
INTRODUCTION
The nucleotide excision repair (NER) pathway is a central DNA repair mechanism eliminating a variety of bulky DNA lesions like pyrimidine photo products [1]. If these sorts of lesions accumulate in the genome, this can result in a cancer prone mutator phenotype as seen in the autosomal recessive model disease xeroderma pigmentosum (XP). Patients present with a high frequency of UV-induced skin tumors as well as a reduced life span [2]. According to the respective disease-causing defective genes (XPA-XPG) seven XP complementation groups (XP-A to XP-G) and a variant form with a mutated translesion polymerase (PolH gene) have been identified so far [3].
NER is characterized by subsequent steps and involves more than 30 proteins. Damage detection can be subdivided into global genome repair (GGR), via the XPC-hHR23B-centrin2 complex together with the DNA damage binding complex (XPE), and transcription coupled repair (TCR), initiated by a stalled RNA polymerase II and transcription factor IIH (TFIIH). Damage demarcation is performed by XPA and XPD followed by DNA unwinding around the damage catalyzed by the ATPase (XPB) and helicase (XPD) activities of TFIIH. The damage is excised by two subsequent incisions performed by XPF/ERCC1 and XPG 5’ and 3’ to the DNA lesion releasing the damage containing oligonucleotide (24–32 nts) from the DNA double helix. Finally, the gap is filled by DNA polymerases (δ, ε, or κ) using the undamaged strand as a template and nick sealing by DNA ligases (I or III) [1]. Furthermore, the endonuclease complex XPF/ERCC1 is also implicated in interstrand cross-link (ICL) repair [4]. In addition, it is known that decreased NER levels are a risk factor for several cancer and tumor entities in the normal population [5]. An implication of the XPF and XPG genes as promising prognostic marker for skin cancer risk as well as for disease outcome is based on genome-wide association studies of single nucleotide polymorphisms (SNPs) and protein expression analyses [6–8].
The human genes for the endonucleases XPF (ERCC4) (OMIM: 278760) and XPG (ERCC5) (OMIM: 278780) encode for 916 and 1186 amino acid (aa) proteins cleaving 5’ and 3’ of UV induced lesions. They are located on chromosomes 16p13.2-p13.1 and 13q32.3-q33.1 [9, 10]. It has been demonstrated that these essential repair genes show a high number of physiologically occurring spontaneous alternatively spliced transcripts with yet unknown functions [11]. Furthermore, the differential expression level of splice variants in several tissues has been shown to be more suitable to distinguish between oncogene and non-oncogene samples than the primary gene transcript itself [12].
In this study we focus on the catalytically active subunits of the NER endonucleases, XPF and XPG. It has been difficult in the past to study the functions of the XPF/ERCC1 complex so far, as there was no appropriate patient cell line without residual repair capability as seen in the XP-G patient XP20BE [13]. We decided to use primary patient fibroblast with a transient transfection approach, as there are no immortalized XP-G patient cells without residual alleles or repair capabilities. Recently, we created a complete XPF knockout (KO) via the CRISPR/Cas9 system [14]. We now demonstrate that there are several spontaneous XPF and XPG mRNA splice variants with residual repair capability and/or inhibitory effects on NER as well as ICL repair in these cells. These XPG splice variants show expression differences in regard to various tissues and are differentially expressed among individuals. Therefore, the variants may present promising prognostic marker candidates for individual cancer risk, disease outcome, or therapeutic success.
