
INTRODUCTION
Epithelial ion transports are critical physiological processes in the human gastrointestinal (GI) tract. Intestinal epithelium either absorbs electrolytes or secretes ions (such as Cl- and HCO3-), which provides the driving force for water absorption or secretion to maintain the liquid homeostasis in the human body. Epithelial ion transports are under control of several neuro-humoral factors, including ACh, 5-HT, PGs, nitric oxide (NO), and capsaicin-sensitive afferent neurons [1]. These neuro-humoral factors mediate epithelial ion transports through three major cellular signaling: Ca2+-, cAMP- and cGMP-dependent pathways. Among them, ACh is one of the major excitatory neurotransmitter in GI system. The chemical analogue of ACh, carbachol (CCh), a muscarinic receptor agonist, is a commonly used Ca2+ mobilizer. Currently, the physiological roles and molecular mechanisms of cAMP- and cGMP-dependent regulation of epithelial Cl- and HCO3- secretion are relatively well defined; however, those mediated by Ca2+ signaling remain poorly understood in small intestinal epithelia [2].
Moreover, although it is known that Ca2+ signaling is critical for intestinal epithelial ion secretion [2], the underlying detailed mechanisms that control cytosolic Ca2+ concentration ([Ca2+]cyt) homeostasis in small intestinal epithelium are not fully understood [3]. It is generally thought that agonists induce Ca2+ signaling via two major processes in non-excitable cells: the IP3-induced release of Ca2+ from intracellular stores, and then an enhanced Ca2+ entry from the extracellular medium [4]. Classically, the Ca2+ entry in non-excitable epithelial cells was thought to occur mainly via so-called capacitative or store-operated Ca2+ channels (SOCs), which activation is entirely dependent on the depletion of intracellular Ca2+ stores [5, 6]. These channels are the Ca2+-release activated Ca2+channels (CRAC) first described in mast cells and Jurkat lymphocytes [7, 8], which are accomplished by the pore forming Ca2+ channel Orai [9, 10]. However, only a few studies have focused on the role of CRAC/Orai channels in polarized epithelial cells, such as intestinal epithelial cell line IEC-6 cells [11] and colonic epithelial cells [12, 13]. So far, the regulatory mechanisms of [Ca2+]cyt homeostasis in native epithelial cells of the small intestine are still unclear.
The major focus of Cl- and of HCO3- secretion in the small intestine is on cAMP- and cGMP-dependent regulatory pathways, which mediates several membrane ion channels to contribute to epithelial ion transports. The cystic fibrosis transmembrane conductance regulator (CFTR) is one of the critical channels in the luminal membrane of enterocytes [14]. CFTR is a cAMP/PKA and cGMP/PKG -dependent channel abundantly expressed in several functionally diverse tissues, such as the pancreas, intestine, kidney, sweat duct, and lung [15, 16]. In intestinal epithelial cells (IEC), it regulates Na+, Cl-, and HCO3- transports [17]. Although CFTR is principally activated by cAMP/PKA and cGMP/PKG pathways, it is also regulated by Ca2+ signaling [18]. However, it is currently unclear for the detailed regulatory mechanisms of CFTR by Ca2+ signaling, particularly it is unknown if Ca2+ signaling per se is able to activate CFTR or is through potentiating cAMP/PKA-mediated CFTR activation.
The duodenal mucosa, due to its strategic location between the stomach and other segments of the small intestine, senses luminal nutrients and regulates duodenal epithelial ion transports, particularly Cl- and HCO3- secretion, which in turn is important for nutrient absorption and mucosal protection from gastric acid [19]. Therefore, in the present study, we sought to investigate Ca2+signaling mediated duodenal epithelial ion secretion and the underlying molecular mechanisms. We found that CCh stimulates extracellular Ca2+ entry likely through SOC/Orai channels mainly from the basolateral membrane of IEC. The increased [Ca2+]cyt per se is enough to trigger duodenal transepithelial secretion through a novel PI3K/AKT/CFTR pathway. This study not only reveals that Ca2+ signaling is critical to activate CFTR-mediated epithelial ion transportsin the small intestine, but also provides novel insights into the detailed mechanisms of Ca2+-dependent transepithelial Cl- and HCO3- secretion.
RESULTS
Activation of muscarinic receptors induced intracellular Ca2+ release and extracellular Ca2+ entry
We applied CCh, a muscarinic receptor agonist, to mobilize intracellular Ca2+. CCh (100 μM) markedly increased duodenal short-circuit current (Isc)(Figure 2A), which was attenuated by atropine(10 μM), a muscarinic receptor antagonist (Supplementary Figure 1A). Activation of muscarinic receptors stimulates the production of inositol 1,4,5-trisphoshate (IP3), leading to intracellular Ca2+ release via IP3 receptors on the endoplasmic reticulum (ER) membrane [20]. We used LiCl(30mM) to inhibit IP3 production and confirmed the involvement of IP3 pathway in the process of muscarinic receptors-mediated intracellular Ca2+ release (Supplementary Figure 1B).
To further test if intracellular Ca2+ release mediates extracellular Ca2+ entry into IEC, CCh-stimulated duodenal Isc was compared in the presence or the absence of extracellular Ca2+ in each side of the Ussing chamber experiments. As shown in Figure 1A and 1B, CCh-stimulated duodenal Isc was significantly attenuated when extracellular Ca2+ was omitted from the serosal side of the duodenal tissues, but not from the mucosal side. Moreover, CCh-stimulated duodenal Isc was not significantly different between Ca2+ omissions from the serosal side only and from both sides of the tissues (Figure 1B). Therefore, CCh may induce intracellular Ca2+ release that further mediates extracellular Ca2+ entry from the serosal side of the duodenal epithelium.
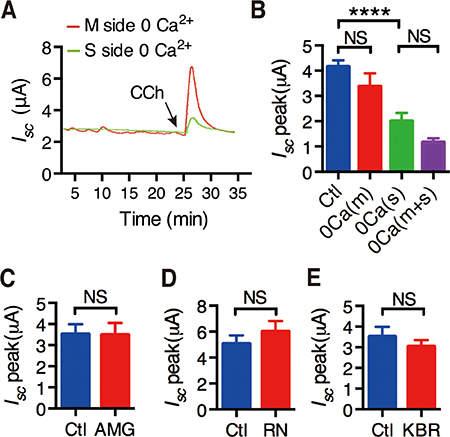
Figure 1: Activation of muscarinic receptors induced extracellular Ca2+ entry through possible pathways. (A) Representative of the time course of CCh-stimulated murine duodenal mucosal Isc after extracellular Ca2+ was omitted from the serosal side or the mucosal side of the duodenal tissues. (B) The summary data of CCh-stimulated duodenal Isc peak after Ca2+ omission from the mucosal side, the serosal side or both sides of duodenal tissues. (C–E) Effects of AMG-517 (AMG, 100 M) (C), RN-1734 (RN, 3 M) (D) and KBR-7943(KBR, 3 M) (E) on CCh-induced duodenal Isc peak after serosal addition. Results are presented as mean SE (n = 6–9 in each series). NS, no significant differences, ****P < 0.0001 vs. control by Student’s t-test.
