
INTRODUCTION
The binding of programmed death ligand 1 (PD-L1) to its receptor PD-1 leads to T cell apoptosis, which, in turn, triggers tumor immune surveillance [1, 2]. PD-L1 overexpression has a known association with poor prognosis in various human cancers, including non-small cell lung cancer (NSCLC), due to tumor immune escape [3]. PD-L1 expression is also higher in patients with epidermal growth factor receptor (EGFR)-mutated NSCLC tumors than with EGFR-wild-type (WT) tumors [4, 5]. A higher PD-L1 expression in EGFR-mutated tumors may alter the tumor immune microenvironment, thereby promoting tumors with a more aggressive phenotype and consequently resulting in poor prognosis [4]. Therefore, we predicted that PD-L1 might make a significantly greater contribution to tumor malignancy in patients with EGFR-mutated NSCLC tumors than with EGFR-WT tumors.
Tumor metastasis and drug resistance represent a crucial limitation in human cancer therapy. EGFR-tyrosine kinase inhibitors (EGFR-TKIs) have known clinical benefits in the treatment of EGFR-mutated NSCLC when compared to EGFR-WT NSCLC [6, 7]. Unfortunately, resistance to TKIs frequently arises in about 9 to 12 months in patients undergoing TKI treatment, and consequently to result in patients’ tumor relapse and death [8–10]. Previously, reduction of FOXO3a, microRNA-200c expression and overexpression of insulin growth factor 1 receptor and phosphorylated EGFR were associated with worse efficacy of EGFR-TKIs in NSCLC patients with EGFR-WT [11–14]. However, the TKI resistance in EGFR-WT NSCLC is still largely unclear. Therefore, discovery of possible mechanisms in EGFR mutation-independent TKI resistance may not only improve TKI resistance in EGFR-mutated NSCLC but also in EGFR-WT NSCLC.
EGFR-TKIs down-regulate PD-L1 expression in EGFR-mutated NSCLC through inhibition of the NF-κB signaling pathway [15]. PD-L1 expression is also downregulated by inhibitors of the MEK/ERK and PI3K/AKT signaling pathways in EGFR-mutated NSCLC cells [16–18]. The decrease in PD-L1 expression by these inhibitors may suppress tumor progression by altering tumor immune surveillance. On the other hand, PD-L1 expression was associated with epithelial-to-mesenchymal transition (EMT), high proliferation activity, and poor prognosis in adenocarcinoma of the lung [19–21]. EMT has known to confer TKI resistance in NSCLC [22]. However, the role of PD-L1 in EGFR mutation-independent TKI resistance in NSCLC remains unclear.
The development of the hypoxic tumor microenvironment, which is associated with tumor progression and metastasis, depends on the presence of myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs) [23, 24], and arises due to the generation of reactive oxygen species (ROS) and subsequent induction of HIF-1α expression [25, 26]. PD-L1, as a direct target of HIF-1α, is blocked under hypoxia, while MDSC-mediated T cell activation is enhanced [27]. High ROS levels are also associated with an increase in YAP1 expression [28]. Increases in PD-L1 expression confer cisplatin resistance in small cell lung cancer and NSCLC cells [29, 30]. We therefore hypothesized that PD-L1 expression might confer EGFR mutation-independent TKI resistance.
Our preliminary data showed that PD-L1, HIF-1α, and YAP1 expressed concomitantly in EGFR-mutated and EGFR-WT NSCLC cells. YAP1 confers chemoresistance in NSCLC cells [31], and YAP1 inhibition restores the sensitivity to EGFR-TKI in TKI-resistant lung adenocarcinoma [32]. Here, we provide evidence that YAP1 expression may be responsible for PD-L1-mediated TKI resistance in NSCLC cells regardless of EGFR mutation.
RESULTS
PD-L1, HIF-1α, and YAP1 concomitantly expressed, and PD-L1 may confer TKI resistance in EGFR-mutated and EGFR-WT cells
Four EGFR-mutated and EGFR-WT cell types were collected to verify whether PD-L1 expression could be associated with HIF-1α and YAP1 expression. Western blotting indicated a higher PD-L1 expression in EGFR-mutated cells than in EGFR-WT cells (Figure 1A). This expression of PD-L1 was consistently observed together with HIF-1α expression in all tested cell types (Figure 1A). PD-L1 expression was manipulated in three cell types with high and one with low expression of PD-L1 and HIF-1α by the use of a small hairpin (sh)RNA and an expression plasmid. A dose-dependent change in PD-L1 expression observed all four tested cell types (Figure 1B, upper panel). Surprisingly, the expression of HIF-1α and YAP1 decreased and increased concomitantly with the PD-L1 expression by these manipulations (Figure 1B, upper panel). The changes induced in YAP1 protein expression by PD-L1 manipulation were consistent with its mRNA levels (Figure 1B, middle panel), suggesting that modulation of YAP1 expression by PD-L1 might occur at the transcriptional level. In addition, the changes in HIF-1α expression following PD-L1 manipulation correlated with ROS levels in these cells (Figure 1B, lower panel). These results suggest that PD-L1 may increase HIF-1α and YAP1 expression by increasing ROS generation in these cells.
