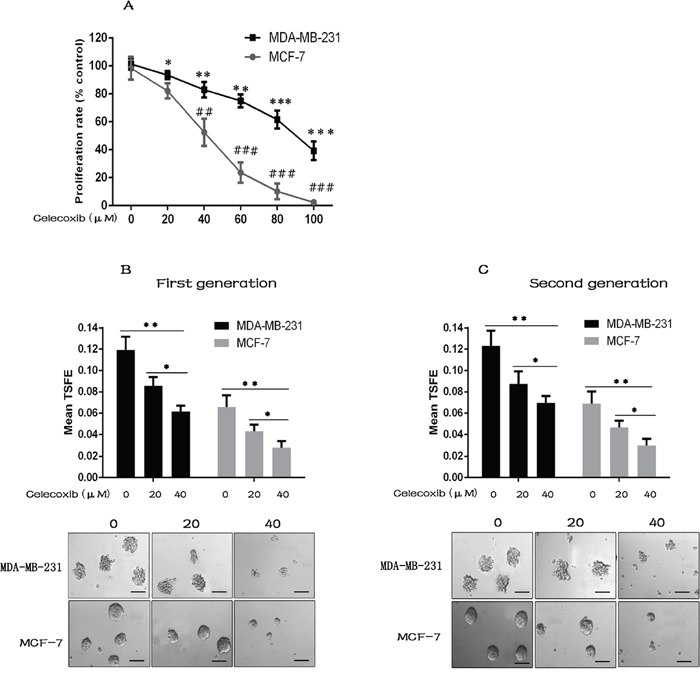
Figure 1: Celecoxib suppresses breast cancer cell proliferation, CSC growth and self-renewal. (A) The cell proliferation of both MCF-7 and MDA-MB-231 cells was decreased by celecoxib treatment in a concentration dependent manner. (B and C) Celecoxib inhibited the first (with celecoxib treatment) and second (without additional celecoxib treatment) generation tumorsphere formation of both MAD-MB-231 and MCF-7 cells. The tumorsphere sizes greater than 100 μm were enumerated, and a representative image of tumorspheres is shown. TSFE: tumorsphere formation efficiency. Scale bar = 100 μm. Data are presented as the means ± SD from three independent experiments; *, P < 0.05; **, ## P < 0.01; ***, ### P < 0.001.
Table 1: IC50 of different drugs / combinations (μM)
Drugs |
MDA-MB-231 |
MCF-7 |
|---|---|---|
Celecoxib |
89.05 |
40.05 |
Cisplatin |
7.95 |
40.63 |
Cisplatin + 20 μM Celecoxib |
4.18 |
21.40 |
5-FU |
444.40 |
78.32 |
5-FU + 20 μM Celecoxib |
112.00 |
20.16 |
Note: IC50 was calculated by nonlinear regression analysis using GraphPad Prism software.
The non-adherent tumorsphere formation assay is commonly used as an in vitro surrogate to quantify the frequency of CSCs [31, 32], and the ability of tumorspheres to be serially passaged at clonal density is an indirect marker of CSC self-renewal [33]. Treatment with celecoxib resulted in a significant reduction of tumorsphere formation efficiency (TSFE) of both MCF-7 and MDA-MB-231 cells in a dose dependent manner (Figure 1B). To evaluate the effect of celecoxib on CSC self-renewal, the primary tumorspheres were collected and dissociated into single cells. The single cells derived from treated or untreated primary tumorspheres were replated without continuous celecoxib exposure. The second generation TSFE was significantly lower in cells derived from celecoxib-treated primary tumorspheres as compared to cells derived from untreated primary tumorspheres (Figure 1C).
Celecoxib sensitizes breast cancer cells to chemotherapeutic drugs by selectively targeting CSCs
CSCs share many features of normal stem cells, including the relative quiescence, resistance to chemotherapeutic drugs and resistance to apoptosis. Conventional chemotherapeutic drugs are successful at killing the differentiated cancer cells but fail to eliminate CSCs, and leading to chemo-resistance and tumor relapse [34]. Therefore, a combination use of drugs targeting both differentiated cancer cells and CSCs was proposed to improve cancer treatment efficacy [35]. In this study, evaluation of chemotherapeutic drugs of 5-FU or cisplatin in combination with 20 μM celecoxib showed that celecoxib sensitized MCF-7 and MDA-MB-231 cells to 5-FU or cisplatin treatment (Figure 2A; Table 1).
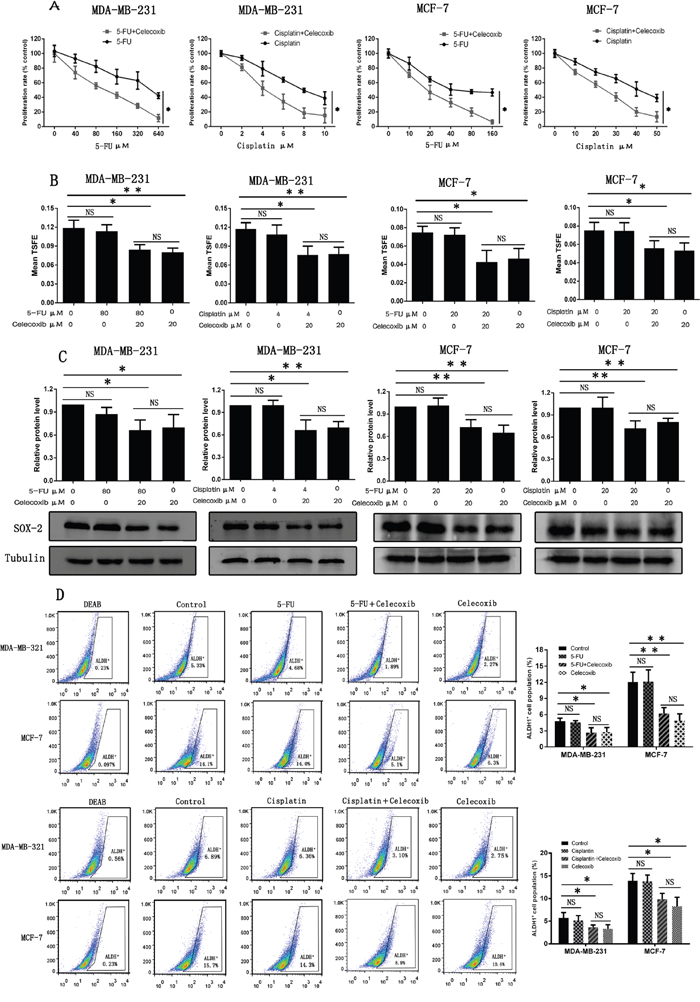
Figure 2: Celecoxib sensitizes breast cancer cells to chemotherapeutic drugs by selectively targeting CSCs. (A) CCK-8 was performed to evaluate cell proliferation of both MDA-MB-231 and MCF-7 cells after being treated with various concentrations of chemotherapeutic drugs (5-FU, cisplatin) alone or in combination with 20 μM celecoxib. (B, C and D) Non-adherent tumorsphere formation assay, western-blot assay and flow cytometry analysis were performed to evaluate the effects of celecoxib on CSCs. MDA-MB-231: 5-FU, 80 μM; cisplatin, 4 μM; Celecoxib, 20 μM. MCF-7: 5-FU, 20 μM; cisplatin, 20 μM; Celecoxib, 20 μM. Data are presented as the means ± SD from three independent experiments; *, P < 0.05; **, P < 0.01; NS, Non statistical significance.