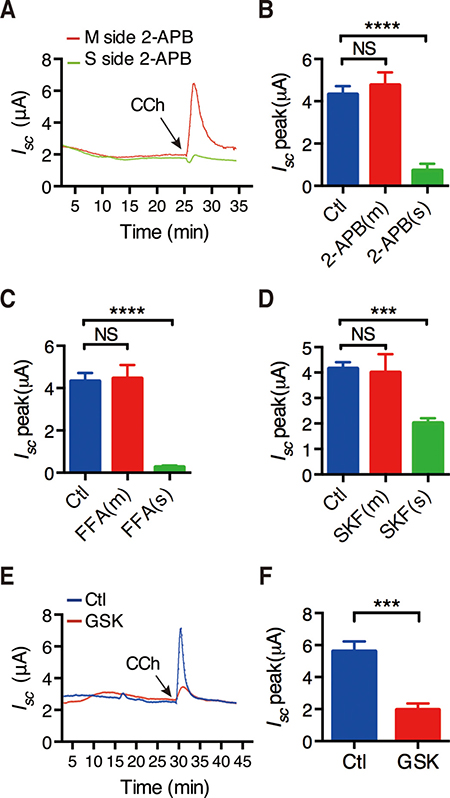
Figure 2: CRAC/Orai channels in the regulation of CCh-stimulated duodenal Isc. (A) Representative of the time course of CCh -stimulated murine duodenal Isc with or without addition of 2-aminoethoxydiphenyl borate (2-APB, 50 M) to the mucosal or serosal side of duodenal tissues. (B) The summary of the effect of 2-APB on CCh-stimulated murine duodenal Isc peak after mucosal or serosal addition. (C–D) Summery effects of the flufenamic acid (FFA, 100 M) (C) or SKF-96365 (SKF, 3 M) (D) on CCh-stimulated duodenal Isc peak after mucosal or serosal addition. (E) Representative of the time course of CCh-stimulated duodenal Isc with or without serosal addition of GSK-7975A (GSK, 100 M). (F) The summary of the effect of GSK-7975A on CCh-stimulated duodenal Isc peak after serosal addition. Results are presented as mean SE (n = 6–9 in each series). NS, no significant differences, ***P < 0.001, ****P < 0.0001 vs. control.
CCh induced SOCE mechanism on the serosal side of the duodenal epithelium
It is well known that intracellular Ca2+ release to deplete the Ca2+ store in the ER would promote extracellular Ca2+ entry, which is the so-called store-operated Ca2+ entry (SOCE). Because it is still uncertain for the functional expression of the voltage-operated Ca2+ channels in small intestinal epithelia, it is believed that SOCE fulfill this function [21]. To test if this mechanism occurs in duodenal epithelium, we used three selective SOCE blockers with different chemical structures. As shown in Figure 2A, addition of 2-Aminoethoxydiphenyl borate (2-APB) (50 μM) to the mucosal side of the duodenal tissues did not affect the time course of CCh-stimulated duodenal Isc, but addition to serosal side significantly suppressed the duodenal Isc. Figure 2B summarizes the effect of 2-APB on duodenal Isc peak after it was added to each side of the duodenal tissues. Similar to 2-APB, both Flufenamic acid (FFA)(100 μM) and SKF-96365 (30 μM) significantly suppressed duodenal Isc peak from serosal side of the duodenal tissues, but not from the mucosal side (Figure 2C and 2D). Therefore, SOCE mechanism occurs exclusively on the serosal side of the duodenal epithelium, which is consistent with the previous findings described above.
CRAC/Orai channels in the regulation of CCh-stimulated duodenal Isc
We screened the molecular candidates of SOCE in duodenal epithelium. We first examined common Ca2+-permeable channels, such as TRPV1, TRPV4 and NCX using their selective blockers AMG-517 (100 μM), RN-1734 (30 μM) and KBR-7943 (30 μM), respectively. As shown in Figure 1C, 1D and 1E, addition of them to the serosal side did not alter CCh-induced duodenal Isc, indicating no functional expression of TRPV1, TRPV4 and NCX to exclude them as the molecular candidates of SOCE in duodenal epithelium.
We further identified the molecular candidates of SOCE using GSK-7975A, a specific CRAC/Orai channel blocker. As shown in Figure 2E, addition of GSK-7975A(100 μM) to serosal side of the duodenal tissues significantly suppressed the time course of CCh-stimulated duodenal Isc. Figure 2F summarizes the inhibitory effect of GSK-7975A on duodenal Isc peak after it was added to serosal side. Therefore, CRAC/Orai channels may represent the molecular candidate of SOCE in duodenal epithelium.
CRAC/Orai channels in CCh-mediated Ca2+ entry into IEC
Since HT-29 cell line is commonly used as a cell model of IEC to study ion absorption and secretion [22–24], and they express the muscarinic receptor [12, 25, 26], we used them to measure [Ca2+]cyt by digital Ca2+ imaging. CCh(100 μM) immediately induced Ca2+ signaling in HT-29 cells (Figure 3A). 2-APB (50 μM) and GSK-7975(30 μM) almost abolished CCh-induced Ca2+ signaling (Figure 3B and 3C). Both peak and the rising slope of CCh-induced [Ca2+]cyt signaling were significantly decreased by 2-APB and GSK-7975 (Figure 3D and 3E).
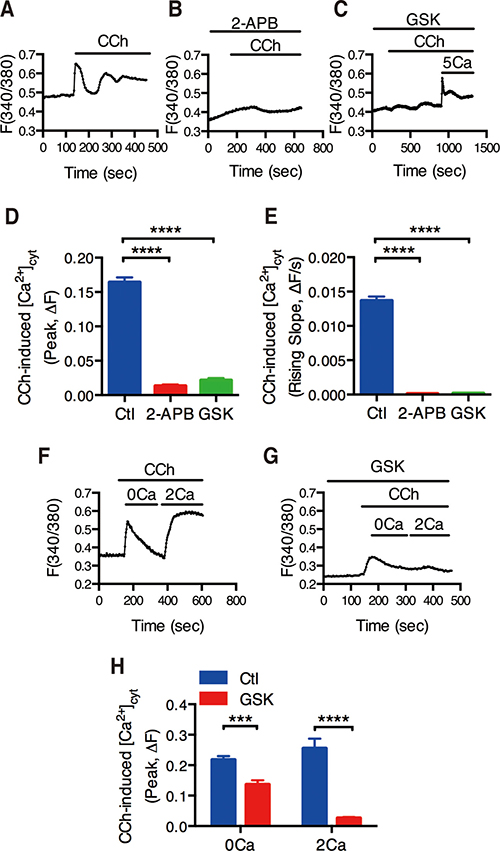
Figure 3: Functional identification of CRAC/Orai channels in CCh-mediated Ca2+ mobilization of intestinal epithelial cells. (A) Time courses showing the effect of CCh (10 M) on basal [Ca2+]cyt of human colon carcinoma HT-29 cells in normal physiological salt solutions. (B–C) Time courses showing the effect of 2-APB (5 M, B) or GSK-7975A (GSK, 3 M, C) on CCh-mediated [Ca2+]cyt mobilization in HT-29 cells. (D–E) The summary of the effect of 2-APB or GSK on the peaks (D) and the rising slopes (F) of CCh-induced [Ca2+]cyt signaling in HT-29 cells. (F) Time courses showing CCh (10 M) first caused a rapid increase in [Ca2+]cyt when HT-29 cells was superfused with Ca2+-free solution (0 Ca) (left). After Ca2+ release from the ER was complete, restoration of extracellular Ca2+ (2 mM Ca) caused an additional increase in [Ca2+]cyt in HT-29 cells (right). (G) Time courses showing the effect of GSK-7975A (GSK, 3 M) on CCh-induced [Ca2+]cyt mobilization in HT-29 cells superfused with Ca2+-free solution (0 Ca) or Ca2+-containing solution (2 Ca). (H) Summary data on the effect of GSK-7975A on CCh-induced [Ca2+]cyt mobilization in HT-29 cells superfused with Ca2+-free solution (0 Ca) or Ca2+-containing solution (2 Ca). Results are presented as mean SE (n = 20-30 cells). ***P < 0.001, ****P < 0.0001vs. control or DMSO.