RESULTS
Protein expression and subcellular localization of XPF and XPG splice variants
All until now known, physiologically occurring spontaneous mRNA splice variants (Figure 1A–1B) of the XPF (XPF-201, XPF-003, XPF-202) (ENA numbers LT827130-LT827133) and XPG (XPG IsoII, XPG IsoIII, XPG IsoIV, XPG IsoV, XPG IsoVI, XPG-201, XPG-202) (ENA numbers LM994821, LM994822, GenBank numbers AH009656, AF255431-AF255442) genes were amplified from wildtype (WT) primary fibroblast or MRC5Vi cDNA, transcribed from mRNA, and cloned into different expression plasmids: pcDNA3.1 (+), pcDNA3.1 (-)mycHisA2, and pcDNA3.1 (+)eGFP (C-terminal tag). All expression cassettes have been validated by Sanger sequence analyses. Figure 1A and 1B illustrate an overview of all XPF and XPG splice variants on protein level depicting domains lost due to truncating mutations. With the aim to analyze transient protein expression, we performed immune blot analyses of the different recombinant XPF and XPG constructs over 72h (Figure 2A–2B). It is shown that all XPF and XPG isoforms were successfully overexpressed over time and resulted in proteins of the calculated size in the case of XPF and its variants. Whilst WT XPF levels peak at 72 h, its isoform XPF-201 shows highest protein levels after 48h. Interestingly, protein levels of the shorter isoforms XPF-202 and XPF-003 decrease over time. WT XPG, XPG IsoII, XPG IsoIV, XPG IsoVI, XPG-201 and XPG-202 as well as the XP20BE patient allele show an increase in expression within the time scale, only expression of XPG IsoIII decreases over time. Only XPG IsoIV presents with a band at the expected size of 35kDa, while XPG IsoV, XPG IsoVI, XPG-201, and XPG-202 travel between 50 and 60 kDa above their expected size.
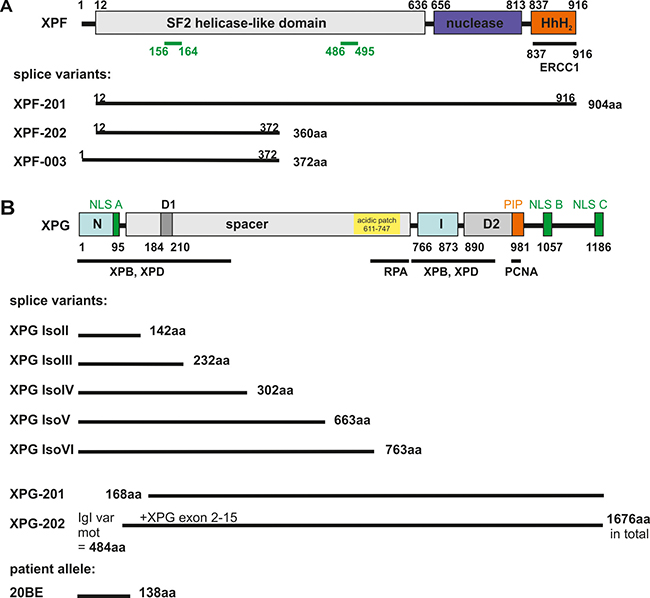
Figure 1: Schematic overview of XPF/XPG domains and their spontaneously and physiologically occurring mRNA splice variants. The colored bars show functional protein domains (e.g. nuclease domain (blue), Helix-hairpin-Helix (HhH)2 motif (orange)). Putative NLS are depicted in green and black lines highlight protein-protein interaction domains. (A) The XPF gene undergoes alternative splicing resulting in C- and/or N-terminally truncated physiologically occurring variants. The isoform XPF-201 only lacks the first 12 aa, while XPF-202 and XPF-003 are severely C-terminally truncated. These two variants lack functional domains, e.g. the nuclease domain. (B) The N and I domains (light blue-green) that form the catalytic center of the XPG endonuclease are separated by a spacer region in the primary sequence, after protein folding they come close together. XPG also has several C-terminally truncated variants (XPG IsoII - IsoVI) missing one or more of the functional domains. On the other hand, variant XPG-201 misses exons one - four (in frame) including one of the nuclease domains (N) and part of an interaction patch with TFIIH. XPG-202 only lacks exon one, but in addition contains the immunoglobulin like variant motif of 484 aa, caused by splicing defects leading to a conjoined gene. The XP20BE patient allele is severely truncated and patient primary fibroblasts were used for functional testing. Modified from [10, 11, 71–74].