We further examined whether celecoxib sensitized both MCF-7 and MDA-MB-231 cells to chemotherapeutic drugs was achieved by selectively targeting CSCs. Treatment with 5-FU or cisplatin alone failed to reduce TSFE of both MCF-7 and MDA-MB-231 cells, but co-treatment with celecoxib or treatment with celecoxib alone decreased the TSFE significantly, while no statistical difference was found between co-treatment and celecoxib treatment alone groups (Figure 2B). Previous studies have reported that SOX-2 is a CSC marker, which is used to identify and characterize CSCs in a variety of tumors including breast tumor [36–38]. Western-blot analysis revealed that 5-FU or cisplatin alone was unable to reduce SOX-2 expressions, but co-treatment with celecoxib or treatment with celecoxib alone led to a significant reduction of SOX-2 expression, while no statistical difference was found between co-treatment and celecoxib treatment alone groups (Figure 2C). In addition, we also identified CSCs based on their expression of high aldehyde dehydrogenase (ALDH) activity [39]. Flow cytometry analysis showed that 5-FU or cisplatin alone was unable to reduce the ALDH-positive cell population, but co-treatment with celecoxib or treatment with celecoxib alone led to a significant reduction of ALDH-positive cell population, while no statistical difference was found between co-treatment and celecoxib treatment alone groups (Figure 2D). Together, these findings indicate that celecoxib sensitizes breast cancer cells to chemotherapeutic drugs by selectively targeting CSCs.
Celecoxib inhibits EMT gene signature
The epithelial to mesenchymal transition (EMT) is a basic process in morphogenesis of various tissues during embryonic development. Recent studies have suggested that EMT is also associated with generation of CSCs [40–43]. MDA-MB-231 cells typically have a spindle-shaped mesenchymal morphology, however, changed into cobble-stone-like epithelial appearance following celecoxib treatment (Figure 3A). To test the hypothesis that celecoxib may inhibit EMT, a group of mesenchymal makers including SNAIL, SLUG and TWIST was analysed by RT-PCR [44]. Our results showed that mRNA expression of these mesenchymal makers was decreased upon celecoxib treatment (Figure 3B). In addition, the expression of two EMT markers of E-cadherin and Vimentin were evaluated by western blot and immunofluorescence staining analysis. Both methods revealed that celecoxib induced an increase of E-cadherin along with a marked decrease of Vimentin in MDA-MB-231 cells (Figure 3C and 3D).
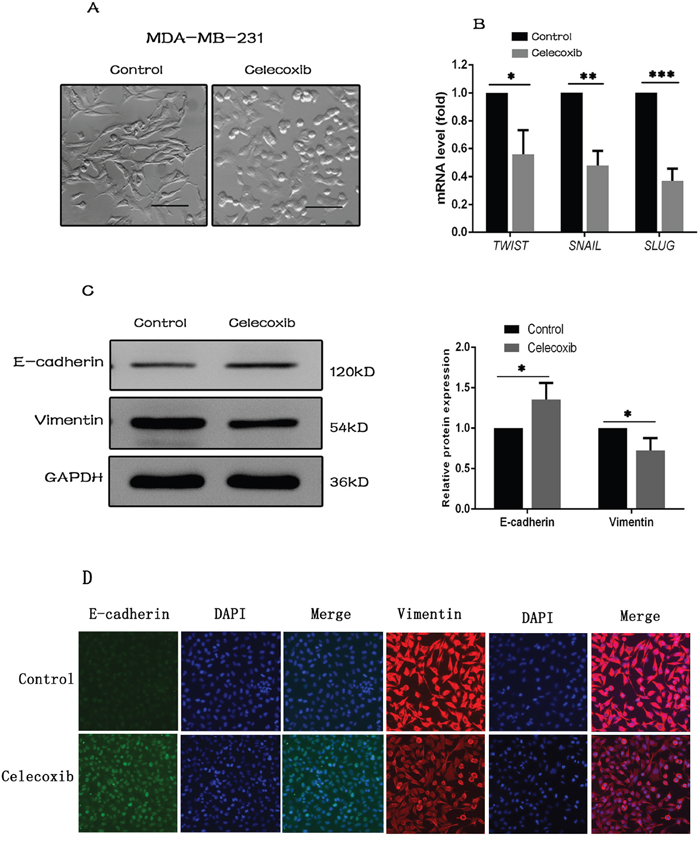
Figure 3: Celecoxib inhibits EMT gene signature. (A) Morphological changes of MDA-MB-231 cells treated with celecoxib (magnification, ×100). (B) Relative mRNA expressions of mesenchymal makers including SNAIL, SLUG, and TWIST following celecoxib treatment. (C and D) The protein expression changes of E-cadherin and Vimentin following celecoxib treatment. Scale bar = 50 μm. Data are presented as the means ± SD from three independent experiments; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Celecoxib attenuates breast cancer cell metastasis
It was reported that CSCs play a critical role in tumor metastasis [45, 46]. MDA-MB-231 cells were used to investigate the effect of celecoxib on breast cancer cell metastasis. Transwell migration assay and wound healing assay are commonly used to determine the metastatic property of cancer cells in vitro. In the transwell assay, celecoxib treatment led to a significant reduction of the number of cells invaded into the lower chambers (Figure 4A). Similarly, in the wound healing assay, celecoxib treatment attenuated the decreased distance between the wounded areas (Figure 4B).
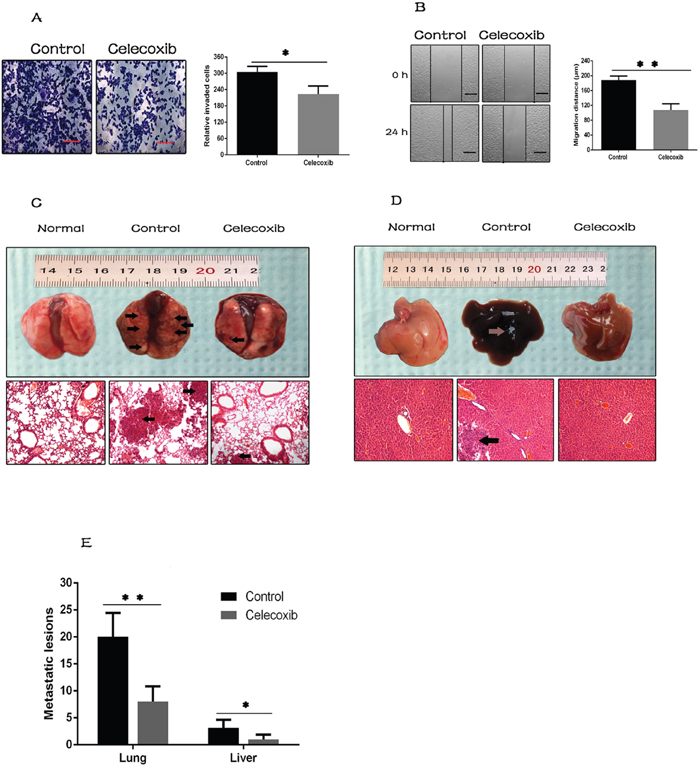
Figure 4: Celecoxib attenuates breast cancer cell metastasis. (A and B) The effect of celecoxib on invasion and migration of MDA-MB-231 cells was measured by the transwell assay and wound healing assay respectively (magnification, ×100). (C and D) The effect of celecoxib on breast cancer cell metastasis was analyzed in vivo. Arrows indicate surface metastatic lesions on the lungs and livers, and metastatic lesions were confirmed by H&E staining (magnification, ×100). Normal lung and liver tissues were used as negative control. (E) Number of visible surface metastatic lesions on lungs and livers of individual mice were counted. Scale bar = 100 μm. Data are presented as mean ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
We further evaluated the effects of celecoxib on breast cancer cell metastasis in vivo by tail vein injection mouse model. Twenty days post injection, the lungs and livers were harvested, and numbers of visible surface metastatic lesions were counted. Metastatic lesions were further confirmed by H&E staining (Figure 4C and 4D). It revealed that the number of surface metastatic lesions of both lungs and livers was significantly lower in celecoxib treated group than in the untreated control (Figure 4E).