We further examined the role of CRAC/Orai channels in SOCE mechanism. In HT-29 cells superfused with Ca2+-free solution (0Ca) first caused a rapid increase in [Ca2+]cyt due to intracellular Ca2+ release from the ER to the cytosol (Figure 3F, left). After Ca2+ release from the ER was complete, restoration of extracellular Ca2+ (2Ca) caused an additional increase in [Ca2+]cyt due to SOCE mechanism (Figure 3F, right). As shown in Figure 3G and H, GSK-7975A (30 μM) significantly inhibited CCh-induced SOCE mechanism. Therefore, these data further support our previous notion that CRAC/Orai channels are the molecular candidate of SOCE in IEC.
Role of CFTR channels in CCh-induced epithelial ion secretion
First, to test if CCh-induced Ca2+ signaling could stimulate duodenal Isc through Ca2+-activated Cl- channels (CaCC), we applied commonly used CaCC blocker niflumic acid (NFA). At the concentrations of 100-300 μM, it did not affect CCh-induced duodenal Isc (Figure 4A). Furthermore, T16Ainh-A01(300 μM), a selective potent CaCC blocker did not affect CCh-induced duodenal Isceither (Figure 4B). These results exclude the role of CaCC in the CCh-induced duodenal epithelial ion secretion.
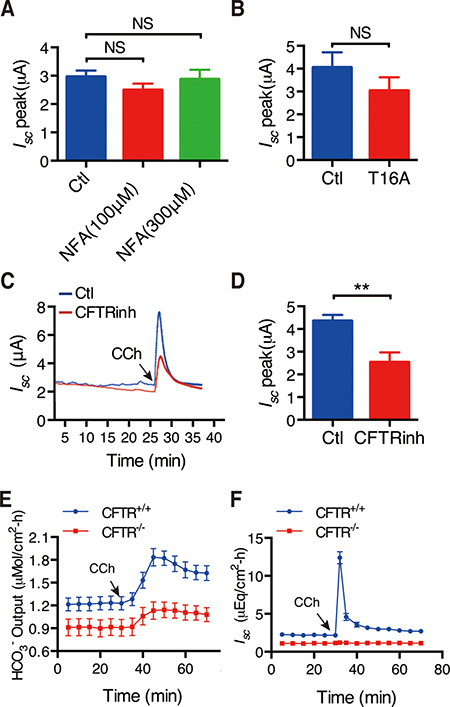
Figure 4: Role of CFTR channels in CCh-induced duodenal epithelial ion secretion. (A–B) Summary data on the effects of NFA (10 M, n = 15 and 3 M, n = 6) (A) and T16Ainh-A01 (T16A, 30 M, n = 11) (B) on CCh-stimulated duodenal Isc peaks after mucosal addition. (C) Representative of the time course of CCh-stimulated duodenal Isc with or without mucosal addition of CFTRinh-172 (CFTRinh, 3 M). (D) Summary on the effect of CFTRinh-172 (CFTRinh, 3 M) on CCh-stimulated duodenal Isc peak (n = 9). (E–F) Summary on the time courses of CCh-stimulated duodenal HCO3- secretion (E) and Isc (F) in CFTR+/+ or CFTR-/- mice (n = 6). Results are presented as mean SE. NS, no significant differences, **P < 0.01vs. control.
Second, we examined the role of CFTR channels in this process since they are critical in epithelial ion secretion stimulated by several secretagogues. As shown in Figure 4C and 4D, CFTRinh-172 (30 μM), a highly potent and specific CFTR inhibitor [27], markedly inhibited CCh-induced duodenal Isc. Further, we applied CCh to CFTR+/+ and CFTR-/- mice, and found that CCh markedly stimulated duodenal Isc and HCO3- secretion in CFTR+/+ mice (Figure 4E and 4F). However, CCh failed to induce duodenal Isc and decreased duodenal HCO3- secretion in CFTR-/- mice. The net peak of CCh-stimulated duodenal HCO3- secretion was reduced by 61% and the net peak of duodenal Isc was reduced by 99%, respectively in CFTR-/- mice (Figure 4E and 4F). We therefore underscored the critical role of CFTR channels in CCh-induced Ca2+-mediated duodenal ion secretion.
CCh-induced duodenal ion secretion was cAMP/PKA-independent
Since Ca2+ signaling is able to activate CFTR-mediated ion secretion through cAMP/PKA pathway in other epithelial tissues, we tested this notion in CCh-induced duodenal epithelial ion secretion. We first determined if there is a cross-talk between Ca2+ and cAMP signaling in the activation of CFTR channels. When low concentrations of cAMP-generating agonist forskolin(0.15 μM) and CCh (30 μM) were added together, a synergistic effect on duodenal Isc was observed (the green line and bar in Supplementary Figure 2A and 2B). However, this synergistic effect was not affected by the pretreatment of H89(20 μM), a commonly used PKA inhibitor (Supplementary Figure 2D–2F).
To exclude the role of cAMP/PKA pathway in CCh-induced duodenal ion secretion, we directly measured cAMP activity. As shown in Supplementary Figure 2C, CCh (100 μM) did not alter cAMP concentration in mouse duodenal epithelium, but forskolin (10 μM) markedly increased it. These results further confirm that [Ca2+]cyt-mediated duodenal ion secretion is cAMP/PKA-independent although a synergy exists between these two signaling pathways.
