
INTRODUCTION
Epithelial ovarian cancer (EOC) is one of the most lethal gynecologic diseases in the world [1]. Although at the initial stage of the disease, the first line chemotherapy like paclitaxel is responsive, the cancer cells acquire drug resistance at their late stage, which leads poor prognosis of EOC patients [2]. Identification of these resistance mechanisms may offer valuable insight to design strategies that modify the chemoresponse of cancer cells and reverse drug resistance. Myriads of regimens have been reported as therapeutic combinations to regulate drug resistance of cancer cells, and several strategies of genetic modulation have been suggested with the potential to switch the resistance [2–4].
Among them, paclitaxel resistance can be explained partially by β-tubulin-dependent mechanism, which is plausible as paclitaxel directly target microtubule and cell cycle [5]. Nevertheless, paclitaxel resistance remains unresolved. It seems to need more details of mechanism until a clinical use. CDKs forms the heterodimeric complex with cyclin proteins and the active conformation directs many transcription factors and leading to regulation of cell division. By the reasons, functional disorder of CDK causes neurodegeneration with abnormal cell death. More frequently, uncontrolled CDK activity is found in cancer cells out of microenvironmental control. A protective role of CDK to suppress unexpected cell death has been reported. CDK1 directly phosphorylates caspase-9 and the phosphorylated caspase-9 at Thr125 turns itself inactive. This link between CDK1 and caspase-9 is thought to suppress unnecessary cell death during eukaryotic cell [6, 7]. It is likely because among twenty CDKs, CDK1 is only essential CDK for cell cycle regulation in mammals, whereas CDK2 and 3, which exhibit high homology with CDK1, are dispensable in control of cell cycle [8]. However, rather high expression of CDK1 expression can cause paclitaxel resistance in cancer cells. A previous study showed that alsterpaullone, a CDK1 inhibitor, reversed drug resistance in vitro and in vivo [9]. Here, we intend to define the details of CDK1-induced drug resistance and trace the molecules that act downstream of CDK1. Although CDK1 was reported as a druggable target to reverse paclitaxel-induced chemoresistance, we newly suggest that caspase-9 is a rate-limiting step to control the acquirement of paclitaxel resistance.
MATERIALS AND METHODS
Cell lines and materials
Ovarian cancer cell lines A2780, HeyA8, SKOV3, A2780-CP, HeyA8-MDR and SKOV3-TR were a kind gift from Dr. Jeong Won Lee (Samsung Medical Center, South Korea). A2780 was established from ovarian endometroid adenocarcinoma tumor and A2780-CR is cisplatin-resistant cell line derived from the parental A2780. HeyA8-MDR is multi drug resistant ovarian cancer cell line induced by paclitaxel. SKOV3, the parental cell of SKOV3-TR was originally derived from ovarian serous cystadenocarcinoma and SKOV3-TR was a taxen-resistant type induced by paclitaxel treatment [10]. All ovarian cancer cells were incubated in RPMI-1640 containing 10% FBS (Gibco) and 250 ng/ml gentamycin. To maintain chemoresistance, SKOV3-TR and HeyA8-MDR cells were supplemented with 150 and 300 ng/mL paclitaxel. Paclitaxel, alsterpaullone, propidium iodide (PI), and Hoechst 33342 were purchased from Sigma Aldrich. Cisplatin was obtained from LC Laboratories (MA, USA).
Cell viability assay
Cell viability was measured by using an MTT assay (Promega, Ltd.) according to the manufacturer’s method. Cells were seeded at 5 × 103 cells in 96-well culture plates and treated with cisplatin for 12 h. The cells were then incubated with 5 mg/mL MTT for 4 h, removed growth medium, 150 μL of solubilization solution were added and incubated for 4 h. The absorbance of each well was measured at 570 nm wavelength. For the cell death analysis, cells were stained with 10 μM Hoechst 33342 and 1 μM PI for 20 min. Cells were then washed twice with serum-free media and stained cells were detected with a fluorescence microscope (Nikon, Japan).