Celecoxib down-regulates the Wnt pathway activity by inhibiting the synthesis of PGE2 and reducing the phosphorylation of GSK-3β
Wnt pathway has been implicated in the maintenance of CSCs in a variety of tumors [2, 17]. To explore whether celecoxib targets breast CSCs via down-regulating Wnt pathway activity, the Wnt pathway was investigated using MDA-MB-231 cells, which is known to express high Wnt pathway activity [47, 48]. Western blot analysis showed that celecoxib treatment decreased the expression of β-catenin, a Wnt pathway effective component, and Wnt pathway target proteins of Survivin and MMP-2 (Figure 5A). In addition, celecoxib decreased mRNA expression levels of Wnt pathway target genes including AXIN2 [49], CYCLIN-D1 [50] and C-MYC [51] (Figure 5B) and their corresponding protein expression levels (Figure 5C). We also use the dual-luciferase reporter assay to investigate the changes of Wnt pathway activity. Celecoxib decreased the luciferase activity of TOP flash reporter, which contains a TCF-binding site. In contrast, the FOP flash reporter containing mutated TCF-binding site, had very low luciferase activity and treatment with celecoxib didn’t decrease the luciferase activity (Figure 5D). Together, these findings suggest that celecoxib down regulated Wnt pathway activity.
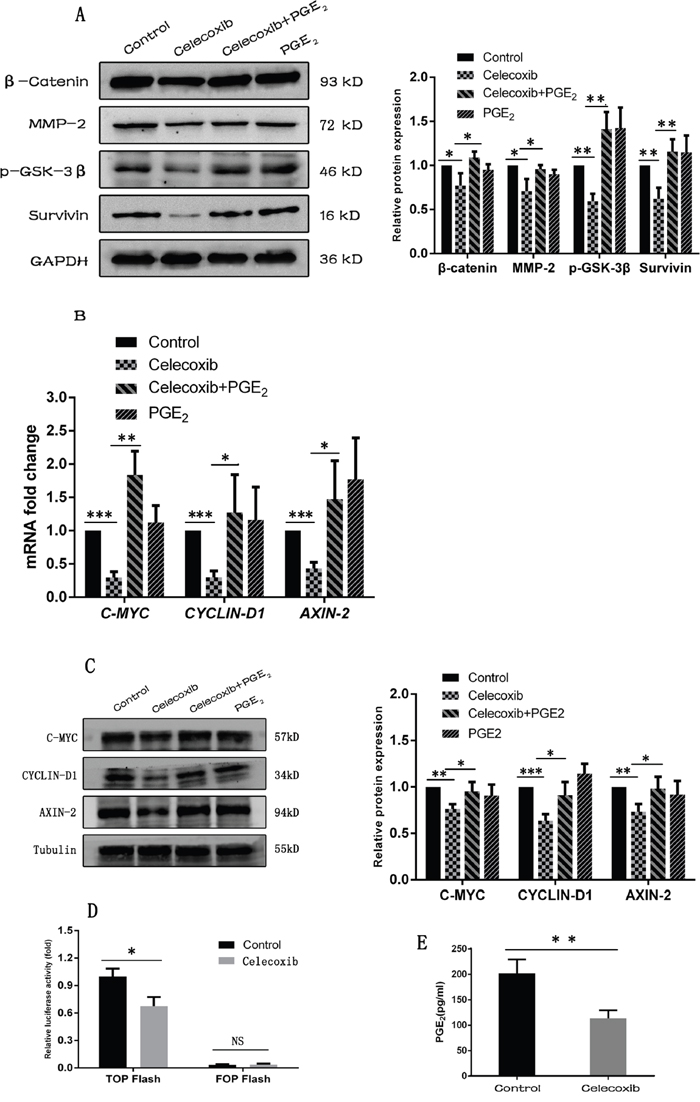
Figure 5: Celecoxib down-regulates the Wnt pathway activity by inhibiting the synthesis of PGE2 and reducing the phosphorylation of GSK-3β. (A, B and C) The Wnt pathway target genes and components were measured by western-blot and RT-PCR. (D) Dual-luciferase reporter assay was carried out to evaluate the changes of Wnt pathway activity. (E) PGE2 levels in the cell culture media was measured by PGE2 ELISA kit. Celecoxib: 20 μM; PGE2: 20 nM. Data are presented as mean ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
More importantly, celecoxib also decreased the synthesis of PGE2 in MDA-MB-231 cells (Figure 5E). To explore whether PGE2 was associated with celecoxib mediated Wnt pathway down-regulation, exogenous PGE2 was added to celecoxib treated MDA-MB-231 cells. As a result, the Wnt pathway activity was partially rescued by exogenous PGE2 addition, which was manifested by partially restored expression of β-catenin and Wnt pathway target genes [49–51] (Figure 5A–5C). Previous studies demonstrated that GSK-3β is a component of the destruction complex that renders β-catenin to proteasomal degradation and maintains Wnt pathway at a low activity. In addition, the destruction complex can be inactivated by the phosphorylation of GSK-3β at Ser9 [16]. In the present study, celecoxib reduced the phosphorylation of GSK-3β, while the addition of exogenous PGE2 increased the phosphorylation of GSK-3β (Figure 5A).
Celecoxib inhibits tumorigenesis in vivo by inhibiting the synthesis of PGE2 and down-regulating the Wnt pathway activity
Previous studies have shown that only CSCs, not the differentiated tumor cells, have the ability to initiate and sustain tumor growth [10, 52]. Xenograft mouse model was used to investigate the growth and tumorigenic property of breast CSCs in vivo. It showed that tumor growth was inhibited by celecoxib treatment, and tumor tissues were confirmed by H&E staining (Figure 6A–6C).