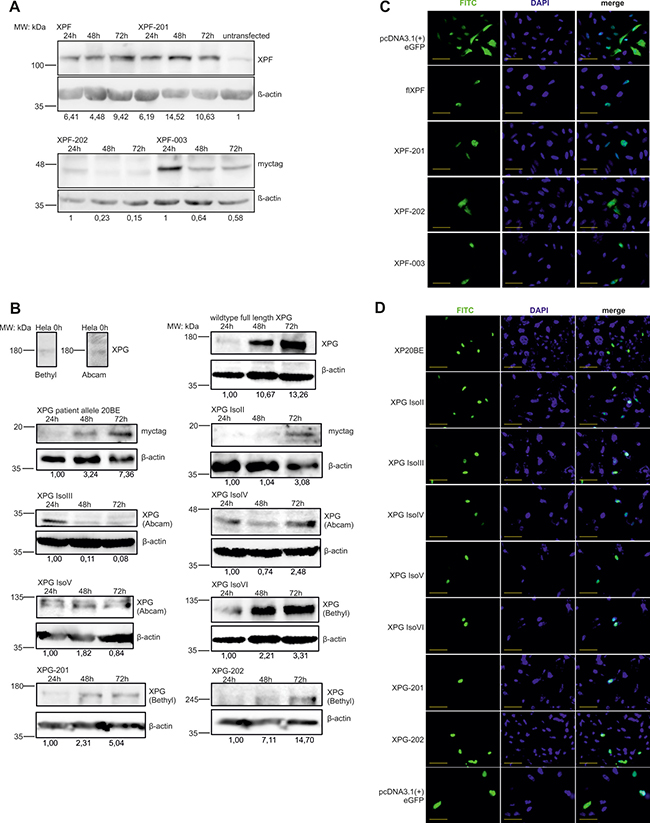
Figure 2: Protein levels of XPF and XPG splice variants over time and subcellular localization of eGFP-tagged isoforms and the XP-G patient allele. To check transfection and overexpression of the splice variants, protein levels in HeLa cells were determined using horizontal SDS Page followed by immunoblotting. (A and B) Cells were transiently transfected with the different constructs harvested after 24 h, 48 h and 72 h, and stained with an anti-XPF/XPG or an antibody directed against the myc-tag. Anti-β-actin staining was used for normalization. Protein levels were quantified in regard to untransfected control cells or to the 24 h value in the case of the myc-tagged constructs as there is no endogenous myc-tag protein. One of three representative experiments is shown. (C and D) For subcellular localization XPF and XPG isoforms as well as the XP20BE patient allele were cloned into an pcDNA3.1(+)eGFP expression vector and overexpressed in HeLa cells for 48 h. Additionally, DAPI staining was performed to visualize the nucleus. Scale bar = 50 μm.
To investigate nuclear localization of the splice variants, the compartment where DNA repair takes place, GFP-fusion proteins of the splice variants were produced in HeLa cells and evaluated by fluorescence microscopy. 48h after overexpression, all XPF and XPG isoforms as well as the WT proteins, could be found in the nucleus, whereas XPF-202 showed a diffuse distribution within the cell (Figure 2C–2D). Theoretically, all isoforms should be able to participate in different DNA repair pathways based on stable protein expression and nuclear localization if they had residual enzymatic or structural functions.