Western blot analysis
Cells were lysed in radioimmunoprecipitation assay buffer containing 20 mM Tris-HCl [pH 7.5], 150 mM sodium chloride, 1 mM disodium ethylenediaminetetraacetic acid, 1 mM ethylene glycol tetraacetic acid, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM sodium orthovanadate and protease inhibitor (Cell Signaling Technology, Inc.). Total cell lysates were denaturated in 5x SDS sample buffer, separated on SDS-PAGE gels and transferred to PVDF membranes. The membranes were incubated with antibodies against caspase-9 (BD Biosciences), phospho-caspase-9/Thr125 (Abcam), CDK1 and β-actin (Santa Cruz Biotechnology).
Animal experiments
All animal experiments were performed in compliance with the institutional guidelines approved by the Institutional Animal Care and Use Committee at Wonkwang University (WK 2014-06-BR-006). Xenograft model was prepared by injection of 2 × 106 HeyA8-MDR cells into the flank of 8-week-old C57BL/6 mice. 3 weeks later, the mice were injected with 5 mg/kg alsterpaullone or 0.9% saline before administration of paclitaxel and administered with 7.5 mg/kg paclitaxel during another 3 weeks.
Immunohistochemistry
To analyze xenografted tumors, tumor tissues were fixed, dehydrated and embedded in paraffin. The paraffin embedded tissues were then sectioned at a thickness of 5 μm and blocked endogenous peroxidase activity with 1% hydrogen peroxide for 5 min. The tissue sections were incubated with antibodies against caspase-9 and caspase-9/Thr125 and developed with DAB (3,3′-diaminobenzidine) for 7 min. Nuclei were counterstained with Mayer’s hematoxylin, slides were mounted and imaged under a microscope (Nikon Eclipse 80).
RESULTS
Relevance of CDK1 in paclitaxel sensitivity
We compared chemosensitive and chemoresistant cells for CDK1 expression levels using western blot analysis. CDK1 expression was commonly upregulated in the paclitaxel-resistant HeyA8-MDR cells as compared with drug-sensitive cells (Figure 1A). As this phenomenon relies on the response of cells to drug treatment, we monitored cells in a time-dependent manner after paclitaxel treatment. Although a fluctuation in CDK1 expression was observed based on drug treatment time, CDK1 basal level was significantly higher in paclitaxel-resistant cells. However, no difference was observed in CDK1 level between cisplatin-sensitive A2780 and cisplatin-resistant A2780-CP cells (Figure 1B). In A2780 cell pairs, CDK2 expression, rather than CDK1, was significantly increased in A2780-CP cells. This result of the CDK1 expression was observed in a dose-dependent manner (Figure 1C and 1D) and the CDK1 and CDK2 protein level was reported to be almost similar during the time course. Taken together, CDK1 appears to be specifically upregulated in paclitaxel-resistant cells.