PI3K/Akt in CCh-induced duodenal ion secretion
Growing evidence suggest that CFTR channels can be activated by Ca2+-dependent PKA, PKC and tyrosine kinase in different epithelial tissues [28]. Here we examined if PI3K/Akt is involved in CCh-induced duodenal ion secretion. As shown in Figure 5A–5D, both selective PI3K inhibitors, wortmannin (0.1 μM) and LY294002 (20 μM), which have been shown to target PI3K activity at these concentrations [29, 30] significantly reduced CCh-stimulated mouse duodenal HCO3- secretion and duodenal Isc. Wortmannin reduced net peak of CCh-stimulated duodenal HCO3- secretion by 49% and duodenal Isc by 42%, respectively (Figure 5A and 5B). LY294002 reduced net peak of CCh-stimulated duodenal HCO3- secretion by 23% and duodenal Isc by 43%, respectively (Figure 5C and 5D).
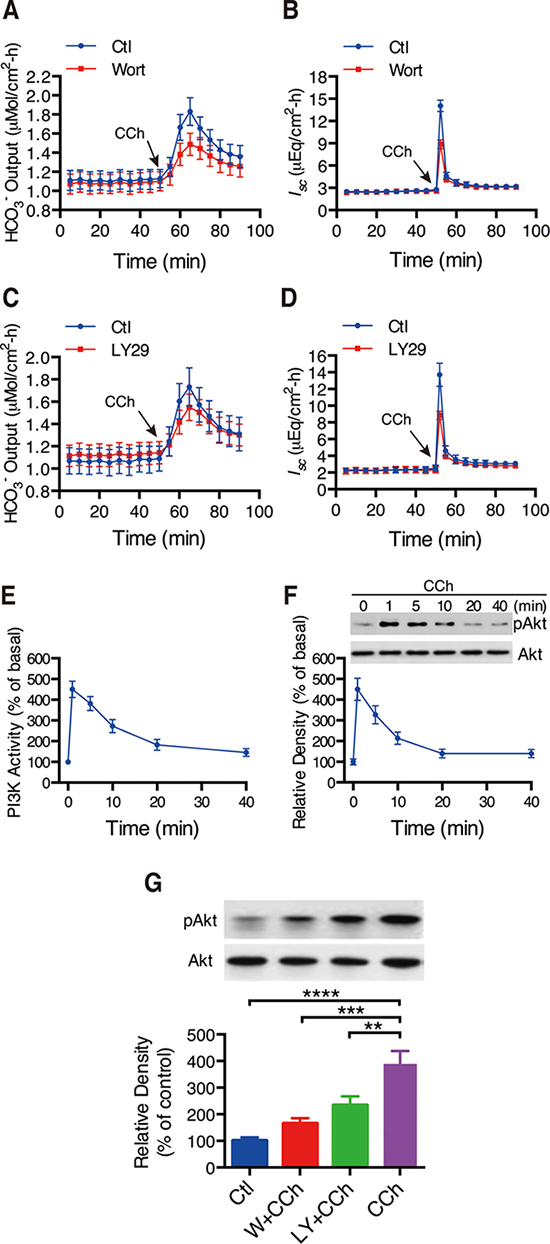
Figure 5: Involvements of PI3K/Akt in CCh-induced duodenal ion secretion. (A–B) Summary on the time courses of CCh-stimulated duodenal HCO3- secretion (A) and Isc (B) with or without wortmannin (Wort, 0.1 M) added to serosal side (n = 7). (C–D) Summary on the time courses of CCh-stimulated duodenal HCO3- secretion (A) and Isc (B) with or without LY294002 (LY29, 2 M) added to serosal side (n = 7). (E) Time course of CCh-stimulated duodenal mucosal epithelial PI3K activity. Murine duodenal mucosa was treated for various periods of time with CCh(10 M). Mucosal extract was immunoprecipitated with anti-PI3K P85 antibody in vitro (n = 4). (F) Time course of CCh-stimulated phosphorylation of Akt. Duodenal mucosae were incubated with CCh for the indicated times and were subjected to Western blot analysis. Top: blots showing Akt phosphorylation. Data are from a single experiment representative of 4 experiments. Bottom: Summary results are expressed as the percentage of basal values (n = 4). (G) Effects of wortmannin (W, 0.1 M), or LY294002 (LY, 2 M) on CCh-stimulated phosphorylation of Akt of murine duodenal mucosa. The summary results are expressed as the percentage of controls (n = 4). Results are presented as mean SE. **p < 0.01, ***P < 0.001, ****P < 0.0001 vs. control.
To confirm the role of PI3K in the regulation of CFTR function, PI3K activity in duodenal epithelium was measured. CCh (100 μM) rapidly stimulated PI3K activity and reached the peak within 1 min (Figure 5E). CCh induced the maximal PI3K activity by 4.5-fold compared with basal levels. Subsequently, we further examined whether CCh induces phosphorylation of Akt, a downstream effector of PI3K. Likewise, CCh caused a rapid phosphorylation of Akt. Notably, the time courses of CCh-stimulated PI3K activity and phosphorylation of Akt in duodenal epitheliumare matchable (Figure 5F). Again, both wortmannin and LY294002 significantly inhibited CCh-stimulated phosphorylation of Akt (Figure 5G). This direct evidence confirms the critical role of PI3K/Akt in CCh-induced Ca2+-mediated activity of CFTR channels.
PI3K/Akt in CCh-mediated phosphorylation and translocation of CFTR channels
The phosphorylation and translocation of CFTR channels to the surface of duodenal villus cells are important indicators of CFTR activity. To provide the confirmatory evidence for the role of PI3K in Ca2+ signaling mediated CFTR activation, we assessed CCh-stimulated translocation and phosphorylation of CFTR channels in duodenal epithelial cells.
We first examined the effect of CCh on translocation of CFTR to the surface of duodenal villus cells in mice. As shown in Figure 6A, CFTR proteins were mainly located in the cytoplasmic compartments of duodenal villus cells in control (a1 in Figure 6A). However, after administration of CCh (100 μM) for 5 min, CFTR proteins were prominent along the plasma membranes of villus cells (a2 in Figure 6A), indicating Ca2+ signaling-stimulated trafficking of CFTR proteins to the plasma membranes of villus cells. Wortmannin(0.1 μM) markedly inhibited CCh-induced CFTR redistribution from the cytoplasmic compartments to the plasma membranes (a3 in Figure 6A). After quantifying the ratio of CFTR fluorescence intensity in plasma membrane and cytoplasm, CCh increased the ratio by 4.4-fold compared with control, but wortmannin decreased CCh-increased ratio by 41% (Figure 6B). Therefore, CCh-induced Ca2+ signaling stimulates translocationof CFTR to the surface of duodenal villus cells, in which PI3K plays a critical role.