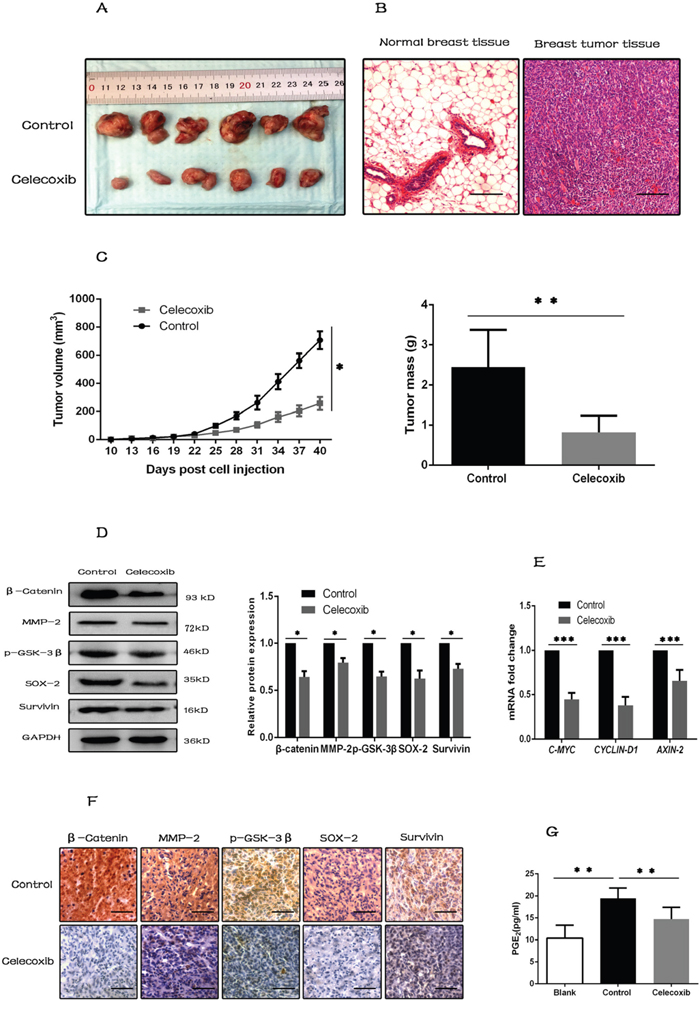
Figure 6: Celecoxib inhibits tumorigenesis in vivo by inhibiting the synthesis of PGE2 and down-regulating the Wnt pathway activity. (A) Photographs of excised tumors from two groups of NOD/SCID mice (N = 6 per group). (B) Tumor tissues were confirmed by H&E staining, and normal breast tissues were used as negative control (magnification, ×100). (C) Tumor growth was measured with a caliper every three days, and tumors were weighted when the mice were sacrificed. (D and E) Proteins and RNAs were extracted from tumor tissues. Wnt pathway components (β-catenin, p-GSK-3β) and target genes (MMP-2, Survivin, AXIN-2, CYCLIN-D1 and C-MYC), and CSC marker (SOX-2) were evaluated by western-blot or RT-PCR. (F) IHC staining for Wnt pathway components, target genes and CSC marker (SOX-2) in sections of tumor tissues (magnification ×200). (G) PGE2 levels in serum of the assayed animals were evaluated by PGE2 ELISA kit. Scale bar = 100 μm. Data are presented as mean ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Celecoxib treatment also decreased the expression levels of Wnt pathway components and target genes, such as β-catenin, p-GSK-3β, MMP-2, Survivin, AXIN2, CYCLIN-D1 and C-MYC, and the CSC marker SOX-2 in the xenograft tumor tissues (Figure 6D and 6E). These changes were also confirmed by immunohistochemistry of tumor tissues (Figure 6F).
Previous studies demonstrated that PGE2 was able to activate Wnt pathway by phosphorylating GSK-3β at Ser9 [28]. Therefore, PGE2 levels in serum of the assayed animals were evaluated by PGE2 ELISA kit. The results showed that PGE2 synthesis was low in serum of blank control mice, but was increased significantly in serum of tumor transplanted mice. However, this increase of PGE2 synthesis was inhibited by celecoxib treatment (Figure 6G).
DISCUSSION
Accumulating evidences have suggested that tumors contain a bulk of heterogeneous tumor cells that derive from a small subset of cell population, which shows the characteristics of stem cells, termed as cancer stem cells (CSCs) [7, 8]. Besides, a critical role of CSCs in tumor initiation, progression, metastasis, and chemo-resistance has been well established in numerous cancer types including breast cancer [9]. Thus, the CSC concept has provided an important milestone in the understanding of chemo-resistance and cancer recurrence. On the basis of their characteristics, targeting and eradicating of CSCs using novel drugs represent a potential strategy for improving cancer treatment outcomes. In fact, previous studies have successfully identified some compounds such as salinomycin, curcumin, resveratrol, and niclosamide, which are able to target breast CSCs through the inhibition of Wnt pathway [53–57]. In addition, multiple lines of evidences demonstrate a significant reduction in breast cancer risk among women with regular NSAIDs use [23, 24]. However, its mechanism has yet been elucidated. In the present study, we demonstrate that celecoxib, a widely used NSAID, targets breast CSCs by inhibiting the synthesis of PGE2 and down-regulating the Wnt pathway activity.
Based on the ability that stem cells grow in serum-free non-adherent conditions, while differentiated cells fail to survive under such conditions. The non-adherent tumorsphere formation assay is commonly used as an in vitro surrogate assay to investigate the growth and tumorigenic property of breast CSCs [31, 32]. Using this assay, we found that celecoxib dramatically inhibited the tumorsphere formation of both MDA-MB-231 and MCF-7 cells (Figure 1B). In addition, the ability of tumorsphere to be serially passaged is an indirect marker of CSCs self-renewal [33]. Previous studies have shown that self-renewal is the key characteristic of breast CSCs, and the tumorsphere formation efficiency is retained as the serial passages [33]. In this study, we found that celecoxib treatment reduced MDA-MB-231 and MCF-7 cells subsequent secondary tumorsphere formation efficiency without additional treatment (Figure 1C). Furthermore, celecoxib completely depleted the tertiary tumorsphere formation (data not shown). These findings suggested that celecoxib is effective in inhibiting the growth, tumorigenesis and self-renewal of breast CSCs.
Previous studies have reported that conventional chemotherapeutic drugs are successful at killing the differentiated cancer cells but fail to eliminate CSCs, and leading to chemo-resistance and tumor relapse [34]. CSCs are resistant to conventional chemotherapeutic drugs via three possible mechanisms: 1) by increasing the expression of ABC transporter genes and consequently excluding chemotherapeutic drugs out of cells; 2) by activating DNA repair capacity and repairing the DNA damages inflicted by chemotherapies; and 3) by dividing infrequently and making them insensitive to antimitotic chemotherapeutic drugs [58, 59]. Based on these findings, we propose that combination use of drugs targeting both differentiated cancer cells and CSCs may improve cancer treatment outcomes [35]. In this study, we found that single use of conventional chemotherapeutic drugs (cisplatin and 5-FU) had a moderate effect on breast cancer cells but combination use of both celecoxib and conventional chemotherapeutic drugs dramatically increased the chemo-sensitivity of breast cancer cells (Figure 2A). This effect may be mediated via an additive mechanism that conventional chemotherapeutic drugs (5-FU and cisplatin) kill the differentiated cancer cells and celecoxib kills the CSCs. Next, we used the non-adherent tumorsphere formation assay and CSC marker SOX-2 to demonstrate that celecoxib decreased tumorsphere formation and SOX-2 expression, while the conventional chemotherapeutic drugs (cisplatin and 5-FU) were unable to do that (Figure 2B and 2C). In addition, previous studies have demonstrated that ALDH-positive cell population is enriched with CSCs [39]. In this study, we found that celecoxib decreased the ALDH-positive CSC population, while the conventional chemotherapeutic drugs (cisplatin and 5-FU) were unable to do that (Figure 2D). A recent study has also demonstrated that celecoxib abrogates chemo-resistance of bladder cancer cells by selectively targeting CSCs [60]. These findings indicate that celecoxib increases the chemo-sensitivity of breast cancer cells by selectively targeting CSCs.