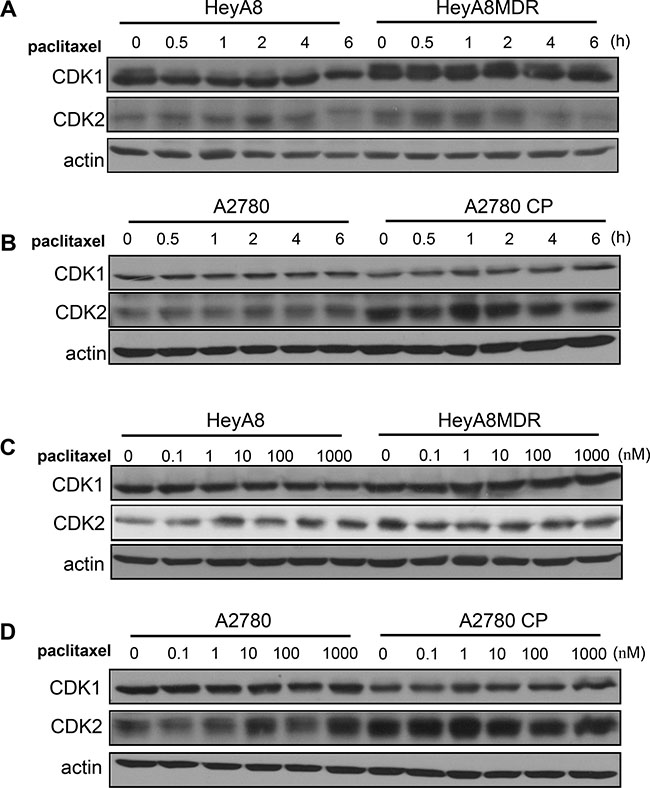
Figure 1: Expression of CDK1/2 in paclitaxel-resistant cells. (A–B) CDK1 and CDK2 levels were analyzed by western blot. Actin was used as a loading control. (C–D) CDK1/2 levels were detected by western blot analysis after treatment of cells with different concentration of paclitaxel.
Paclitaxel resistance is mediated by the phosphorylation of caspase-9
The phosphorylation of caspase-9 via CDK1 kinase activity mediates the suppression of cell death during mitosis [11]. Therefore, we investigated whether the reversal of paclitaxel resistance by alsterpaullone treatment occurs through the modulation of capase-9 phosphorylation. Basal levels of phosphorylated caspase-9 were higher in paclitaxel-resistant cells, HeyA8-MDR
and SKOV3-TR, as compared to their chemosensitive counterparts (Figure 2A). Repeated western blot analysis showed a low intensity band, which may be attributed to the phosphorylation of caspase-9 during the metaphase of cell cycle. Thus, we first attempted to synchronize the cell cycle in G2/M phase by nocodazole treatment. Analysis of cells during metaphase revealed a successful increase in the phosphorylation levels of caspase-9 (Figure 2B, third lane of each panel). In the absence of nocodazole, phospho-caspase-9 levels failed to show any significant change even after alsterpaullone treatment. Thus, CDK1 inhibitor alsterpaullone inhibited the phosphorylation of caspase-9 in chemoresistant HeyA8-MDR and SKOV3-TR cells during nocodazole-pretreated metaphase (Figure 2B). We tried to validate whether the reversal of paclitaxel resistance in the alsterpaullone-pretreated cells is caused by cell population in metaphase. During metaphase, most cells display a rounded morphology. Based on these morphological changes, we confirmed the cells arrested in metaphase. After treatment with nocodazole, the population of floating cells increased to 58.9%. Upon treatment with paclitaxel, the population of dead cells increased from 9.8% to 38.1%, while the proportion of floating cells sharply decreased from 58.9% to 33.8%. The proportion of adhesive cells was unchanged (Figure 2C and 2D). This data support our hypothesis that alsterpaullone suppresses the activity of CDK1 in metaphase cells.
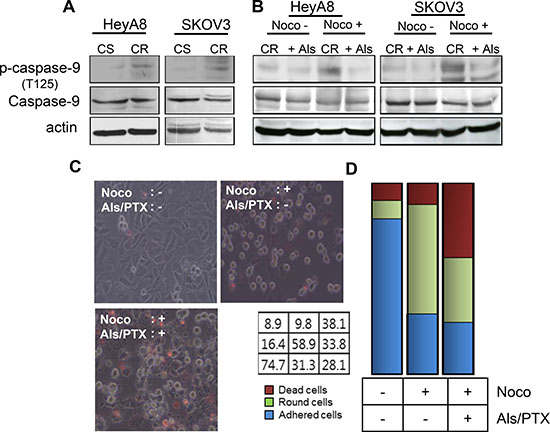
Figure 2: CDK1 mediates paclitaxel-induced chemoresistance through caspase-9. (A) Total caspase-9 and phospho-caspase-9 levels were analyzed by western blot. Actin was used as a loading control. CS, chemosenstive; CR, chemoresistant. (B) HeyA8-MDR and SKOV3-TR cells were treated with nocodazole to arrest cells in their mitosis. Noco, nocodazole; Als, alsterpaullone; PTX, paclitaxel. (C) Cell death was compared between three experimental groups: alsterpaullone-pretreated cells (upper left), alsterpaullonepretreated cells with added nocodazole (upper right), and alsterpaullone-pretreated cells with added paclitaxel and nocodazole (lower left). Administration of paclitaxel was followed by nocodazole treatment. (D) Populations of adherent (blue) and floating (green) cells are shown. Dead cells were stained with red dye.