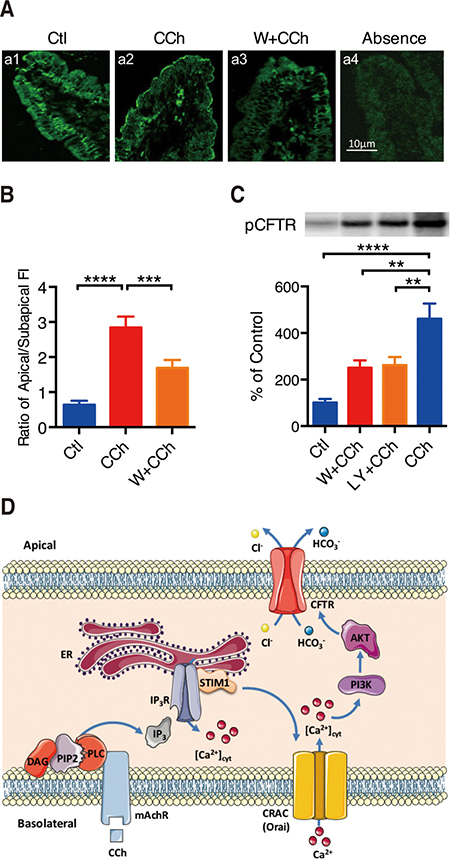
Figure 6: PI3K/Akt in CCh-mediated phosphorylation and translocation of CFTR channels. (A) Representative showing effect of wortmannin (W, 0.1 M) on CCh-induced CFTR trafficking to the plasma membranes of duodenal villus cells. a1: distribution of CFTR in unstimulated control vehicle-treated duodenal villus cells. a2: distribution of CFTR in duodenal villus cells at 5 min after stimulation using CCh (100 M). a3: effect of wortmannin (W, 0.1 M) on CCh-induced distribution of CFTR in duodenal villus cells. a4: control with CCh in the absence of primary anti-CFTR antibody. Magnification, 600; calibration bar, 10 m. (B) Summary data on the ratio of plasma membrane to cytoplasm CFTR fluorescence intensity (FI). Values are mean SE (n = 120 cells in 4 animals, 30 cells from each tissue). (C) Summary data on the effects of wortmannin (W, 0.1 M), LY294002 (LY, 2 M) on CCh-induced CFTR phosphorylation in duodenal mucosal epithelial cells. Top: autoradiographs showing 32P-phosphorylated CFTR. Data are from a single representative experiment of 4 experiments in each series. Bottom: The summary results are expressed as a percentage of controls. Values are mean SE (n = 4). (D) Schematic diagram depicting the proposed mechanisms of Ca2+-mediated duodenal epithelial HCO3- and Cl- secretion mediated by CCh.
We further examined the role of CCh-induced Ca2+ signaling in CFTR phosphorylation of mouse duodenal epithelia. The upper panel in Figure 6C illustrates a representative autoradiograph of CFTR phosphorylation after pretreatment with CCh and CCh plus either wortmannin (0.1 μM) or LY294002(20 μM). The lower panel in Figure 6C summarizes the intensity of CFTR phosphorylation. Compared with control, CCh increased CFTR phosphorylation by 4.6-fold. However, wortmannin and LY294002 inhibited CCh-induced CFTR phosphorylation by 46% and 43%, respectively. Therefore, these data not only confirm CCh-induced Ca2+ signaling stimulates CFTR phosphorylation, but also indicate the important role of PI3K in the activation of duodenal epithelial CFTR channels.
DISCUSSION AND CONCLUSIONS
Although it is known for the importance of Ca2+ signaling in ion transports in the salivary gland, pancreatic ducts and colonic epithelia, the detailed role of [Ca2+]cyt in the modulation of samll intestinal epithelial ion secretion and the underlying molecular mechanisms are not fully understood. In the present study, using native mouse duodenal epithelium with preserved polarity we reveal that: 1) activation of muscarinic receptors induces epithelial ion secretion via a pure Ca2+ signaling rather than a combination to cAMP/PKA pathway; 2) muscarinic receptor-induced Ca2+ signaling is via the SOCE mechanism on the serosal side of epithelial cells; 3) CRAC/Orai channels may serve as SOCE mechanism to mediate Ca2+-dependent ion secretion; 4) Ca2+ signaling regulates ion secretion via a novel PI3K/Akt-mediated activity of CFTR channels. Therefore, our results not only indicate that Ca2+ signaling per se plays a critical role in the regulation of small intestinal ion transports, but also provide a novel insight into the molecular mechanisms of Ca2+-mediated epithelial ion secretion.
[Ca2+]cyt acts as an universal second messenger to regulate many different cellular functions in a variety of cells [31], including epithelial cells, and [Ca2+]cyt is considered as an important regulator of intestinal ion transports [2, 32, 33]. In excitable cells, Ca2+ entry is mainly mediated via voltage-gated Ca2+ channels (VGCC). However, little is known about Ca2+ entry pathways in nonexcitable intestinal epithelial cells, since they may not express functional VGCC [34]. There was essentially less information available about the specific Ca2+ entry pathways existing in native small intestinal epithelium to mediate ion secretion. Therefore, in the present study, we firstly confirmed the critical role of pure Ca2+ signaling in duodenal ion secretion. Since intestinal epithelium is polarized in nature, we then revealed that CCh-induced external Ca2+ entry is just from serosal side of duodenal epithelial cells rather than from the lumen. We also examined the potential molecular candidate of SOCE mechanisms in regulating duodenal ion transport, and identified CRAC/Orai channels as the most likely molecular candidate, which is partially consistent with a previous report that STIM1/Orai may regulate Ca2+ influx across the apical and basolateral membrane in rat colonic epithelium [12]. However, it is currently unknown why CCh induces Ca2+ influx across the basolateral membrane only in mouse duodenal epithelium, which needs further investigation.
cAMP and [Ca2+]cyt signaling are pleiotropic primary second messengers that regulate all secretory epithelia functions. Since cAMP pathway has been extensively studied, it is questioned if pure Ca2+ signaling is critical for intestinal ion secretion. Moreover, mutual regulation of cAMP and Ca2+ signaling is referred to as crosstalk, while integration of their effects can result in an additive or synergistic physiological response [35]. So far, the synergism of cAMP and Ca2+ signaling has been demonstrated for ion secretion in salivary gland and pancreatic ducts [36], but not in small intestinal epithelium. The available findings suggest that the IP3R binding protein released with inositol 1,4,5-trisphosphate (IRBIT) protein is a central component of the mechanism mediating the synergism between cAMP and Ca2+ by functioning as a third messenger that translocates between the IP3R and target proteins [37, 38]. Both cAMP and PKA are critical stimulators for this synergistic process. However, in the present study, although we observed the synergism of cAMP- and Ca2+-mediated ion secretion, we did not detect any change in cAMP concentrations induced by CCh in duodenal epithelium. Furthermore, the synergistic effect was not affected by PKA inhibition. Therefore, our data indicate that CCh induced Ca2+-dependent but cAMP/PKA-independent ion secretionin duodenal epithelium, confirming the critical role of pure Ca2+ signaling in this process.
We further elucidated the underlying molecular mechanisms by how Ca2+ signaling regulates duodenal ion secretion. Although CaCC is critical for Ca2+-mediated ion secretion in other epithelial tissues, we did not detect their functional expression in duodenal epithelium. Therefore, the present study focused on the CFTR-mediated ion secretion since we underscored the importance of these channels in the process. Growing evidence suggests that Ca2+ signaling can stimulate CFTR channels by activating Ca2+-dependent adenylyl cyclase via cAMP/PKA pathway. However, our study ruled out this possibility in the duodenum because cAMP/PKA pathway is not involved in this CCh-induced Ca2+-mediatedion secretion. Furthermore, we revealed that Ca2+ signaling regulates duodenal ion secretion via a novel PI3K/Akt-mediated activity of CFTR channels.