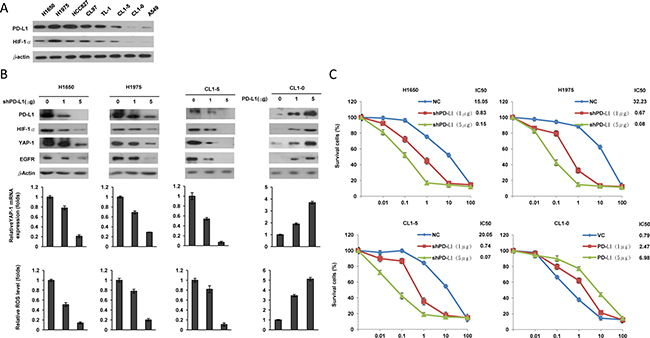
Figure 1: PD-L1, HIF-1α, and YAP1 are concomitantly expressed, and PD-L1 may confer TKI resistance in EGFR-mutated and EGFR-WT cells. (A) Western blottingnanalysis was performed to evaluate PD-L1 expressions in different lung cancer cell lines. (B) H1650, H1975 and CL1-5 cells were transfected with PD-L1 shRNA, while CL1-0 cells were transfected with a PD-L1 expressing plasmid for 48 h. The expression of PD-L1 and YAP1 in these cells was evaluated by western blotting. The mRNA level was determined by real-time PCR. ROS production in response to Cells with transfected with vector control or PD-L1 expression vector/shRNA was determined by a flow cytometry following ROS detection kit. The relative ROS levels are shown in the lower panel. (C) Cells were treated with six concentrations of gefitenib to calculate the IC50 value from the dose-response survival curve determined by the MTT assay.
We next examined the possibility that PD-L1 could be responsible for TKI resistance in NSCLC cells. Two EGFR-mutated and EGFR-WT cell types collected for manipulation of PD-L1 expression to test this possibility. The MTT assay indicated that the inhibitory concentration of gefitinib yielding 50% cell viability (IC50) decreased markedly by PD-L1 silencing in H1650, H1975, and CL1-5 cells. The IC50 values were gradually increased by ectopic PD-L1 expression in CL1-0 cells (Figure 1C). These results suggest that PD-L1-mediated TKI resistance might occur by an increase in HIF-1α and YAP1 expression due to enhanced ROS generation.
PD-L1 may induce HIF-1α by ROS generation, and in turn, upregulate YAP1 expression in NSCLC cells regardless of EGFR mutation
We next examined whether PD-L1 may induce HIF-1α expression through ROS generation, thereby upregulating YAP1 expression at the transcription level. H1975 and CL1-0 cells were collected to manipulate PD-L1 expression and/or treatment with a ROS inducer (pyocyanin) or a ROS scavenger (N-acetylcysteine; NAC). Western blotting showed a concomitant decrease in expression of HIF-1α and YAP1 following shPD-L1 transfection or treatment with NAC. However, the decrease in HIF-1α and YAP1 expression by PD-L1 silencing reversed by pyocyanin treatment in both cell types (Figure 2A, upper panel).
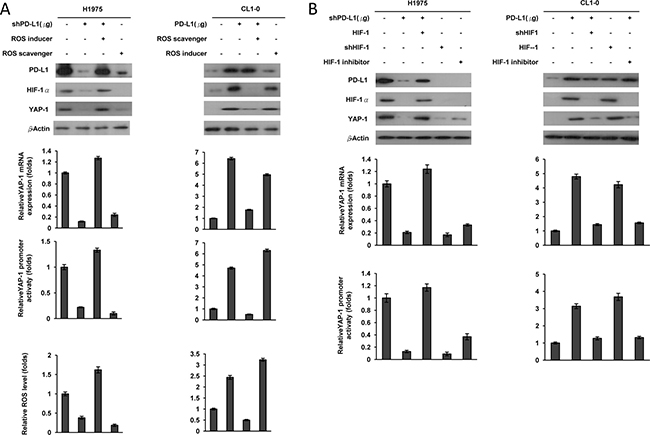
Figure 2: PD-L1 may induce HIF-1α by ROS generation, and in turn, upregulate YAP1 expression in NSCLC cells. (A) ROS inducer (pyocyanin), ROS scavenger (NAC), PD-L1 plasmid and/or PD-L1 shRNA were treated with H1975 and CL1-0 cells for 12 h. The expression of PD-L1 and YAP1 in these cells was evaluated by western blotting. The mRNA level was determined by real-time PCR. The changes in YAP1 promoter activity were evaluated by the luciferase reporter assay. ROS production in response to cells were determined by a flow cytometry following ROS detection kit. (B) H1975 cells were transfected with PD-L1 shRNA, HIF-1 shRNA, HIF-1 expression plasmids or HIF-1 inhibitor; CL1-0 cells were transfected with a PD-L1, HIF-1 expressing plasmids, HIF-1 shRNA or HIF-1 inhibitor for 48 h. The expression of PD-L1, HIF-1, and YAP1 in these cells was evaluated by western blotting using their specific antibodies. The mRNA level was determined by real-time PCR. The changes in YAP1 promoter activity were evaluated by the luciferase reporter assay.