Correlation between caspase-9 phosphorylation and paclitaxel-induced cell death
Although CDK1 may induce phosphorylation of caspase-9, no correlation was observed between CDK1 expression level and survival of patients with ovarian cancer. We tested if the expression of CDK1 may shorten patient survival, given its close association with drug resistance. Gene expression data were selected from 567 patients who had a survival record from The Cancer Genome Atlas (TCGA). Based on CDK1 expression level, the data were divided into 85 patients in top 15% and another 85 patients in bottom 15%. In contrast to our expectation, no significant difference was observed between the two groups (Supplementary Figure 1).
To determine if the phosphorylation level of caspase-9 correlates with the resistance to cell death, we treated drug-sensitive HeyA8 cells with 10 μM (low dose) paclitaxel. In these cells, caspase-9 phosphorylation was induced by paclitaxel at different degrees. We numbered the degree of phosphorylation from 1 to 7 according to the measured density (Figure 3A), 1 being smaller than 7 at phosphorylation.
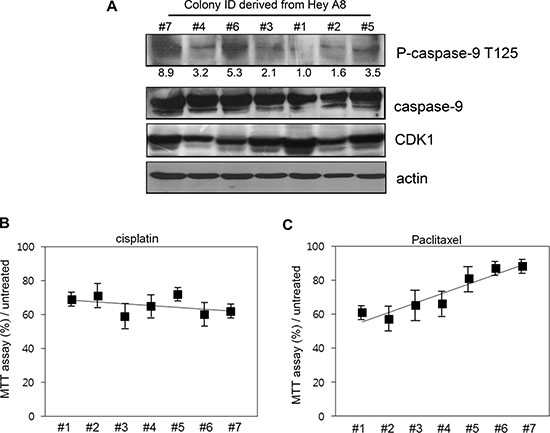
Figure 3: Correlation between caspase-9 phosphorylation level and resistance to cell death. (A) The colonies with different phosphorylation level were selected after low-dose paclitaxel treatment and cultured for 7 additional days. The ID number was conferred based on the phosphorylation level detected with western blot analysis. Relative density of phospho-caspase-9 was analyzed by Image J 3.0. (B) Different cells were treated with cisplatin for 12 h and cell viability assessed using MTT assay. (C) Cells were treated with paclitaxel and cell death assessed using MTT assay.
We compared the cell propensity in the presence of 200 μM (high dose) paclitaxel. The same number of cells was spread on the plates and the cell death was analyzed. Although cell death was similar between the cells upon cisplatin treatment (Figure 3B), MTT assay percent value showed a negative correlation with the phosphorylation level of caspase-9 (Figure 3C).
Caspase-9 is critical for paclitaxel-induced cell death
To directly evaluate whether caspase-9 is critical for alsterpaullone-mediated reversal of paclitaxel chemoresistance, we targeted caspase-9 with siRNA in HeyA8-MDR and SKOV3-TR cells. Suppression of caspase-9 with siRNA compensated for alsterpaullone-mediated chemoresistance reversal by 20% and 14% in HeyA8-MDR and SKOV3-TR cells, respectively (Figure 4A and 4B). We used T125A site mutant vector of caspase-9, which shows defective phosphorylation. T125A-transfected cells showed significantly increased cell death upon alsterpaullone/paclitaxel treatment in HeyA8 and SKOV3 cells (Figure 4C and 4D).