The involvement of XPF and XPG splice variants in NER and ICL repair
As illustrated in Figure 1, XPF and XPG isoforms either show C-or N-terminal truncations, leading to full or partial loss of protein domains, which are important for enzymatic activity or protein-protein interactions. We applied the functional Host Cell Reactivation (HCR) assay to investigate residual repair capability. The assay was originally applied to study the repair of UV induced DNA lesions (6–4photoproducts (6–4PPs) and cyclobutane pyrimidine dimers (CPDs)) repaired by NER. We also adapted the assay to the repair of intrastrand and interstrand crosslinks by treating the firefly reporter plasmid with cis-diammineplatinum (II) dichloride (CP) and 4,5’,8-trimethylpsoralen (TMP) plus UVA light as described in Lehmann et al. 2017 [14]. NER and ICL repair activity of all XPG (in XP20BE cells) and XPF isoforms (in MRC5Vi XPF KO Cells) was determined (Figure 3A–3F) in comparison to primary WT fibroblasts and WT MRC5Vi cells. Primary WT fibroblasts exhibited a NER repair capability of 15% (15.72 ± 0.96%), while XP20BE patient cells show no residual repair (0.09% ± 0.03) (see Figure 3A). This effect could only be rescued by complementation with full-length XPG (flXPG) or XPG isoforms V and VI in comparison to the empty vector control (XPG: 4.9 ± 1.22%, IsoV: 0.40 ± 0.07%, IsoVI: 0.78 ± 0.12%) close to WT levels of repair (15.72 ± 0.96%). For ICL repair, WT repair levels of about 15% (CP: 13.92 ± 1.71%, TMP + UVA: 17.25 ± 3.34%) were measured (Figure 3B–3C). Interestingly, co-transfection with flXPG was closer to WT levels than observed for UVC induced lesions (CP: 11.61 ± 2.75%, TMP + UVA: 12.71 ± 1.55%). Again, isoform V and VI showed a significant increase in repair capability (IsoV: CP 5.98 ± 0.98%, TMP + UVA 8.55 ± 1.54% and IsoVI: CP 4.57 ± 0.76%, TMP + UVA 9.94 ± 2.32%), while the other isoforms did not have a significant effect. Overall basic levels of repair were higher than for UVC (CP 1.98 ± 0.19%, TMP + UVA 5.12 ± 0.43%), suggesting that the involvement of XPG is more important in NER than ICL repair.
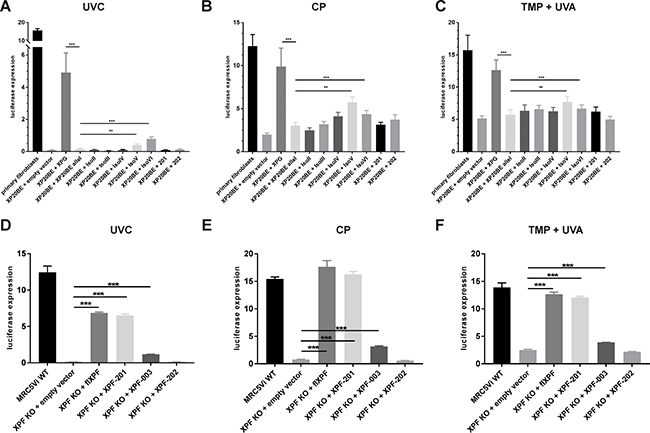
Figure 3: Reactivation of a reporter gene after treatment with UVC, cisplatin, or trimethylpsoralen activated by UVA light in XPF KO cells or XP-G patient cells complemented with XPF or XPG splice variants. Firefly plasmids were treated with (A and D) UVC irradiation, (B and E) cisplatin (intrastrand crosslinks), or (C and F) trimethylpsoralen activated by 1 J/cm2 UVA irradiation (interstrand crosslinks), to induce specific lesions, transfected into MRC5Vi WT/XP20BE primary fibroblasts (XP-G patient cells) or XPF KO cells, and complemented with plasmids coding for full-length proteins and the different splice variants. The relative repair capability is calculated as the percentage (luciferase expression) of the reporter gene activity (firefly luciferase) compared to the untreated plasmid, after normalization to an internal co-transfected control (Renilla luciferase). Data are presented as the mean ± SEM. The one-tailed, unpaired student’s t-test was applied, *** P < 0.001 or ** P < 0.01. At least four independent experiments in triplicates were performed.
Residual repair capabilities of XPF splice variants were studied in the same way in the complete XPF KO [14] cells compared to MRC5Vi WT cells (12.45 ± 0.86%). XPF KO cells show a nearly complete loss of repair capabilities (0.15 ± 0.02%), but repair can be rescued by complementation with full-length XPF (6.87 ± 0.13%) (Figure 3D–3F). Two XPF splice variants exhibited significant residual repair capabilities in NER as well as ICL repair (XPF-201: UVC 6.50 ± 0.22%, CP 16.28 ± 0.51%, TMP + UVA 12.06 ± 0.22% and XPF-003: UVC 1.15 ± 0.02%, CP 3.17 ± 0.10%, TMP + UVA 3.89 ± 0.03%). As shown in Figure 1B XPF-201 only lacks the first 12 aa of the protein, whereas XPF-003 is severely C-terminally truncated, lacking the endonuclease domain. Interestingly, XPF-202, another variant, that only differs from XPF-003 in the first 12 aa did not display any significant repair capability at all (UVC 0.15 ± 0.02%, CP 0.58 ± 0.03%, TMP + UVA 2.17 ± 0.06%) (Figure 3D–3F). This prompts towards a high importance of those 12 N-terminal aa of the XPF protein, e.g. due to an undescribed essential interaction domain of the protein, which may be necessary for structural or catalytic roles of the protein.