The results from real-time PCR, luciferase reporter assays, and flow cytometry analysis confirmed that PD-L1 expression correlated with HIF-1α expression, with YAP1 mRNA expression and reporter activity, and with ROS levels in both cell types, as determined by PD-L1 manipulation and/or ROS inducer or ROS scavenger treatment (Figure 2A, lower panel). The HIF-1α expression and the YAP1 mRNA expression and reporter activity correlated with the ROS levels generated following PD-L1 manipulation plus ROS inducer or scavenger treatments (Figure 2A, lower panel). We also used H1975 and CL1-0 cells in which we manipulated PD-L1 expression. These cells were transfected with an HIF-1α expression plasmid (shHIF-1α) or treated with an HIF-1α inhibitor (2-methoxyestrodiol) to confirm that PD-L1 induced HIF-1α expression and, in turn, upregulated YAP1 transcription (Figure 2B). These results clearly indicate that PD-L1 may stimulate ROS production, which induces HIF-1α expression, and consequently, upregulates YAP1 transcription in NSCLC cells regardless of EGFR mutation.
PD-L1-mediated YAP1 expression is responsible for EGFR mutation-indepdendent TKI resistance in NSCLC cells
We further examined the possibility that PD-L1-mediated YAP1 expression could be responsible for EGFR mutation-independent TKI resistance in NSCLC cells. EGFR-mutated H1975 and EGFR-WT CL1-0 cells were collected for manipulation of PD-L1 expression and/or transfection with HIF-1α, a YAP1 expression plasmid, shHIF-1α, or shYAP1. The co-expression of PD-L1, HIF-1α, and YAP1 modulated by these gene manipulations was confirmed in both cell types by western blotting (Figure 3A, upper left and right panels).
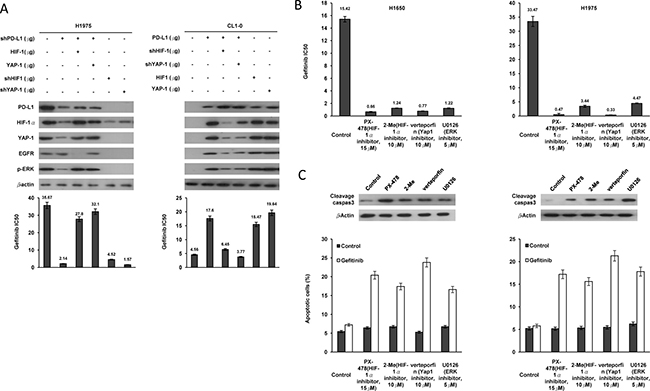
Figure 3: PD-L1-mediated YAP1 expression is responsible for TKI resistance in NSCLC cells. (A) H1975 cells were transfected with PD-L1 shRNA, HIF-1 shRNA, YAP1 shRNA, HIF-1 and YAP1 expression plasmids; CL1-0 cells were transfected with PD-L1, HIF-1, YAP1 expressing plasmids, PD-L1 shRNA and HIF-1 shRNA for 48 h. The expression of PD-L1, HIF-1, and YAP1 in these cells was evaluated by western blotting using their specific antibodies. Cells were treated with six concentrations of gefitenib to calculate the IC50 value from the dose-response survival curve determined by the MTT assay. (B) H1650 and H1975 cell were treated with PX-478 (15 μM), 2-Me(10 μM), Verteorfin (10 μM), U0126(5 μM) and six concentrations of gefitenib to calculate the IC50 value from the dose-response survival curve determined by the MTT assay. (C) The expression of cleavage caspase3 in these cells was evaluated by western blotting using their specific antibodies. The cells were then subjected to annexin V and PI staining, followed by a flow cytometry. Percentage of apoptotic cells including with the Annexin V+/PI− population (early apoptosis) plus Annexin V+/PI+ (late apoptosis/secondary necrosis) was summarized by a flow cytometric analysis.
The MTT assay indicated that the greatest change in the IC50 value for gefitinib was obtained with YAP1 silencing, followed, in decreasing size of the response, by PD-L1 silencing and HIF-1α silencing in H1975 cells; however, the decrease in the IC50 value obtained by PD-L1 silencing was reversed by ectopic expression of YAP1 or HIF-1α (Figure 3A lower left panel). Conversely, the IC50 value was markedly increased by ectopic expression of PD-L1, HIF-1α, or YAP1 in CL1-0 cells when compared with CL1-0 cells transfecting an empty vector (VC) (Figure 3A, lower right panel). An increase in the IC50 value by ectopic PD-L1 expression was reversed by HIF-1α or YAP1 silencing (Figure 3A, lower right panel).