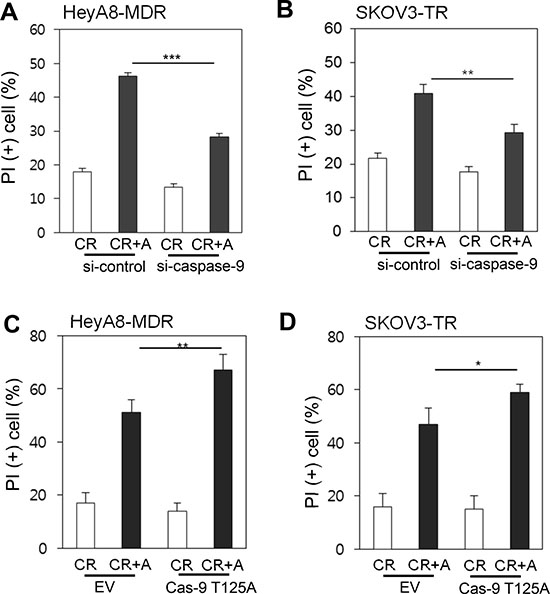
Figure 4: Caspase-9-dependent cell death (A–B) siRNA against caspase-9 was administrated in chemoresistant paired cell lines, HeyA8 and SKOV3. A mixture of random siRNAs was used as a control. (C–D) Cells were transfected with T125Amutant caspase-9, followed by treatment with alsterpaullone and paclitaxel. *, p < 0.05;**, p < 0.01;***, p < 0.001.
In vivo validation of alsterpaullone-mediated chemoresistance reversal
We injected HeyA8-MDR cells in the flank region of mice. Three weeks before administration of paclitaxel, either 0.9% saline or alsterpaullone (5 mg/kg) was intraperitoneally injected every other day. At week 7, the tumors were isolated and subjected to immunohistochemical analysis. Phospho-caspase-9 level was lower in tumors treated with alsterpaullone as compared with the untreated tumors (Figure 5).
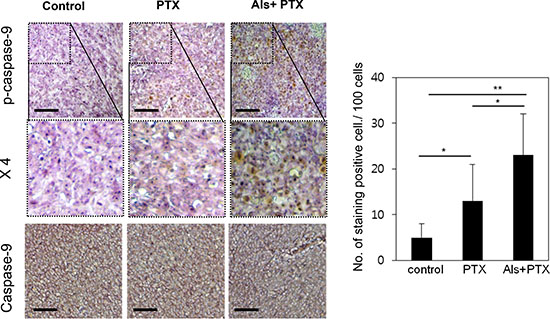
Figure 5: In vivo validation of alsterpaullone-mediated chemoresistance reversal. Immunohistochemical analysis of molecular markers in control, paclitaxel-treated, and alsterpaullone-pretreated tumors. Representative images of tumors in each treatment group. PTX, paclitaxel; AP, alsterpaullone.*,p < 0.05;**, p < 0.01.
Inhibition of caspase-9 phosphorylation by alsterpaullone
The activity of CDK1 is reported to increase aberrantly as cancer cells acquire resistance to paclitaxel. During this process, caspase-9 is readily phosphorylated, thereby blocking paclitaxel-mediated cell death (Figure 6, upper panel). Continuous alsterpaullone treatment for more than 2 weeks reduced CDK1 activity. As a consequence, cells again showed a partial response to paclitaxel (Figure 6, lower panel).