XPG and XPF splice variants exhibit inhibitory effects on NER
In order to investigate the interference of XPG and XPF spontaneous mRNA splice variants with endogenous DNA repair, exemplarily NER, cell lines stably overexpressing these variants were generated in MRC5Vi WT cells. To test the effect on endogenous NER ability of MRC5Vi WT cells, three positive clones with a similar level of overexpression were selected for each variant or full-length protein, and repair capabilities were then averaged for statistical purposes. Figure 4A and 4B exemplarily show the stable overexpression of the full-length proteins (XPG 180kDa, XPF 112kDa) as well as their splice variants (XPG IsoV 100kDa, XPF-201 112kDa) in MRC5Vi WT cells, displaying levels of overexpression between 1.13–9.65 fold. XPG-201, a variant N-terminally lacking the first four exons in frame resulting in a loss of part of XPG’s endonuclease domains (N-domain), showed a significant dominant negative effect on NER (10.75 ± 0.50%) compared to MRC5Vi WT cells (14.17 ± 1.20%) (see Figure 4C). The same effect could be observed for XPG IsoVI, the variant exhibiting the highest residual repair capability in XPG deficient cells, lacking the endonucleolytic I-domain (7.85 ± 0.72%). In turn, all three XPF splice variants, as well as the full-length protein, significantly reduced repair capabilities of MRC5Vi WT cells (XPF: 9.19 ± 0.83%, XPF-201: 5.26 ± 0.80%, XPF-003: 6.90 ± 0.06%, XPF-202: 8.08 ± 0.73%). Figure 4D shows that individual clones with different levels of overexpression have a different effect on influencing repair capability. Anyhow, the average of three clones presents a balanced and averaged measurement for inhibitory effects. In summary, two XPG splice variants (XPG IsoVI and XPG-201) as well as WT XPF and all three splice variants (XPF-201, XPF-003, XPF-202) inhibited the endogenous NER capabilities of MRC5Vi WT cells.
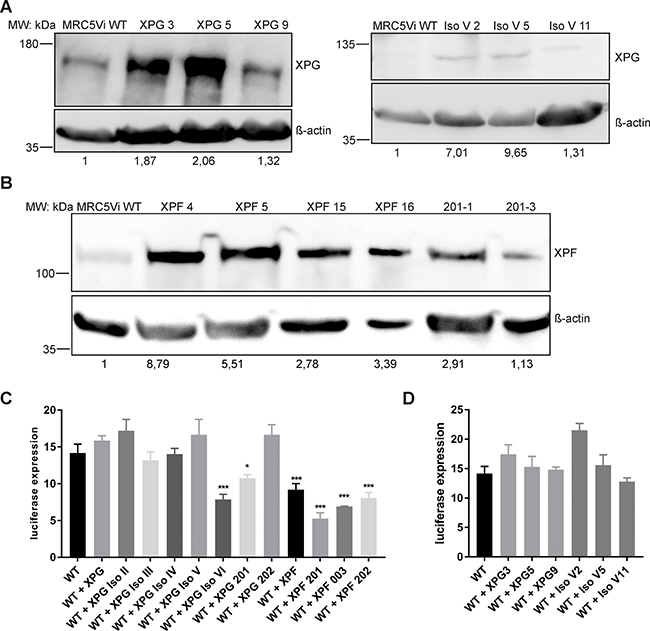
Figure 4: Inhibitory effects of the overexpression of XPG, XPF, or respective splice variants in MRC5Vi WT cells. Immunoblot analyses of single clones and reactivation of a reporter gene after treatment with UVC in MRC5Vi WT cells and single clones. Antibodies for (A) XPG or (B) XPF were used and β-actin staining was applied for normalization. Representatively, one of three independent experiments is shown. Protein levels in MRC5Vi WT cells were set to one. (C and D) For functional analyses, firefly plasmids were treated with UVC irradiation, transfected into MRC5Vi WT or single clones overexpressing XPG, XPF, or respective splice variants. The relative repair capability is calculated as the percentage (luciferase expression) of the reporter gene activity (firefly luciferase) compared to the untreated plasmid, after normalization to an internal co-transfected control (Renilla luciferase). Data are presented as the mean ± SEM. The one-tailed, unpaired student’s t-test was applied; significances are displayed with regard to WT cell repair capability, * P < 0.05 or *** P < 0.001. At least four independent experiments in triplicates were performed.