Compelling evidence shows that cAMP-induced epithelial ion secretion is regulated mainly through PKA-mediated CFTR phosphorylation, which in turn stimulates both channel gating and trafficking to apical plasma membrane of polarized cells, leading to the increase of ion secretion [39, 40]. Previous studies reported the requirement of PI3K for glucagon-induced trafficking of aquaporin-8 [41] and angiotensin II-induced trafficking of Na+/H+ exchanger to plasma membrane, indicating that PI3K is involved in exocytotic insertion of proteins into plasma membrane [42]. Therefore, we further examined whether intracellular trafficking of CFTR is stimulated by CCh-induced Ca2+ signaling. Indeed, CCh stimulated CFTR trafficking to the plasma membranes of villus cells in duodenal epithelia, which was significantly inhibited by PI3K inhibitors. Together, our results obtained from functional, biochemical, and morphological studies have demonstrated the importance of PI3K in the regulation of CCh-mediated duodenal epithelial CFTR channel activity. To provide direct evidence for the role of PI3K in the regulation of duodenal epithelial CFTR function, we further examined PI3K activity and phosphorylation of Akt, a downstream effector of PI3K, in murine duodenal epithelium. Our results indicate that CCh-induced Ca2+ signal activates PI3K/Akt in duodenal epithelium.
In conclusion, we underscored the critical role of Ca2+ signaling in small intestinal epithelial ion secretion via the SOCE mechanism on the serosal side of epithelial cells. We identified that CRAC/Orai channels may serve as SOCE mechanism to mediate Ca2+-dependent epithelial ion secretion. We also demonstrated the molecular mechanisms of Ca2+ signaling in CFTR-mediated ion secretion via a novel PI3K/Akt pathway rather than the well-known cAMP/PKA pathway. A scheme summarizes our findings in Figure 6D. A full understanding of Ca2+-mediated intestinal epithelial Cl- and HCO3- secretion and the precise modulatory mechanisms of this process will greatly enhance our knowledge about ion transports in GI tract. Our findings suggest new perspectives for potential drug targets to protect the upper GI tract through promoting epithelial HCO3- secretion and to control intestinal liquid homeostasis through modulating epithelial Cl- secretion.
MATERIALS AND METHODS
Cell culture
The HT-29 cell lines used in this work were obtained from the American Type Culture Collection (Rockville, MD) and were cultured at 37°C under 5% CO2 in RPMI 1640 medium supplemented with antibiotics (100 U/ml of penicillin and 100μg/ml of streptomycin) and 10% heat-inactivated fetal bovine serum. After the cells had grown to confluence, they were replated onto 12-mm round coverslips (Warner Instruments Inc., Hamden, CT) and incubated for at least 24 h before use for cytosolic Ca2+ concentration([Ca2+]cyt).
Animal preparation
All studies were approved by Committees on Investigations Involving Animals in Xinqiao Hospital of Third Military Medical University, China and the University of California, San Diego. Experiments were performed with on 6–12 wk male Harlan C-57 black mice; homozygous CFTR knockout (CFTR-/-) mice and their wild-type littermates (CFTR+/+), which were established as described previously [43].
Ussing chamber experiments
Ussing chamber experiments were performed as previously described [44]. The duodenal tissue from each animal was stripped of seromuscular layers, divided, and examined in four chambers (window area, 0.1 cm2). Experiments were performed under continuous short-circuited conditions (Voltage-Current Clamp, VCC MC6; Physiologic Instruments, San Diego, CA), and luminal pH was maintained at 7.40 by the continuous infusion of 0.5 mM HCl under the automatic control of a pH-stat system (PHM290, pH-Stat controller; Radiometer Copenhagen). The volume of the titrant infused per unit time was used to quantitate HCO3- secretion. These measurements were recorded at 5-min intervals. The rate of luminal HCO3- secretion is expressed as micromoles per centimeter squared per hour. Transepithelial short-circuit current (Isc; reported as μA or eq cm-2 h-1) was measured via an automatic voltage clamp. After a 15-30 min measurement of basal parameters, inhibitors were added to the tissues for 10-20 min, as dictated by the experimental design, followed by addition of carbachol (100 μM) or forskolin (10 μM) to the serosal side of tissue.
Measurement of [Ca2+]cyt by digital Ca2+ imaging
Ca2+ imaging experiments were performed as previously described [45]. HT-29 cells cultured on coverslips were loaded with 5μM Fura-2 AM (Invitrogen, NY, USA) in physiological salt solution (PSS) at 37°C for 50 min and then washed with PSS or PPS with 2-APB (Tocris Bioscience, Minneapolis, MN, USA), a CRAC channel blockers (50μM); or GSK-7975A (Tocris Bioscience, Minneapolis, MN, USA), for 30 min. Then, cells on coverslips were placed in a standard perfusion chamber on the stage of an inverted fluorescence microscope (Nikon, Japan). For the Ca2+-free solution, Ca2+ was omitted and 0.5 mM EGTA was added to prevent possible Ca2+ contamination.
Phosphate labeling and immunoprecipitation of CFTR
The mice were prepared as described above. Segments of proximal duodenum (~5 mm) opened along the mesenteric border were placed in modified buffered Ringer solution at 37°C gassed with 95% O2-5% CO2 for incubation. After stabilization for 20 min, CCh (100 μM) or control vehicle was added to the bathing solution for 5 min of incubation. When wortmannin (0.1 μM) or LY294002 (20 μM) was used, it was added at 30 min before the agents above. Phosphate labeling and immunoprecipitation of CFTR was performed as described [43]. Briefly, CFTR was immunoprecipitated from the supernatant with CFTR polyclonal antibody (CFTR H-182; Santa Cruz Biotechnology) for 1h at 4°C. The samples were centrifuged, and the supernatants were analyzed by 6% SDS-polyacrylamide gel electrophoresis (SDS- PAGE). 32P-phosphorylated CFTR was visualized by autoradiography and quantified by scintillation counting of excised bands.
Immunoprecipitation of PI3K and ELISA for detection of PI3K and cAMP activity
Segments of murine duodenum were incubated for different time points as described above. Immunoprecipitation of PI3K was performed as described [44]. PI3K activity was measured in vitro using a competitive ELISA format (Echelon Biosciences, Salt Lake City, UT) and cAMP activity was measured in vitro using Mouse/Rat cAMP assay(FGE012B) from R&D Systems Inc. (Minneapolis, MN, USA) according to the manufacturer’s instructions. Enzyme activity was estimated by comparing the values from samples containing enzymatic reaction products to the values in the standard curve.