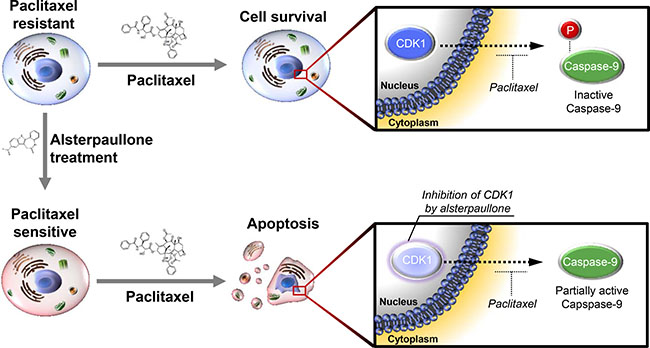
Figure 6: Schematic diagram explaining the mode of action of alsterpaullone. Phosphorylation of caspase-9 indicates its inactive form. In the lower panel, the light color of CDK1 is intended to express the relatively reduced activity of CDK1.
DISCUSSION
Cancer may be curable, if cancer cells fail to acquire drug resistance. Ovarian cancer is one of the most referred cancer types to demonstrate drug resistance. Of the various resistance-causing drugs, we studied the effect of paclitaxel as a combinatory or single chemical drug for clinical treatment of ovarian cancer [12, 13]. Paclitaxel inhibits the disassembly of microtubules during mitosis and is a naturally attractive target for anticancer therapy. In ovarian cancer, paclitaxel resistance is easily observed in clinics and is the cause of poorer survival. In our previous study, we suggested that CDK1 inhibitor alsterpaullone may reverse paclitaxel resistance. During mitosis, CDK1 phosphorylates and cleaves caspase-9, leading to its inactivation and activation of caspase 3 and 7 [14]. Thus, the overexpression of caspase-9 even in drug-sensitive cancer cells deems cells vulnerable to aberrant apoptosis under normal condition. Otherwise, suppression of caspase-9 inhibits apoptosis (Figure 4A and 4B). Thus, caspase-9 must be a critical factor for the initiation of cell death. Cancer cells with acquired resistance may show different phases of cell death. In paclitaxel-resistant cells, caspase-9 seems inefficient to induce caspase cascade for cell death. As shown in Figure 3A, the total caspase-9 level was not proportional to phosphorylated caspase-9 level and cellular viability. In addition, patients with high expression of caspase-9 showed a short survival (p = 0.1; Supplementary Figure 1B). Although CDK1 expression level failed to show precise correlation with drug resistance, the level of phospho-caspase-9 is a better predictor of paclitaxel resistance. Thus, phosphorylation of caspase-9 is a good marker of paclitaxel resistance.
Phosphorylation of caspase-9 at Thr125 may be also mediated by extracellular signal-regulated kinase 1 (ERK1) [15]. These results suggest that the phosphorylation of Thr125 on caspase-9 may be an important mechanism through which growth factors and survival signals activate the ERK–mitogen-activated protein kinase (MAPK) pathway. This phenomenon may explain the constitutive overexpression of Erk1 observed in tumorigenesis. Thus, phosphorylation of caspase-9 at Thr125 may be the target of various oncogenic kinases [16] and a valuable marker for the prediction of cancer patient prognosis. Although alsterpaullone was selected as an effective adjuvant therapy for paclitaxel-treated patients, other phosphatases for caspase-9 may be more effective for their anti-cancer or drug resistance reversal effect.
ACKNOWLEDGMENTS AND FUNDING
This study was supported by a grant from the National R&D Program for Cancer Control, Ministry of Health and Welfare (grant no. 1720130) and by the Basic Science Research Program, Ministry of Education (grant no. NRF-2016R1D1A1B03930490), Republic of Korea.
CONFLICTS OF INTEREST
Authors declare no conflicts of interest.
REFERENCES
1. Wingo PA, Tong T, Bolden S. Cancer statistics, 1995. CA Cancer J Clin. 1995; 45:8–30.
2. Agarwal R, Kaye SB. Ovarian cancer: strategies for overcoming resistance to chemotherapy. Nat Rev Cancer. 2003; 3:502–16. https://doi.org/10.1038/nrc1123.