A reduction in the IC50 value for gefitinib was observed in H1650 and H1975 cell types subjected to inhibitors of HIF-1α (2-methoxyestrodiol and PX478), YAP1 (verteporfin), and ERK (U0126), when compared to both cell types without these inhibitor treatments (Figure 3B upper panel). Interestingly, the expression of cleaved caspase-3, evaluated by western blotting, was increased markedly by these inhibitor treatments (Figure 3B middle panel). Annexin V-PI staining indicated that the percentage of apoptotic cells showed the greatest increase in response to verteporfin, followed, in decreasing size of the response, by PX-478, 2-methoxyestradiol, and U0126 treatment in H1650 and H1975 cells (Figure 3B lower panel). These results clearly indicated that YAP1 expression might be responsible for PD-L1-mediated TKI resistance in EGFR-mutated and –WT NSCLC cells via modulating apoptotic pathway.
Tumor response to TKI therapy in patients with NSCLC was associated with expression of PD-L1 and YAP1
We enrolled 46 tumors from surgically resected patients with NSCLC who had received TKI therapy to examine the possibility of an association between the expression of PD-L1 and YAP1 and with tumor response to TKI therapy in these patients. All patients were adenocarcinoma and unknown EGFR mutation status. The mRNA expression levels of both genes in the lung tumor samples evaluated by real-time PCR. The median expression values for both genes then used as cutoff points to divide tumors into “high” and “low” subgroups. The PD-L1 mRNA expression level was not associated with any clinical parameters in these 46 NSCLC patients (Supplementary Table 1); however, a positive correlation was observed between PD-L1 and YAP1 mRNA expression in this study population, as high-PD-L1 mRNA tumors were frequently also high-YAP1 mRNA tumors (69.6% vs. 30.4%, P = 0.008; Table 1). Interestingly, tumors expressing high levels of both PD-L1 and YAP1 mRNA were more likely to show an unfavorable response to TKI therapy, when compared with tumors that expressed low levels of mRNA for both these genes (66.7% vs. 36.0%, P = 0.038 for PD-L1; 81.0% vs. 24.0%, P = 0.010 for YAP1; Table 2). Moreover, all patients with high-PD-L1/high-YAP1 expressing tumors exhibited an unfavorable response to TKI therapy (Table 2). These results from patients appeared to support the mechanism of action proposed for the cell model and suggested that PD-L1-mediated YAP1 expression may have the potential to predict an unfavorable response to TKI therapy in patients with NSCLC regardless of EGFR mutation.
Table 1: Relationships of PD-L1andYAP1 mRNA expression in NSCLC cancer patients
Characteristics |
Patient No. |
PD-L1 |
P value |
|
|---|---|---|---|---|
Low (%) |
High (%) |
|||
Total patients |
46 |
23 (50.0) |
23 (50.0) |
|
YAP1 |
||||
Low |
23 |
16 (69.6) |
7 (30.4) |
0.008 |
High |
23 |
7 (30.4) |
16 (69.6) |
|
Table 2: Association of PD-L1 and YAP1 mRNA expression with tumor response to EGFR-TKI therapy in NSCLC patients
Patient No. |
Tumor Response |
P |
||
|---|---|---|---|---|
Unfavorable (%) |
Favorable (%) |
|||
PD-L1 |
||||
Low |
23 |
9 (36.0) |
16 (64.0) |
0.038 |
High |
23 |
14 (66.7) |
7 (33.3) |
|
YAP1 |
||||
Low |
23 |
6 (24.0) |
19 (76.0) |
0.010 |
High |
23 |
17 (81.0) |
4 (19.0) |
|
PD-L1/YAP1 |
||||
Low/Low |
16 |
6 (37.5) |
10 (62.5) |
0.001 |
Low/High |
7 |
3 (42.9) |
4 (57.1) |
|
High/Low |
7 |
0 (0.0) |
7 (100.0) |
|
High/High |
16 |
16 (100.0) |
0 (0.0) |
|
PD-L1/YAP1 |
||||
Others |
9 (30.0) |
21 (70.0) |
||
High/High |
16 (100.0) |
0 (0.0) |
||
PD-L1 and YAP1 mRNA levels in lung tumors were determined by real-time PCR analysis. The median value of PD-L1 (0.92) and YAP1 mRNA (1.43) in this study population was used as a cutoff point to divide patients into “low” and “high” subgroup.
Unfavorable response to TKI: patients with stable or progression disease.
Favorable response to TKI: patients with complete or partial response.
DISCUSSION
PD-L1 is a novel direct target of HIF-1α and its blockade under hypoxia enhances MDSC-mediated T cell activation [27]. HIF-1α expression, mediated by ROS generation, plays a central role in the inflammatory tumor microenvironment [24]. Hypoxia modulates SIAH2 ubiquitin E3 ligase, which enhances the formation of a complex between YAP1 and HIF-1α, which increases HIF-1α stability [33]. Hypoxia also increases YAP1 gene expression [34]. The evidence provided here demonstrates that upregulation of YAP1 expression by PD-L1 may confer TKI resistance by increasing ROS generation and therefore promoting HIF-1α expression. This finding strongly supports our other study indicating that DDX3 confers cetuximab resistance by inducing KRAS transcription, which increases ROS-induced HIF-1α and subsequent upregulation of YAP1 expression [35]. Nevertheless, to the best of our knowledge, this is the first study to reveal transcriptional regulation of YAP1 expression by HIF-1α expression induced by PD-L1.