The epithelial to mesenchymal transition (EMT) is a basic process in the morphogenesis of various tissues during embryonic development. It is defined that epithelial cells loss epithelial traits, acquire mesenchymal characteristics and show reduced intercellular adhesion and increased cell motility. Previous studies have suggested that EMT also associates with the generation of CSCs [40–43]. For example, the induction of EMT results in tumor aggressiveness, chemo-resistance and recurrence that are tightly linked with the characteristics of CSCs. In addition, various genes that induce EMT are also related to the expression of CSC markers. Furthermore, a high-throughput drug screening has identified salinomycin as a drug that specifically kills CSCs through the inhibition of EMT [53]. In this study, we observed that celecoxib induced morphological changes of MDA-MB-231 cells from spindle-shape to cobble-stone-like (Figure 3A). On the other hand, celecoxib decreased the mRNA expression of a panel of mesenchymal marker genes including SNAIL, SLUG and TWIST (Figure 3B). Moreover, western blot and immunofluorescence staining analysis demonstrated that celecoxib increased the level of epithelial marker protein of E-cadherin and decreased the level of mesenchymal marker protein of Vimentin (Figure 3C and 3D). These findings demonstrate that celecoxib is able to inhibit EMT, which is a property of breast CSCs.
Tumor metastasis is a complex process requiring the most aggressive tumor cells to survive the long time in the circulation system and form metastatic lesions in distance through invasion and migration. Previous studies have reported that CSCs display a mesenchymal morphology, show reduced intercellular adhesion and increased migration and invasion ability, therefore, promote tumor metastasis [45, 46]. Metastasis is a key feature of breast cancer, and extensive studies have demonstrated that breast CSCs play an important role in forming metastatic lesions. In the present study, we found that celecoxib dramatically inhibited both invasion and migration of MDA-MB-231 cells in vitro (Figure 4A and 4B). We also found that celecoxib reduced formation of metastatic lesions in lungs and livers in vivo (Figure 4C–4E). Given the critical role that CSCs play in tumor metastasis, it seems that celecoxib reduces breast cancer cell metastasis at least partially through the inhibition of breast CSCs.
It is well established that Wnt pathway regulates the self-renewal of stem cells in various organs, including mammary gland [15]. Ectopic activation of Wnt pathway in mouse models results in mammary carcinogenesis. In addition, Wnt pathway deregulation has been reported in breast cancer patients [17]. Most importantly, previous studies have shown that Wnt pathway plays a critical role in the maintenance of CSCs [18]. PGE2 is an inflammation inducer and is synthesized by COX-2. Previous studies have reported that PGE2 can enhance the expansion of stem cells in the hematopoietic system and CSCs in colorectal tumors through the up-regulation of Wnt pathway activity [25–30]. Another study also reported that paracrine and autocrine synthesized PGE2 induced the formation of CSCs through activation of Wnt pathway [61]. In this study, we demonstrate that celecoxib targets breast CSCs by inhibiting the synthesis of PGE2 and down-regulating the Wnt pathway activity.
In summary, we have used various approaches to comprehensively analyze the role of celecoxib plays in the inhibition of breast CSCs. Moreover, we have uncovered the molecular mechanism that celecoxib targets breast CSCs by inhibiting the synthesis of PGE2 and down-regulating the Wnt pathway activity. It conceptually advances the current understanding of the molecular mechanisms by which celecoxib acts on cancer prevention. Furthermore, gave the critical role that CSCs play in tumor initiation, progression, chemo-resistance and recurrence, it is interesting to speculate that celecoxib is useful not only in cancer prevention but also as an adjuvant drug to improve cancer treatment outcomes by targeting CSCs.
MATERIALS AND METHODS
Reagents and antibodies
Celecoxib, cisplatin, 5-fluorouracil (5-FU) and prostaglandin E2 (PGE2) were purchased from Sigma-Aldrich. Each compound was prepared as 1 mM stock solution in dimethylsulfoxide (DMSO) for dilution into various concentrations. The following mouse or rabbit monoclonal primary antibodies were used: anti-E-cadherin (Abcam), anti-MMP-2 (Abcam), anti-Vementin (Abcam), anti-c-MYC (Abcam), anti-Axin-2 (Abcam), anti-Cyclin-D1 (Abcam), anti- β-catenin (Cell Signaling Technology), anti-Survivin (Cell Signaling Technology), anti-SOX-2 (Cell Signaling Technology), anti-p-GSK-3β (Cell Signaling Technology), anti-GAPDH (Beyotime Biotechnology) and anti-Tubulin (Beyotime Biotechnology). HRP conjugated goat anti-mouse or anti-rabbit secondary antibodies were purchased from Beyotime Biotechnology.
Monolayer cell culture
MDA-MB-231 and MCF-7 cells were purchased from American Type Culture Collection (ATCC) where they were characterized by mycoplasma detection, DNA Fingerprinting, isozyme detection and cell vitality detection. For monolayer Cell culture, human breast cancer cell lines MDA-MB-231 and MCF-7 were cultured in DMEM (Gibco) and RPMI-1640 (Gibco) respectively, which were supplemented with 10% fetal bovine serum (FBS) (Gibco), 100 U/ml penicillin and 100 U/ml streptomycin at 37°C in a humidified 5% CO2 incubator.
Tumorsphere formation assay
For tumorsphere formation, single MDA-MB-231 and MCF-7 cells were suspended in serum-free DMEM-F12 medium, which was supplemented with B-27 (Invitrogen), 20 ng/ml EGF (Invitrogen), 20 ng/ml FGF (Invitrogen), 4 μg/ml heparin (Sigma-Aldrich) and plated at 2000 cells per well in a 96-well non-attachment plate (Thermo Fisher).
Primary tumorspheres were centrifuged (500 rcf), dissociated with trypsin, and then sieved through a 40 μm cell strainer to obtain single cell suspensions. These dissociated single cells were replated at 2000 cells per well in a 96-well non-attachment plate to form secondary tumorspheres.
Tumorspheres were cultured for seven days before counting the numbers. Individual tumorspheres were counted under an inverted microscope at ×100 magnification using the NIS-Elements imaging software. The percentage of cells capable of forming tumorspheres, termed as tumorsphere formation efficiency (TSFE), was calculated as follows: number of tumorspheres formed/number of single cells plated.
Cell proliferation assay
The CCK-8 detection kit was used to assess cell proliferation according to the manufacturer’s instructions. Briefly, cells were seeded in a 96-well plate at 3000 cells per well. After incubation for 24 h, cells were treated with various concentrations of celecoxib for 24 h. Subsequently, CCK-8 solution was added and the plate was incubated at 37°C for 2.5 h. The number of viable cells was measured at a wavelength of 450 nm using a Versamax microplate reader.
For the evaluation of chemo-sensitivity, cells were treated with increasing concentrations of Cisplatin and 5-FU or in combination with 20 μM celecoxib. After 24 h of incubation, cell proliferation was assessed by CCK-8 detection kit as described above.
Wound healing and transwell assay
Cell migration was determined by using a wound healing assay. Briefly, MDA-MB-231 cells were plated as monolayer at a density of 5x105 cells per well in a 6-well plate and grown to confluence, then the monolayer cells were scratched with a 10-μL micropipette tip and then replaced with fresh medium supplemented with 20 μM celecoxib. After incubation for 24 h, the wound distances were measured using NIS-Elements imaging software.