Differential expression of functionally relevant XPG splice variants in tissues and healthy individuals
The physiological relevance of XPG isoforms with repair catalyzing functions (isoform V and VI), was analyzed via detection of specific mRNA expression in 20 different healthy human tissues. Unfortunately, the material was only sufficient to analyze XPG, but not XPF, variants. Figure 5A shows the comparison of the three XPG species in the 20 different tissues. WT XPG, as well as both functionally relevant isoforms were expressed in all tissues. Heart, liver and skeletal muscle exhibited very low expression levels of all three XPG transcripts (light blue arrows), while the expression of both isoforms, V and VI, was high in spleen, testes, and thymus compared to the primary transcript (dark blue arrows). Furthermore, to compare human inter-individual expression levels of the three XPG transcripts, blood samples from 20 healthy donors (age 22–61, 7 male and 13 female) were compared (see Figure 5B). We could identify three different patterns. The expression profile either displayed high or low expression of all three transcripts (middle blue arrows), expression of a predominant primary transcript (dark blue arrows), or predominance of the two isoforms V and VI (light blue arrows). All in all, isoform VI, containing the highest residual catalytic activity in the functional experiments, was predominantly expressed in blood samples of 80% of the investigated subjects. Relations of expression between age and sex could not be observed in this small collective.
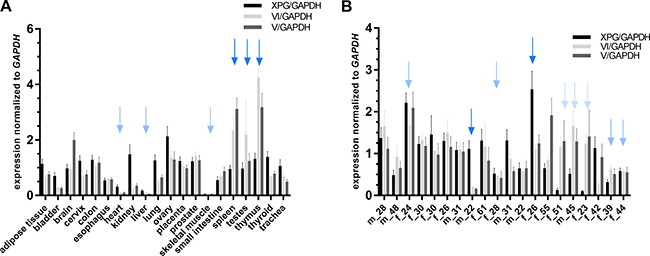
Figure 5: Analysis of XPG expression in different tissues and human blood samples. RNA samples from 20 tissues (A) and 20 unrelated human individuals (B) were transcribed into cDNA and subject to qRT-PCR measurements. Expression levels are shown as absolute values in relation to GAPDH used as the internal control to normalize the expression of the target gene or isoform. Results are shown as mean values from three independent experiments in duplicates. Data are presented as mean ± SEM. Colored arrows indicate groups with similar expression patterns.
DISCUSSION
The two endonucleases XPG and XPF are involved in several essential cellular processes like NER and ICL repair, in addition to basal transcription. They exert catalytic as well as structural functions. Mutations in these two genes result in complex genotype-phenotype correlations. Notably, patients with pathological XPG splice variants that still contain residual functions show a milder form of the XP/Cockayne syndrome (CS) complex phenotype and a longer skin cancer free survival time [10]. More precisely, a splice variant of a 28-year-old patient with minimal residual function has led to a meaningful prolongation of his life [15]. On the other hand, a dominant negative splice variant of an XP-C patient, that was still functionally active, reduced NER activity by approximately 50% [16]. Eventually, this variant was identified in about 40% of normal individuals and could be associated with a two-fold increased melanoma risk [17].