YAP1 is known to confer cancer drug resistance [35]. For example, YAP1 acts as a parallel survival input to promote resistance to RAF or MEK inhibitor therapy, and similarly, combined suppression of YAP and RAF or MEK enhances the treatment response and patient survival [36]. A similar finding of resistance to a MAPK inhibitor was observed in melanoma [37]. A functional genomic screen identified YAP1 as a key determinant that conferred resistance to EGFR-TKI in lung cancer cells [38]. In the present study, PD-L1 not only increased YAP1 expression, but it also elevated the expression of EGFR and p-ERK in EGFR-mutated and –WT NSCLC cells (Figures 1 and 3). A similar YAP1 upregulation of EGFR overexpression confers chemoresistance in esophageal cancer [31]. NF-κB activation may play a role in inducing PD-L1 expression and TKI resistance [15]. Our previous studies indicated that PAK1 and PD-L1 expression might confer EGFR mutation-independent TKI resistance in NSCLC via persistent activation of PI3K/AKT and MEK/ERK signaling pathways [39, 40]. TKI inhibited EGFR phosphorylation, but activation of downstream gene phosphorylation of AKT and ERK in TKI resistant EGFR-WT NSCLC cells [14]. In addition, another report indicated that NF-κB-driven suppression of FOXO3a contributed to EGFR-mutation- independent gefitinib resistance in NSCLC cells [11]. We therefore suggest that PD-L1 confers EGFR mutation-independent TKI resistance through a YAP1/EGFR/ERK/NF-κB feedback loop in NSCLC cells. A possible pathway for PD-L1-mediated TKI resistance in NSCLC cells is proposed in Figure 4.
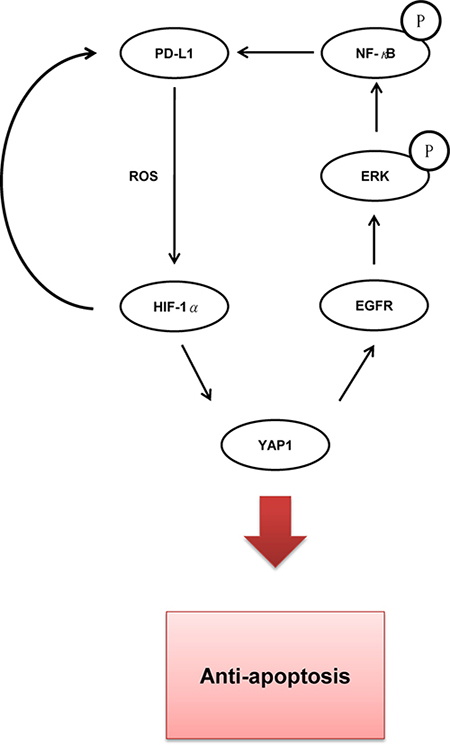
Figure 4: The possible route for the mechanistic action of PD-L1-induced YAP1 expression in TKI resistance of lung cancer.
In summary, we provided evidence that PD-L1 may confer TKI resistance in NSCLC by generating ROS to induce the expression of HIF-1α, which then upregulates YAP1 transcription. This proposed mechanism of action of PD-L1-mediated TKI resistance further validated by a retrospective study of TKI response in a small subset of NSCLC patients. Therefore, we suggest that PD-L1 immunotherapy, singly or in combination with a YAP1 inhibitor, may not only reduce tumor aggressiveness but it could also overcome TKI resistance in NSCLC, particularly in patients with tumors that express both PD-L1 and YAP1.
MATERIALS AND METHODS
Study subjects
Lung tumor specimens were collected from 140 patients who underwent primary NSCLC surgical resection at the Department of Thoracic Surgery, Taichung Veterans General Hospital (Taichung, Taiwan) between 1998 and 2004. Patients were asked to submit written informed consent; the study was approved by the Institutional Review Board (TMUH No. 201301051). The tumor type and stage of each collected specimen were histologically determined in accordance with the World Health Organization classification system. Among these patients, 46 patients were received TKI therapy when these patients were occurred tumor relapse. The tumor response to TKI therapy collected from chart review.
Cell lines
TL-1 cells was kindly provided by Dr. Y.-W. Cheng (Graduate Institute of Cancer Biology and Drug Discovery, Taipei Medical University, Taipei, Taiwan) [36]. H1650, H1975, HCC827, and A549 cells were obtained from the Bioresource Collection and Research Center, the Food Industry Research and Development Institute (Hsinchu, Taiwan). CL1-0 and CL1-5 cells were kindly provided by Professor P.-C. Yang (Department of Internal Medicine, National Taiwan University Hospital, Taipei, Taiwan). TL-1, H1650, H1975, HCC827, CL1-5, and CL 1-0 cancer cell lines were maintained in RPMI-1640 medium (HyClone, Logan, UT). A549 cancer cell lines were maintained in DMEM medium (HyClone, Logan, UT). The media contained 10% fetal bovine serum (FBS) supplemented with penicillin (100 U/mL) and streptomycin (100 mg/mL). The cells were cultured according to the suppliers’ instructions. Once resuscitated, the cell lines were routinely authenticated (once every 6 months; the cells were last tested in December 2012) by cell morphology monitoring, growth curve analysis, species verification via isoenzymology and karyotyping, identity verification via short tandem repeat profiling analysis, and contamination checks.