Cell invasion was conducted by using Transwell Chambers (Corning). The upper chamber was coated with 100 μL Matrigel (Invitrogen), and the lower chamber was filled with 500 μL DMEM supplemented with 10% FBS. MDA-MB-231 cells were plated at 3000 cells per chamber in the upper chamber containing 20 μM celecoxib. The transwell chambers were incubated at 37°C and 5% CO2 for 24 h. Cells on the upper surface of the insert were removed using a cotton swab, and cells that had migrated to the lower surface were stained with 2% crystal violet (Beyotime Biotechnology) for 30 min. Images of migrated cells were taken and the number of migrated cells was counted under a microscope in three randomly selected fields (magnification 100x).
Quantitative real-time PCR assay
To assess the expression levels of SNAIL, SLUG, TWIST, CYCLIN-D1, C-MYC and AXIN-2, total RNA was extracted from cultured cells or xenografted tumor tissues using the Trizol reagent (Life Technology) according to the manufacturer’s instructions. The quantity of RNA was determined by the spectrophotometer Nanodrop2000 (Thermal Fisher, USA). cDNA was prepared from 1 μg of total RNA using a Prim Script RT reagent kit (Takara, Japan) according to the manufacturer’s instructions. Primers for these genes were listed in Table 2. The relative changes in gene expression data were calculated by the 2ΔΔCT method, β-actin was used as an internal control.
Table 2: The primers used for quantitative real-time PCR
Primers |
Forward |
Reverse |
|---|---|---|
SNAIL |
5′-GCCTAGCGAGTGGTTCTTCTGC-3′ |
5′-TGGTCGTAGGGCTGCTGGAA-3′ |
SLUG |
5′-CCCTGGTTGCTTCAAGGACACA-3′ |
5′-GCTACACAGCAGCCAGATTCCT-3′ |
TWIST |
5′-GGA GTCCGCAGTCTTACGAG-3′ |
5′-TCTGGAGGACCTGGTAGAGG-3′ |
CYCLIN-D1 |
5′-CGATGCCAACCTCCTCAACGA-3′ |
5′-TCCTCCTCGCACTTCTGTTCCT-3′ |
C-MYC |
5′-CCCGCTTCTCTGAAAGGCTCTC-3′ |
5′-TCTGCTGCTGCTGCTGGTAGA-3′ |
AXIN-2 |
5′-GACCAAGCAGACGACGAAGCAT-3′ |
5′-CGTGCCTTTCCCATTGCGTTTG-3′ |
β-ACTIN |
5′-CTGGAACGGTGAAGGTGACA-3′ |
5′-AAGGGACTTCCTGTAACAATGCA-3′ |
Protein isolation and western blot assay
Cells or xenografted tumor tissues were lysed in lysis buffer (60 mM Tris-HCl, pH 6.8, 5% glycerol, 2% SDS and 1 mM PMSF). Protein concentrations were determined by BCA Protein Assay Kit (Beyotime Biotechnology). 30 μg of total proteins were separated by SDS-PAGE gels and transferred onto PVDF membranes. For immunoblotting, membranes were incubated with the following primary monoclonal antibodies: anti-β-catenin, anti-E-cadherin, anti-Vimentin, anti-MMP-2, anti-p-GSK-3β, anti-SOX-2, anti-c-MYC, anti-Axin-2, anti-Cyclin-D1 and anti-Survivin. Antibodies against Tubulin or GAPDH were used as internal controls. After washing three times in TBST, membranes were incubated with the corresponding HRP-conjugated secondary antibodies and visualized by the Gene Gnome imaging system. Relative protein expression quantities were analyzed by Quantity One software.
PGE2 synthesis assay
The amounts of synthesized PGE2 in cell culture supernatants and serum of the assayed animals were measured by a commercial PGE2 detection kit (Cayman Chemicals) according to the manufacturer’s instructions.
Immunohistochemistry assay and H&E staining
Formalin-fixed, and paraffin-embedded tumor, lung and liver tissues were sliced into 4 μm thick sections for immunohistochemistry assay or H&E staining. Endogenous peroxidase was inhibited by incubating sections with 3% H2O2 for 15 min and non-specific binding was blocked with 10% goat serum for 30 min at room temperature. Sections were incubated with specific primary antibodies at 4°C overnight, washed with PBST, and incubated with corresponding secondary antibodies for 1 h at room temperature. The sections were stained with diaminobenzidine (DAB) before observation under a microscope. The immunohistochemistry (IHC) intensity was scored by standard methods as described in previously published study [62].
Immunofluorescence staining assay
MDA-MB-231 cells were grown in 24-well plates, and grown to 70% confluence prior to the treatment with 20 μM celecoxib or DMSO for 24 h. Cells were then fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100 and blocked with 10% goat serum for 1 h at room temperature. The cells were then incubated with the primary antibodies (mouse monoclonal antibody) anti-E-cadherin or anti-vimentin for overnight at 4°C. Alexa Fluor 488 or 647 conjugated goat anti-mouse were used as the secondary antibody. Finally the cells were washed 3 times in PBS and incubated with DAPI as the nuclear counterstain. Images were acquired at 100×magnification using a fluorescence microscope (Nikon, TE-2000U).
Aldefluor assay
The Aldefluor assay was carried out according to the manufacturer’s instructions (Stem Cell Technologies, Canada). Briefly, MDA-MB-231 and MCF-7 cells were grown to 70% confluence prior to the treatment with 20 μM celecoxib or DMSO for 24 h. Single cells were suspended in Aldefluor assay buffer containing ALDH substrate and incubated for 45 minutes at 37°C. For negative control, half of the cell suspensions from each sample were incubated under the same conditions in the presence of DEAB (diethylaminobenzaldehyde). Flow cytometry was carried out to measure the ALDH-positive cell population. The data was analyzed by Flow Jo software.
Dual-luciferase reporter assay
The MDA-MB-231 cells were grown to 80% confluence and transiently transfected with 1 μg of either TOP flash or FOP flash luciferase reporter vector and 20 ng of Renilla as internal control (Millipore Corporation) using 2 μL Lipo6000TM reagent (Beyotime Biotechnology) in each well of a 12-well plate. Five hours after transfection, the medium was replaced with completed cell culture medium containing 20 μM celecoxib or DMSO. Twenty-four hours later, cells were lysed in lysis buffer (Dual-luciferase Reporter Assay System, Promega, USA), and 20 μL of each sample was monitored for luciferase activity using a luminometer.
Animal study
All animal experiments were performed according to protocols approved by the Committee of Care and Use of Laboratory Animals of Wenzhou Medical University, China. MDA-MB-231 cells (5 x 106 per mouse) were transplanted into the cleared mammary fat pad of 8-week old NOD/SCID female mice (Shanghai Laboratory Animal Co., Shanghai, China). When tumors reached a palpable size, a total of 12 mice were randomly assigned to either control group or treatment group, six mice per group. Celecoxib dissolved in PBS (30 mg/kg/day) was administered by oral gavage every day for 30 days to the treatment group, and the same volume of PBS was given to the control group at the same time. In addition, three NOD/SCID female mice were used as blank control, which were neither transplanted with MDA-MB-231 cells nor administered with celecoxib. Tumor volumes were measured once every three days using a caliper, and calculated based on the following formula: volume = length×(width)2/2. All mice were euthanized at 30 days after the first drug treatment and the tumor masses were excised.