3. Ali AY, Farrand L, Kim JY, Byun S, Suh JY, Lee HJ, Tsang BK. Molecular determinants of ovarian cancer chemoresistance: new insights into an old conundrum. Ann N Y Acad Sci. 2012; 1271:58–67. https://doi.org/10.1111/j.1749-6632.2012.06734.x.
4. Saraswathy M, Gong S. Different strategies to overcome multidrug resistance in cancer. Biotechnol Adv. 2013; 31:1397–407. https://doi.org/10.1016/j.biotechadv.2013.06.004.
5. Kavallaris M, Kuo DY, Burkhart CA, Regl DL, Norris MD, Haber M, Horwitz SB. Taxol-resistant epithelial ovarian tumors are associated with altered expression of specific beta-tubulin isotypes. J Clin Invest. 1997; 100:1282–93. https://doi.org/10.1172/JCI119642.
6. Monaco EA 3rd, Vallano ML. Cyclin-dependent kinase inhibitors: cancer killers to neuronal guardians. Curr Med Chem. 2003; 10:367–79.
7. Monaco EA 3rd, Vallano ML. Role of protein kinases in neurodegenerative disease: cyclin-dependent kinases in Alzheimer’s disease. Front Biosci. 2005; 10:143–59.
8. Diril MK, Ratnacaram CK, Padmakumar VC, Du T, Wasser M, Coppola V, Tessarollo L, Kaldis P. Cyclin-dependent kinase 1 (Cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration. Proc Natl Acad Sci U S A. 2012; 109:3826–31. https://doi.org/10.1073/pnas.1115201109.
9. Bae T, Weon KY, Lee JW, Eum KH, Kim S, Choi JW. Restoration of paclitaxel resistance by CDK1 intervention in drug-resistant ovarian cancer. Carcinogenesis. 2015; 36:1561–71. https://doi.org/10.1093/carcin/bgv140.
10. Han LY, Landen CN, Trevino JG, Halder J, Lin YG, Kamat AA, Kim TJ, Merritt WM, Coleman RL, Gershenson DM, Shakespeare WC, Wang Y, Sundaramoorth R, et al. Antiangiogenic and antitumor effects of SRC inhibition in ovarian carcinoma. Cancer Res. 2006; 66:8633–9. https://doi.org/10.1158/0008-5472.CAN-06-1410.
11. Allan LA, Clarke PR. Phosphorylation of caspase-9 by CDK1/cyclin B1 protects mitotic cells against apoptosis. Mol Cell. 2007; 26:301–10. https://doi.org/10.1016/j.molcel.2007.03.019.
12. Sawada S, Mese H, Sasaki A, Yoshioka N, Matsumura T. Combination chemotherapy of paclitaxel and cisplatin induces apoptosis with Bcl-2 phosphorylation in a cisplatin-resistant human epidermoid carcinoma cell line. Cancer Chemother Pharmacol. 2003; 51:505–11. https://doi.org/10.1007/s00280-003-0614-z.
13. Yamamoto K, Kikuchi Y, Kudoh K, Nagata I. Modulation of cisplatin sensitivity by taxol in cisplatin-sensitive and – resistant human ovarian carcinoma cell lines. J Cancer Res Clin Oncol. 2000; 126:168–72.
14. Sherr CJ. Cancer cell cycles. Science. 1996; 274:1672–7.
15. Allan LA, Morrice N, Brady S, Magee G, Pathak S, Clarke PR. Inhibition of caspase-9 through phosphorylation at Thr 125 by ERK MAPK. Nat Cell Biol. 2003; 5:647–54. https://doi.org/10.1038/ncb1005.
16. Allan LA, Clarke PR. Apoptosis and autophagy: Regulation of caspase-9 by phosphorylation. FEBS J. 2009; 276:6063–73. https://doi.org/10.1111/j.1742-4658.2009.07330.x.