Real-time PCR analysis
The expression of PD-L1 mRNA and YAP1 mRNA levels in patients’ tumors was determined by real-time PCR analysis as described previously [39].
Plasmid construction and transfection
shRNA was purchased from the National RNAi Core Facility, Academia Sinica, Taiwan. Plasmids containing the PD-L1 expression construct were constructed by cloning the full-length human PD-L1 cDNA (GenBank accession number NM_014143) into the pcDNA3.1 eukaryotic expression vector, which also expresses a neomycin (Neo) resistance gene. YAP1 and HIF1 overexpression plasmids were provided from Origene (Rockville, MD). The transfection and stable clone selection procedures have been described previously [39]. All data were collected from three independent experiments.
3-(4,5-cimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) cytotoxicity assay
The cell lines were cultured in 96-well flat-bottomed microtiter plates supplemented with RPMI 1640 and DMEM containing 10% heat-inactivated fetal bovine serum, 100 units/mL penicillin, and 100 units/mL streptomycin in a humidified atmosphere containing 95% air and 5% CO2 at 37°C in a humidified incubator. Before TKI treatment (0, 0.01, 0.1, 1, 10, 100 μM), the cells cultured in the exponential growth phase were pretreated with shRNAs, PD-L1 and BAG-1 overexpression plasmid for 24 h. After 48 h incubation, the in vitro cytotoxic effects of these treatments were determined by MTT assay (at 570 nm). The data were obtained from three independent experiments.
Annexin-V/PI staining
The cells were collected by trypsinization and centrifugation at 1,000 g for 5 minutes. Following resuspension in binding buffer (10 mmol/L HEPES-NaOH, 140 mmol/L NaCl, 2.5 mmol/L CaCl2) at a final cell density of 1 to 2*106 cells/mL, 100 μL of a single-cell suspension (1-2*105 cells) was incubated with 5 μL Annexin-V–FITC and 5 μL propidium iodide (PI) for 15 minutes at room temperature in the dark. After addition of 400 Ml of binding buffer, the samples were analyzed with a BD FACS Calibur flow cytometer (BD Biosciences) within 1 hour. For each sample, 10,000 events were counted.
Statistical analysis
Statistical analysis was calculated using by the SPSS statistical software program (Version 15.0; SPSS Inc.).
CONFLICTS OF INTEREST
The authors disclose no conflicts of interests.
FUNDING
This work was jointly supported by grants from Tung’s Taichung Metro-Harbor Hospital (TTM-TMU-106-02), Taipei Medical University (104-6602) and the Ministry of Science and Technology (MOST103 - 2320 - B - 038 - 036 - MY2), Taiwan.
REFERENCES
1. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, Lennon VA, Celis E, Chen L. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002; 8: 793–800.
2. Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A. 2002; 99: 12293–12297.
3. Konishi J, Yamazaki K, Azuma M, Kinoshita I, Dosaka-Akita H, Nishimura M. B7-H1 expression on non-small cell lung cancer cells and its relationship with tumor-infiltrating lymphocytes and their PD-1 expression. Clin Cancer Res. 2004; 10: 5094–5100.
4. Azuma K, Ota K, Kawahara A, Hattori S, Iwama E, Harada T, Matsumoto K, Takayama K, Takamori S, Kage M, Hoshino T, Nakanishi Y, Okamoto I. Association of PD-L1 overexpression with activating EGFR mutations in surgically resected nonsmall-cell lung cancer. Ann Oncol. 2014; 25: 1935–1940.
5. Chen J, Jiang CC, Jin L, Zhang XD. Regulation of PD-L1: a novel role of pro-survival signalling in cancer. Ann Oncol. 2016; 27: 409–416.
6. Takano T, Fukui T, Ohe Y, Tsuta K, Yamamoto S, Nokihara H, Yamamoto N, Sekine I, Kunitoh H, Furuta K, Tamura T. EGFR mutations predict survival benefit from gefitinib in patients with advanced lung adenocarcinoma: a historical comparison of patients treated before and after gefitinib approval in Japan. J Clin Oncol. 2008; 26: 5589–5595.
7. Tomizawa Y, Iijima H, Sunaga N, Sato K, Takise A, Otani Y, Tanaka S, Suga T, Saito R, Ishizuka T, Dobashi K, Minna JD, Nakajima T, et al. Clinicopathologic significance of the mutations of the epidermal growth factor receptor gene in patients with non-small cell lung cancer. Clin Cancer Res. 2005; 11: 6816–6822.