To investigate the effect of celecoxib on tumor metastasis, MDA-MB-231 cells (2 x 106 per mouse) were injected into the tail vein of 12 NOD/SCID female mice. After injection, the mice were randomly assigned to the control or treatment group, six mice per group. Celecoxib was administered as described above. The mice were sacrificed at 20 days after first drug treatment and the lungs and livers were collected. Metastatic lesions on the surface of lungs and livers were confirmed by H&E staining and the number of lesions was counted.
Statistical analysis
All data were presented as mean ± SD from three sets of independent experiments and analyzed using statistical software GraphPad Prism. Statistical differences between groups were determined by unpaired Student’s t-test or One-way Analysis of Variance (ANOVA); P < 0.05 was considered as statistical significance.
Abbreviations
CSCs: cancer stem cells; EMT: epithelial to mesenchymal transition; 5-FU: 5-fluorouracil; PGE2: prostaglandin E2; DMSO: dimethylsulfoxide; TSFE: tumorsphere formation efficiency.
Author contributions
C.L.H. conceived and wrote the manuscript, C.L.H., Y.C., H.L., J.Y., X.S., J.Z. and N.H. conducted the experiments, C.J.Z. and Y.W. provided reagents and advices on Wnt signaling experiments. C.J.H. and Q.D. supervised the project and edited the paper. All authors reviewed the manuscript.
CONFLICTS OF INTEREST
The authors declared no conflicts of interest.
FINANCIAL SUPPORT
This work was supported by funding from the National Natural Science Foundation of China (No. 81373031, No.81572730). C.J.Z. is supported by NIH grants (1R01DE021696& 1R01DE026737).
REFERENCES
1. Howell A, Anderson AS, Clarke RB, Duffy SW, Evans DG, Garcia-Closas M, Gescher AJ, Key TJ, Saxton JM, Harvie MN. Risk determination and prevention of breast cancer. Breast Cancer Res. 2014.
2. Takebe N, Harris PJ, Warren RQ, Ivy SP. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol. 2011; 8:97-106.
3. Ferlay J, Héry C, Autier P, Sankaranarayanan R. Global Burden of Breast Cancer. In: Li C, ed. Breast Cancer Epidemiology. 2010; 1-19.
4. Coates AS, Winer EP, Goldhirsch A, Gelber RD, Gnant M, Piccart-Gebhart M, Thürlimann B, Senn HJ, André F, Baselga J, Bergh J, Bonnefoi H, Burstein H, et al. Tailoring therapies—improving the management of early breast cancer: St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2015. Ann Oncol. 2015; 26:1533-1546.
5. Marshall DC, Webb TE, Hall RA, Salciccioli JD, Ali R, Maruthappu M. Trends in UK regional cancer mortality 1991–2007. Br J Cancer. 2016; 114:340-347.
6. Gunsoy NB, Garcia-Closas M, Moss SM. Estimating breast cancer mortality reduction and overdiagnosis due to screening for different strategies in the United Kingdom. Br J Cancer. 2014; 110:2412-2419.
7. Pece S, Tosoni D, Confalonieri S, Mazzarol G, Vecchi M, Ronzoni S, Bernard L, Viale G, Pelicci PG, Di Fiore PP. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. 2010; 140:62-73.
8. Stingl J, Caldas C. Molecular heterogeneity of breast carcinomas and the cancer stem cell hypothesis. Nat Rev Cancer. 2007; 7:791-799.
9. Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell. 2012; 10:717-728.
10. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003; 100:3983-3988.
11. Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, Wicha M, Clarke MF, Simeone DM. Identification of pancreatic cancer stem cells. Cancer Res. 2007; 67:1030-1037.
12. Chai S, Xu X, Wang Y, Zhou Y, Zhang C, Yang Y, Yang Y, Xu H, Xu R, Wang K. Ca2+/calmodulin-dependent protein kinase IIγ enhances stem-like traits and tumorigenicity of lung cancer cells. Oncotarget. 2015; 6:16069-16083. https://doi.org/10.18632/oncotarget.3866.
13. Zhang X, Hua R, Wang X, Huang M, Gan L, Wu Z, Zhang J, Wang H, Cheng Y, Li J, Guo W. Identification of stem-like cells and clinical significance of candidate stem cell markers in gastric cancer. Oncotarget. 2016; 7:9815-9831. https://doi.org/10.18632/oncotarget.6890.
14. Wang J, Lei Z, Guo Y, Wang T, Qin Z, Xiao H, Fan L, Chen D, Bian X, Liu J, Wang B. miRNA-regulated delivery of lincRNA-p21 suppresses β-catenin signaling and tumorigenicity of colorectal cancer stem cells. Oncotarget. 2015; 6:37852-37870. https://doi.org/10.18632/oncotarget.5635.
15. Valenta T, Degirmenci B, Moor AE, Herr P, Zimmerli D, Moor MB, Hausmann G, Cantu C, Aguet M, Basler K. Wnt ligands secreted by subepithelial mesenchymal cells are essential for the survival of intestinal stem cells and gut homeostasis. Cell Rep. 2016; 15:911-918.
16. MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev cell. 2009; 17:9-26.
17. Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006; 127:469-480.
18. Yang K, Wang X, Zhang H, Wang Z, Nan G, Li Y, Zhang F, Mohammed MK, Haydon RC, Luu HH, Bi Y, He TC. The evolving roles of canonical WNT signaling in stem cells and tumorigenesis: implications in targeted cancer therapies. Lab Invest. 2016; 96:116-136.
19. Cuzick J, Otto F, Baron JA, Brown PH, Burn J, Greenwald P, Jankowski J, La Vecchia C, Meyskens F, Senn HJ, Thun M. Aspirin and non-steroidal anti-inflammatory drugs for cancer prevention: an international consensus statement. Lancet Oncol. 2009; 10:501-507.
20. Rothwell PM, Wilson M, Elwin CE, Norrving B, Algra A, Warlow CP, Meade TW. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet. 2010; 376:1741-1750.
21. Rothwell PM, Fowkes FG, Belch JF, Ogawa H, Warlow CP, Meade TW. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet. 2011; 377:31-41.
22. Algra AM, Rothwell PM. Effects of regular aspirin on long-term cancer incidence and metastasis: a systematic comparison of evidence from observational studies versus randomised trials. Lancet Oncol. 2012; 13:518-527.
23. Fraser DM, Sullivan FM, Thompson AM, McCowan C. Aspirin use and survival after the diagnosis of breast cancer: a population-based cohort study. Br J Cancer. 2014; 111:623-627.
24. Harris RE, Chlebowski RT, Jackson RD, Frid DJ, Ascenseo JL, Anderson G, Loar A, Rodabough RJ, White E, McTiernan A. Breast cancer and nonsteroidal anti-inflammatory drugs: prospective results from the Women’s Health Initiative. Cancer Res. 2003; 63:6096-6101.
25. Goessling W, North TE, Loewer S, Lord AM, Lee S, Stoick-Cooper CL, Weidinger G, Puder M, Daley GQ, Moon RT, Zon LI. Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell. 2009; 136:1136-1147.
26. Goessling W, Allen RS, Guan X, Jin P, Uchida N, Dovey M, Harris JM, Metzger ME, Bonifacino AC, Stroncek D, Stegner J, Armant M, Schlaeger T, et al. Prostaglandin E2 enhances human cord blood stem cell xenotransplants and shows long-term safety in preclinical nonhuman primate transplant models. Cell Stem Cell. 2011; 8:445-458.