Western blot analysis for measurement of Akt phosphorylation
Segments of murine duodenum were incubated with CCh (100 μM), or control vehicle for different time points as described above. At the end of incubation, the tissue was processed as described in detection of PI3K activity above. Protein concentrations were determined using a Bradford protein assay (BioRad, Hercules, CA, USA). A total of 50 μg protein aliquots were separated by SDS-PAGE electrophoresis and blotted using a V3 Western Workflow system (BioRad, Hercules, CA, USA) according to the manufacturer’s instructions. PVDF membranes were incubated with primary antibodies: anti-phospho-Akt S473 or anti-Akt (diluted 1:1,000; Cell Signaling) overnight at 4°C. All results were measured by densitometry and presented as relative expression to tubulin as a reference protein.
Immunohistochemistry
Segments of murine duodenum were incubated as described above. Immunofluorescence labeling was performed as described [43]. Sections were incubated in a moist chamber overnight with primary anti-CFTR antibody (CFTR H-182, 1:50 dilution in 1% PBS-BSA) at 4°C, washed three times for 15 min each and incubated with FITC-conjugated secondary antibody (affinity purified donkey anti-rabbit IgG, 1:400 dillution in 1% PBS-BSA), for 1 h at room temperature to detect CFTR staining. Confocal microscopy and images analysis of CFTR fluorescence intensities were performed as described by Ameen et al. [46, 47] using a confocal microscope (TCS SP2 AOBS; Leica, Wetzlar, Germany) equipped with image analysis software. Data were collected from an average of 30 cells in random sections labeled for CFTR (average 10 sections) from each tissue examined (4 animals in each series) and is expressed as the ratio of plasma membrane to cytoplasm fluorescence intensity.
Statistics
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology [48]. All results are means ± SE. Net peaks for duodenal HCO3- secretion and Isc refer to stimulated peak responses minus basal levels. Data were analyzed using one-way analysis of variance followed by the Newman-Keul post hoc test or, when appropriate, by the two-tailed Student’s t-tests. P<0.05 was considered statistically significant.
Materials
The following drugs were used: carbachol, Forskolin, wortmannin, LY294002, and rapamycin from Sigma; CFTRinh-172 from Calbiochem; [32P]orthophosphate from Amer-sham; Anti-PI3K p85 was obtained from Upstate Biotechnology; Anti-phospho-Akt S473 antibody and anti-Akt antibody were obtained from Cell Signaling; All other chemicals in solutions were obtained from Sigma and Calbiochem.
Abbreviations
CCh: carbachol; CFTR: cystic fibrosis transmembrane conductance regulator; GI: gastrointestinal; CaCCs: Ca2+-activated Cl- channels; 2-APB: 2-aminoethoxydiphenyl borate; SOCE: store-operated Ca2+ entry; CRAC: Ca2+ release-activated Ca2+ channels; IP3: inositol 1,4,5-trisphoshate; [Ca2+]cyt: cytosolic Ca2+ concentration; IEC: intestinal epithelial cells; Isc: short-circuit current; FFA: Flufenamic acid; ER: endoplasmic reticulum; NFA: niflumic acid; VGCC: voltage-gated Ca2+ channels; IRBIT: IP3R binding protein released with inositol 1,4,5-trisphosphate.
Author contributions
H.D. designed the experiments. X.Y. did the Ca2+ imaging and Elisa. G.R.W. and B.G.T. did the immunoprecipitation, Western blot and immunohistochemistry. X.Y. and F.L.Z. did the Ussing chamber experiments. H.X.W. and J.L.H. did the cell culture. X.Y. and H.D. wrote the manuscript. H.D. and S.M.Y. revised and finalized the manuscript. All authors reviewed the manuscript.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
These studies were supported by research grants from the National Key Research and Development Program of China (No. 2016YFC1302200 to HD) and the National Natural Science Foundation of China (No. 81570477 and No. 31371167 to HD).
REFERENCES
1. Flemstrom G, Garner A. Gastroduodenal HCO3(-) transport: characteristics and proposed role in acidity regulation and mucosal protection. Am J Physiol. 1982; 242:G183-193.
2. Jung J, Lee MG. Role of calcium signaling in epithelial bicarbonate secretion. Cell Calcium. 2014; 55:376–384.
3. Xie R, Dong X, Wong C, Vallon V, Tang B, Sun J, Yang S, Dong H. Molecular mechanisms of calcium-sensing receptor-mediated calcium signaling in the modulation of epithelial ion transport and bicarbonate secretion. J Biol Chem. 2014; 289:34642–34653.
4. Putney JW Jr. Recent breakthroughs in the molecular mechanism of capacitative calcium entry (with thoughts on how we got here). Cell Calcium. 2007; 42:103–110.
5. Putney JW Jr. A model for receptor-regulated calcium entry. Cell Calcium. 1986; 7:1–12.
6. Putney JW Jr. Capacitative calcium entry revisited. Cell Calcium. 1990; 11:611–624.
7. Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992; 355:353–356.
8. Hoth M, Penner R. Calcium release-activated calcium current in rat mast cells. J Physiol. 1993; 465:359–386.
9. Mignen O, Thompson JL, Yule DI, Shuttleworth TJ. Agonist activation of arachidonate-regulated Ca2+-selective (ARC) channels in murine parotid and pancreatic acinar cells. J Physiol. 2005; 564:791–801.
10. Molnar T, Yarishkin O, Iuso A, Barabas P, Jones B, Marc RE, Phuong TT, Krizaj D. Store-Operated Calcium Entry in Muller Glia Is Controlled by Synergistic Activation of TRPC and Orai Channels. J Neurosci. 2016; 36:3184–3198.
11. Rao JN, Rathor N, Zou T, Liu L, Xiao L, Yu TX, Cui YH, Wang JY. STIM1 translocation to the plasma membrane enhances intestinal epithelial restitution by inducing TRPC1-mediated Ca2+ signaling after wounding. Am J Physiol Cell Physiol. 2010; 299:C579-588.
12. Onodera K, Pouokam E, Diener M. STIM1-regulated Ca2+ influx across the apical and the basolateral membrane in colonic epithelium. J Membr Biol. 2013; 246:271–285.
13. Lefkimmiatis K, Srikanthan M, Maiellaro I, Moyer MP, Curci S, Hofer AM. Store-operated cyclic AMP signalling mediated by STIM1. Nat Cell Biol. 2009; 11:433–442.
14. Seidler U, Blumenstein I, Kretz A, Viellard-Baron D, Rossmann H, Colledge WH, Evans M, Ratcliff R, Gregor M. A functional CFTR protein is required for mouse intestinal cAMP-, cGMP- and Ca(2+)-dependent HCO3- secretion. J Physiol. 1997; 505:411–423.
15. Bradbury NA. Intracellular CFTR: localization and function. Physiol Rev. 1999; 79:S175–191.
16. Sheppard DN, Welsh MJ. Structure and function of the CFTR chloride channel. Physiol Rev. 1999; 79:S23–45.
17. Banks MR, Farthing MJ. Fluid and electrolyte transport in the small intestine. Curr Opin Gastroenterol. 2002; 18:176–181.