8. de Bruin EC, Cowell C, Warne PH, Jiang M, Saunders RE, Melnick MA, Gettinger S, Walther Z, Wurtz A, Heynen GJ, Heideman DA, Gomez-Roman J, Garcia-Castano A, et al. Reduced NF1 expression confers resistance to EGFR inhibition in lung cancer. Cancer Discov. 2014; 4: 606–619.
9. Oxnard GR, Arcila ME, Sima CS, Riely GJ, Chmielecki J, Kris MG, Pao W, Ladanyi M, Miller VA. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: distinct natural history of patients with tumors harboring the T790M mutation. Clin Cancer Res. 2011; 17: 1616–1622.
10. Suda K, Murakami I, Katayama T, Tomizawa K, Osada H, Sekido Y, Maehara Y, Yatabe Y, Mitsudomi T. Reciprocal and complementary role of MET amplification and EGFR T790M mutation in acquired resistance to kinase inhibitors in lung cancer. Clin Cancer Res. 2010; 16: 5489–5498.
11. Chiu CF, Chang YW, Ku KT, Shen YS, Liu CY, Yu YH, Cheng CC, Lee KY, Chen FC, Hsu MK, Kuo TC, Ma JT, Su JL. NF-κB-driven suppression of FOXO3a contributes to EGFR mutation-independent gefitinib resistance. Proc Natl Acad Sci USA. 2016; 113: E2562–2535.
12. Li J, Li X, Ren S, Chen X, Zhang Y, Zhou F, Zhao M, Zhao C, Chen X, Cheng N, Zhao Y, Zhou C, Hirsch FR. MiR-200c overexpression is associated with better efficacy of EGFR-TKIs in non-small cell lung cancer patients with EGFR wild-type. Oncotarget. 2014; 5:7902–7916. http://doi.org/10.18632/oncotarget.2302.
13. Yang H, Wang R, Peng S, Chen L, Li Q, Wang W. Hepatocyte growth factor reduces sensitivity to the epidermal growth factor receptor-tyrosine kinase inhibitor, gefitinib, in lung adenocarcinoma cells harboring wild-type EGFR. Oncotarget. 2016; 7:16273–16281. http://doi.org/10.18632/oncotarget.7586.
14. Wang F, Wang S, Wang Z, Duan J, An T, Zhao J, Bai H, Wang J. Phosphorylated EGFR expression may predict outcome of EGFR-TKIs therapy for the advanced NSCLC patients with wild-type EGFR. J Exp Clin Cancer Res. 2012; 31: 65–74.
15. Lin K, Cheng J, Yang T, Li Y, Zhu B. EGFR-TKI down-regulates PD-L1 in EGFR mutant NSCLC through inhibiting NF-kappaB. Biochem Biophys Res Commun. 2015; 463: 95–101.
16. Chen N, Fang W, Zhan J, Hong S, Tang Y, Kang S, Zhang Y, He X, Zhou T, Qin T, Huang Y, Yi X, Zhang L. Upregulation of PD-L1 by EGFR Activation Mediates the Immune Escape in EGFR-Driven NSCLC: Implication for Optional Immune Targeted Therapy for NSCLC Patients with EGFR Mutation. J Thorac Oncol. 2015; 10: 910–923.
17. Karakhanova S, Meisel S, Ring S, Mahnke K, Enk AH. ERK/p38 MAP-kinases and PI3K are involved in the differential regulation of B7-H1 expression in DC subsets. Eur J Immunol. 2010; 40: 254–66.
18. Ota K, Azuma K, Kawahara A, Hattori S, Iwama E, Tanizaki J, Harada T, Matsumoto K, Takayama K, Takamori S, Kage M, Hoshino T, Nakanishi Y, et al. Induction of PD-L1 Expression by the EML4-ALK Oncoprotein and Downstream Signaling Pathways in Non-Small Cell Lung Cancer. Clin Cancer Res. 2015; 21: 4014–4021.
19. Chen J, Gibbons DL, Goswami S, Cortez MA, Ahn MA, Byers LA, Zhang X, Yi X, Dwyer D, Lin W, Diao L, Wang J, Roybal J, et al. Metastasis is required via microRNA-200/ZEB1 axis control of tumor cell PD-L 1 expression and intratumoral immunosuppression. Natl Commun. 2014; 5:5241–5253.
20. Shimoji M, Shimizu S, Sato K, Suda K, Kobayashi Y, Tomizawa K, Takemoto T, Mitsudomi T. Clinical and pathological features of lung cancer expressing programmed cell death ligand 1 (PD-L1). Lung Cancer. 2016; 98: 69–75.
21. Ren S, Su C, Wang Z, Li J, Fan L, Li B, Li X, Zhao C, Wu C, Hou L, He Y, Gao G, Chen X, et al. Epithelial phenotype as a predictive marker for response to EGFR-TKIs in non-small cell lung cancer patients with wild-type EGFR. Int J Cancer. 2014; 135:2962–2971.