27. North TE, Goessling W, Walkley CR, Lengerke C, Kopani KR, Lord AM, Weber GJ, Bowman TV, Jang IH, Grosser T, Fitzgerald GA, Daley GQ, Orkin SH, Zon LI. Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis. Nature. 2007; 447:1007-1011.
28. Li L, Kim HT, Nellore A, Patsoukis N, Petkova V, McDonough S, Politikos I, Nikiforow S, Soiffer R, Antin JH, Ballen K, Cutler C, Ritz J, Boussiotis VA. Prostaglandin E2 promotes survival of naive UCB T cells via the Wnt/beta-catenin pathway and alters immune reconstitution after UCBT. Blood Cancer J. 2014; 4:e178.
29. Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science. 2005; 310:1504-1510.
30. Wang D, Fu L, Sun H, Guo L, DuBois RN. Prostaglandin E2 promotes colorectal cancer stem cell expansion and metastasis in mice. Gastroenterology. 2015; 149:1884-1895.e1884.
31. Sheridan C, Kishimoto H, Fuchs RK, Mehrotra S, Bhat-Nakshatri P, Turner CH, Goulet R Jr, Badve S, Nakshatri H. CD44+/CD24- breast cancer cells exhibit enhanced invasive properties: an early step necessary for metastasis. Breast Cancer Res. 2006; 8:R59.
32. Chen YC, Ingram PN, Fouladdel S, McDermott SP, Azizi E, Wicha MS, Yoon E. High-throughput single-cell derived sphere formation for cancer stem-like cell identification and analysis. Sci Rep. 2016; 6:27301.
33. Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, Wicha MS. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003; 17:1253-1270.
34. Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005; 5:275-284.
35. Liu S, Wicha MS. Targeting breast cancer stem cells. J Clin Oncol. 2010; 28:4006-4012.
36. Leis O, Eguiara A, Lopez-Arribillaga E, Alberdi MJ, Hernandez-Garcia S, Elorriaga K, Pandiella A, Rezola R, Martin AG. Sox2 expression in breast tumours and activation in breast cancer stem cells. Oncogene. 2012; 31:1354-1365.
37. Tam WL, Ng HH. Sox2: masterminding the root of cancer. Cancer Cell. 2014; 26:3-5.
38. Boumahdi S, Driessens G, Lapouge G, Rorive S, Nassar D, Le Mercier M, Delatte B, Caauwe A, Lenglez S, Nkusi E, Brohee S, Salmon I, Dubois C, et al. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature. 2014; 511:246-250.
39. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu SL, Schott A, Hayes D, Birnbaum D, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007; 1:555-567.
40. Nieto MA, Huang RY, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016; 166:21-45.
41. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008; 133:704-715.
42. Morel AP, Lievre M, Thomas C, Hinkal G, Ansieau S, Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One. 2008; 3:e2888.
43. Luo M, Brooks M, Wicha MS. Epithelial-mesenchymal plasticity of breast cancer stem cells: implications for metastasis and therapeutic resistance. Curr Pharm Des. 2015; 21:1301-1310.
44. Huang RY, Kuay KT, Tan TZ, Asad M, Tang HM, Ng AH, Ye J, Chung VY, Thiery JP. Functional relevance of a six mesenchymal gene signature in epithelial-mesenchymal transition (EMT) reversal by the triple angiokinase inhibitor, nintedanib (BIBF1120). Oncotarget. 2015; 6:22098-22113. https://doi.org/10.18632/oncotarget.4300.
45. Velasco-Velazquez MA, Popov VM, Lisanti MP, Pestell RG. The role of breast cancer stem cells in metastasis and therapeutic implications. Am J Pathol. 2011; 179:2-11.
46. Balic M, Lin H, Young L, Hawes D, Giuliano A, McNamara G, Datar RH, Cote RJ. Most early disseminated cancer cells detected in bone marrow of breast cancer patients have a putative breast cancer stem cell phenotype. Clin Cancer Res. 2006; 12:5615-5621.
47. Dey N, Barwick BG, Moreno CS, Ordanic-Kodani M, Chen Z, Oprea-Ilies G, Tang W, Catzavelos C, Kerstann KF, Sledge GW Jr, Abramovitz M, Bouzyk M, De P, Leyland-Jones BR. Wnt signaling in triple negative breast cancer is associated with metastasis. BMC Cancer. 2013; 13:537.
48. Bilir B, Kucuk O, Moreno CS. Wnt signaling blockage inhibits cell proliferation and migration, and induces apoptosis in triple-negative breast cancer cells. J Transl Med. 2013; 11:280.
49. Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol. 2002; 22:1172-1183.
50. Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999; 398:422-426.
51. He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science. 1998; 281:1509-1512.
52. Huang Z, Wu T, Liu AY, Ouyang G. Differentiation and transdifferentiation potentials of cancer stem cells. Oncotarget. 2015; 6:39550-39563. https://doi.org/10.18632/oncotarget.6098.
53. Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, Lander ES. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009; 138:645-659.
54. Lu D, Choi MY, Yu J, Castro JE, Kipps TJ, Carson DA. Salinomycin inhibits Wnt signaling and selectively induces apoptosis in chronic lymphocytic leukemia cells. Proc Natl Acad Sci U S A. 2011; 108:13253-13257.
55. Kakarala M, Brenner DE, Korkaya H, Cheng C, Tazi K, Ginestier C, Liu S, Dontu G, Wicha MS. Targeting breast stem cells with the cancer preventive compounds curcumin and piperine. Breast Cancer Res Treat. 2010; 122:777-785.
56. Wang YC, Chao TK, Chang CC, Yo YT, Yu MH, Lai HC. Drug screening identifies niclosamide as an inhibitor of breast cancer stem-like cells. PLoS One. 2013; 8:e74538.
57. Chin YT, Yang SH, Chang TC, Changou CA, Lai HY, Fu E, HuangFu WC, Davis PJ, Lin HY, Liu LF. Mechanisms of dihydrotestosterone action on resveratrol-induced anti-proliferation in breast cancer cells with different ERα status. Oncotarget. 2015; 6:35866-35879. https://doi.org/10.18632/oncotarget.5482.
58. Charafe-Jauffret E, Monville F, Ginestier C, Dontu G, Birnbaum D, Wicha MS. Cancer stem cells in breast: current opinion and future challenges. Pathobiology. 2008; 75:75-84.
59. Franqui-Machin R, Wendlandt EB, Janz S, Zhan F, Tricot G. Cancer stem cells are the cause of drug resistance in multiple myeloma: fact or fiction? Oncotarget. 2015; 6:40496-40506. https://doi.org/10.18632/oncotarget.5800.
60. Kurtova AV, Xiao J, Mo Q, Pazhanisamy S, Krasnow R, Lerner SP, Chen F, Roh TT, Lay E, Ho PL, Chan KS. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature. 2015; 517:209-213.
61. Li HJ, Reinhardt F, Herschman HR, Weinberg RA. Cancer-stimulated mesenchymal stem cells create a carcinoma stem cell niche via prostaglandin E2 signaling. Cancer Discov. 2012; 2:840-855.
62. Cai J, Feng D, Hu L, Chen HY, Yang GZ, Cai QP, Gao CF, Wei D. FAT4 functions as a tumour suppressor in gastric cancer by modulating Wnt/beta-catenin signalling. Br J Cancer. 2015; 113:1720-1729.