18. Chao AC, de Sauvage FJ, Dong YJ, Wagner JA, Goeddel DV, Gardner P. Activation of intestinal CFTR Cl- channel by heat-stable enterotoxin and guanylin via cAMP-dependent protein kinase. EMBO J. 1994; 13:1065–1072.
19. Kaji I, Akiba Y, Kaunitz JD. Digestive physiology of the pig symposium: involvement of gut chemosensing in the regulation of mucosal barrier function and defense mechanisms. J Anim Sci. 2013; 91:1957–1962.
20. Lindqvist SM, Sharp P, Johnson IT, Satoh Y, Williams MR. Acetylcholine-induced calcium signaling along the rat colonic crypt axis. Gastroenterology. 1998; 115:1131–1143.
21. Mei FC, Qiao J, Tsygankova OM, Meinkoth JL, Quilliam LA, Cheng X. Differential signaling of cyclic AMP: opposing effects of exchange protein directly activated by cyclic AMP and cAMP-dependent protein kinase on protein kinase B activation. J Biol Chem. 2002; 277:11497–11504.
22. Perego S, Del Favero E, De Luca P, Dal Piaz F, Fiorilli A, Cantu L, Ferraretto A. Calcium bioaccessibility and uptake by human intestinal like cells following in vitro digestion of casein phosphopeptide-calcium aggregates. Food Funct. 2015; 6:1796–1807.
23. Dong X, Ko KH, Chow J, Tuo B, Barrett KE, Dong H. Expression of acid-sensing ion channels in intestinal epithelial cells and their role in the regulation of duodenal mucosal bicarbonate secretion. Acta Physiol (Oxf). 2011; 201:97–107.
24. Bajwa PJ, Lee JW, Straus DS, Lytle C. Activation of PPARgamma by rosiglitazone attenuates intestinal Cl- secretion. Am J Physiol Gastrointest Liver Physiol. 2009; 297:G82-89.
25. Himmerkus N, Vassen V, Sievers B, Goerke B, Shan Q, Harder J, Schroder JM, Bleich M. Human beta-defensin-2 increases cholinergic response in colon epithelium. Pflugers Arch. 2010; 460:177–186.
26. De La Fuente R, Namkung W, Mills A, Verkman AS. Small-molecule screen identifies inhibitors of a human intestinal calcium-activated chloride channel. Mol Pharmacol. 2008; 73:758–768.
27. Ma T, Thiagarajah JR, Yang H, Sonawane ND, Folli C, Galietta LJ, Verkman AS. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J Clin Invest. 2002; 110:1651–1658.
28. Billet A, Hanrahan JW. The secret life of CFTR as a calcium-activated chloride channel. J Physiol. 2013; 591:5273–5278.
29. Powis G, Bonjouklian R, Berggren MM, Gallegos A, Abraham R, Ashendel C, Zalkow L, Matter WF, Dodge J, Grindey G, Vlahos CJ. Wortmannin, a potent and selective inhibitor of phosphatidylinositol-3-kinase. Cancer Res. 1994; 54:2419–2423.
30. Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J Biol Chem. 1994; 269:5241–5248.
31. Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003; 4:517–529.
32. Chew CS, Safsten B, Flemstrom G. Calcium signaling in cultured human and rat duodenal enterocytes. Am J Physiol. 1998; 275:G296–304.
33. Flemstrom G, Isenberg JI. Gastroduodenal mucosal alkaline secretion and mucosal protection. News Physiol Sci. 2001; 16:23–28.
34. Parekh AB, Penner R. Store depletion and calcium influx. Physiol Rev. 1997; 77:901–930.
35. Ahuja M, Jha A, Maleth J, Park S, Muallem S. cAMP, Ca(2)(+) signaling in secretory epithelia: crosstalk and synergism. Cell Calcium. 2014; 55:385–393.
36. Park S, Shcheynikov N, Hong JH, Zheng C, Suh SH, Kawaai K, Ando H, Mizutani A, Abe T, Kiyonari H, Seki G, Yule D, Mikoshiba K, Muallem S. Irbit mediates synergy between ca(2+) and cAMP signaling pathways during epithelial transport in mice. Gastroenterology. 2013; 145:232–241.
37. Ando H, Kawaai K, Mikoshiba K. IRBIT: a regulator of ion channels and ion transporters. Biochim Biophys Acta. 2014; 1843:2195–2204.
38. Ando H, Mizutani A, Kiefer H, Tsuzurugi D, Michikawa T, Mikoshiba K. IRBIT suppresses IP3 receptor activity by competing with IP3 for the common binding site on the IP3 receptor. Mol Cell. 2006; 22:795–806.
39. Bertrand CA, Frizzell RA. The role of regulated CFTR trafficking in epithelial secretion. Am J Physiol Cell Physiol. 2003; 285:C1-18.
40. Guggino WB, Stanton BA. New insights into cystic fibrosis: molecular switches that regulate CFTR. Nat Rev Mol Cell Biol. 2006; 7:426–436.
41. Gradilone SA, Carreras FI, Lehmann GL, Marinelli RA. Phosphoinositide 3-kinase is involved in the glucagon-induced translocation of aquaporin-8 to hepatocyte plasma membrane. Biol Cell. 2005; 97:831–836.
42. du Cheyron D, Chalumeau C, Defontaine N, Klein C, Kellermann O, Paillard M, Poggioli J. Angiotensin II stimulates NHE3 activity by exocytic insertion of the transporter: role of PI 3-kinase. Kidney Int. 2003; 64:939–949.
43. Tuo B, Wen G, Zhang Y, Liu X, Wang X, Liu X, Dong H. Involvement of phosphatidylinositol 3-kinase in cAMP- and cGMP-induced duodenal epithelial CFTR activation in mice. Am J Physiol Cell Physiol. 2009; 297:C503-515.
44. Tuo B, Wen G, Wang X, Xu J, Xie R, Liu X, Dong H. Estrogen potentiates prostaglandin E-stimulated duodenal mucosal HCO(3)(-) secretion in mice. Am J Physiol Endocrinol Metab. 2012; 303:E111–121.
45. Wan H, Xie R, Xu J, He J, Tang B, Liu Q, Wang S, Guo Y, Yang X, Dong TX, Carethers JM, Yang S, Dong H. Anti-proliferative Effects of Nucleotides on Gastric Cancer via a Novel P2Y6/SOCE/Ca2+/beta-catenin Pathway. Sci Rep. 2017; 7:2459.
46. Ameen NA, Martensson B, Bourguinon L, Marino C, Isenberg J, McLaughlin GE. CFTR channel insertion to the apical surface in rat duodenal villus epithelial cells is upregulated by VIP in vivo. J Cell Sci. 1999; 112:887–894.
47. Ameen NA, Marino C, Salas PJ. cAMP-dependent exocytosis and vesicle traffic regulate CFTR and fluid transport in rat jejunum in vivo. Am J Physiol Cell Physiol. 2003; 284:C429-438.
48. Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA, Gilchrist A, Hoyer D, Insel PA, Izzo AA, Lawrence AJ, MacEwan DJ, Moon LD, et al. Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol. 2015; 172:3461–3471.