22. Chang TH, Tsai MF, Su KY, Wu SG, Huang CP, Yu SL, Yu YL, Lan CC, Yang CH, Lin SB, Wu CP, Shih JY, Yang PC. Slug confers resistance to the epidermal growth factor receptor tyrosine kinase inhibitor. Am J Respir Crit Care Med. 2011; 83: 1071–1079.
23. Noman MZ, Messai Y, Carre T, Akalay I, Meron M, Janji B, Hasmim M, Chouaib S. Microenvironmental hypoxia orchestrating the cell stroma cross talk, tumor progression and antitumor response. Crit Rev Immunol. 2011; 31: 357–377.
24. Taddei ML, Giannoni E, Comito G, Chiarugi P. Microenvironment and tumor cell plasticity: an easy way out. Cancer Lett. 2013; 341: 80–96.
25. Barsoum IB, Smallwood CA, Siemens DR, Graham CH. A mechanism of hypoxia-mediated escape from adaptive immunity in cancer cells. Cancer Res. 2014; 74: 665–74.
26. Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, Schumacker PT. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem. 2000; 275: 25130–25138.
27. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, Bronte V, Chouaib S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014; 211: 781–790.
28. Dixit D, Ghildiyal R, Anto NP, Sen E. Chaetocin-induced ROS-mediated apoptosis involves ATM-YAP1 axis and JNK-dependent inhibition of glucose metabolism. Cell Death Dis. 2014; 5: e1212.
29. Yan F, Pang J, Peng Y, Molina JR, Yang P, Liu S. Elevated Cellular PD1/PD-L1 Expression Confers Acquired Resistance to Cisplatin in Small Cell Lung Cancer Cells. PLoS One. 2016; 11: e0162925.
30. Zhang P, Ma Y, Lv C, Huang M, Li M, Dong B, Liu X, An G, Zhang W, Zhang J, Zhang L, Zhang S, Yang Y. The up-regulation of PD-L1 promotes the resistant response in non-small cell lung cancer patients with neo-adjuvant chemotherapy. Cancer Sci. 2016; 107:1563–1571.
31. Song S, Honjo S, Jin J, Chang SS, Scott AW, Chen Q, Kalhor N, Correa AM, Hofstetter WL, Albarracin CT, Wu TT, Johnson RL, Hung MC, et al. The Hippo Coactivator YAP1 Mediates EGFR Overexpression and Confers Chemoresistance in Esophageal Cancer. Clin Cancer Res. 2015; 21:2580–90.
32. Lee JE, Park HS, Lee D, Yoo G, Kim T, Jeon H, Yeo MK, Lee CS, Moon JY, Jung SS, Kim JO, Kim SY, Park DI, et al. Hippo pathway effector YAP inhibition restores the sensitivity of EGFR-TKI in lung adenocarcinoma having primary or acquired EGFR-TKI resistance. Biochem Biophys Res Commun. 2016; 474: 154–60.
33. Ma B, Chen Y, Chen L, Cheng H, Mu C, Li J, Gao R, Zhou C, Cao L, Liu J, Zhu Y, Chen Q, Wu S. Hypoxia regulates Hippo signalling through the SIAH2 ubiquitin E3 ligase. Nat Cell Biol. 2015; 17: 95–103.
34. Li Q, Tsuneki M, Krauthammer M, Couture R, Schwartz M, Madri JA. Modulation of Sox10, HIF-1alpha, Survivin, and YAP by Minocycline in the Treatment of Neurodevelopmental Handicaps following Hypoxic Insult. Am J Pathol. 2015; 185:2364–2378.
35. Keren-Paz A, Emmanuel R, Samuels Y. YAP and the drug resistance highway. Nat Genet. 2015; 47: 193–194.
36. Lin L, Sabnis AJ, Chan E, Olivas V, Cade L, Pazarentzos E, Asthana S, Neel D, Yan JJ, Lu X, Pham L, Wang MM, Karachaliou N, et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat Genet. 2015; 47: 250–256.
37. Hugo W, Shi H, Sun L, Piva M, Song C, Kong X, Moriceau G, Hong A, Dahlman KB, Johnson DB, Sosman JA, Ribas A, Lo RS. Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell. 2015; 162: 1271–1285.
38. Cheng H, Zhang Z, Rodriguez-Barrueco R, Borczuk A, Liu H, Yu J, Silva JM, Cheng SK, Perez-Soler R, Halmos B. Functional genomics screen identifies YAP1 as a key determinant to enhance treatment sensitivity in lung cancer cells. Oncotarget. 2016; 7:28976–28988. http://doi.org/10.18632/oncotarget.6721.
39. Wu DW, Wu TC, Chen CY, Lee H. PAK1 is a novel therapeutic target in tyrosine kinase inhibitor-resistant lung adenocarcinoma activated by the PI3K/AKT signaling regardless of EGFR mutation. Clin Cancer Res. 2016; 22: 5370–5382.
40. Lin PL, Wu TC, Wu DW, Wang L, Chen CY, Lee H. An increase in BAG-1 by PD-L1 confers resistance to tyrosine kinase inhibitor in non-small cell lung cancr via persistent activation of ERK signaling. Eur J Cancer. 2017; 85: 95–105.