Cloned XPF and XPG splice variants produce stable proteins and localize to the nucleus
A prerequisite for analyses of XPF and XPG splice variants was to generate artificial constructs for further functional in vitro experiments. The generated constructs lead to stable overexpression of proteins (see Figure 2A–2B). In the case of XPF, protein products of the expected size were produced. XPG shows a band around 180kDa, which is above the calculated size of 136kDa, caused by its acidic patch in the spacer region (amino acids 611–747, see Figure 1B). Due to an abnormal mass-to-charge ratio the migration behavior in SDS-Page is changed [18–20]. This is also the case for the larger splice variants (XPF IsoV-VI, XPG-201, XPG-202). The acidic patch is not present in the rudimentary patient allele 20BE or in the short XPG IsoII, but they do not travel according to their size due to the myc/HisA2-tag, which is positively charged and binds more than the statistical amount of SDS. Additionally, the splice variants exhibit different peak protein level over time. These differences in expression could be explained by proteolytic degradation [21] or triggered by toxic effects of shorter isoforms possibly constituting inhibitory effects on DNA repair.
The nucleus is the cellular compartment where DNA repair takes place making it necessary that all proteins involved are either passively or actively imported. Some of the truncated splice variants (XPF-003 and XPF-202, as well as XPG II-VI) lack the C-terminal nuclear localization signal (NLS) (XPF aa486–495, XPG aa1057–1074 and aa1171–1185) (see Figure 1A). The smaller variants (XPG-II-IV) should be able to passively diffuse into the nucleus due to their size, whereas XPF-003, XPF-202, XPG-V, XPG-VI, XPG-201, and XPG-202 depend on a NLS, suggesting a less efficient transport for these variants as they only contain one of two (XPF) or three (XPG) NLS. Fluorescent analysis (see Figure 2C–2D) showed that after 48 h all isoforms had been imported to the nucleus, meaning that the activity of one NLS is sufficient and apparently a prerequisite for catalytic activity of the splice variants. Only XPF-202 displayed a rather diffuse cellular distribution with an accumulation in the nucleus. As seen in an XFE patient, dysfunctional or unstable XPF proteins exhibit an abnormal subcellular localization, which may indicate that XPF-202 is not involved in functional DNA repair [22, 23] (see Figure 3D–3F).
Splice variants of the two endonucleases XPF and XPG show residual repair capabilities in NER and ICL repair
Physiologically occurring, spontaneous mRNA splice variants may contain residual catalytic or putative structural DNA repair functions. XPG splice variants II-VI lack the endonuclease activity containing I region (see Figure 1), anyhow XPG IsoV and VI contain residual repair capabilities as well as two XPF splice variants (XPF-201 and XPF-003). Interestingly, the isoform XPF-202 that only differs from XPF-003 in the first 12 aa did not show any complementation.
Complementation of XPG- or XPF-deficient cells with full-length proteins only resulted in partial restoration of repair capability (XPG 4.90%, XPF 6.87%) in comparison to WT levels (primary fibroblasts 15.72%, MRC5Vi WT 12.45%), caused by transfection efficiencies of only 50%. Moreover, both proteins are involved in basal transcription making the HCR assay a qualitative rather than quantitative read-out of repair capability. Different damage loads can be excluded in this experimental setting due to the fact that i) the level of WT MRC5Vi cells is comparable for all three lesions, as well as for WT fibroblasts; ii) the results are always presented as percent expression of untreated vector within one group of DNA damages, and iii) a batch treatment of plasmids was done and used for all respective experiments. Splice variants do even exhibit a smaller complementation effect, XPG isoform V and VI as well as XPF-003 only elicit minor relative increases in NER and ICL repair capability, yet, clearly statistically significant in comparison to mock vector transfectants (see Figure 3). Initially this seems to be a marginal increase, but regarding a patient’s survival or skin cancer free life period the effect might be tremendous. As mentioned before, these isoforms contain only one NLS, decreasing nuclear import activity, explaining the low increase in repair capability in partial. A stronger effect might be observable if these variants could be transported into the nucleus more efficiently. In a patient with nearly normal XPG mRNA expression Emmert et al. found one allele with an early stop codon mutation, inherited from her father, while the maternal allele only showed a single base missense mutation. This resulted in a residual repair activity of 10% and the 14 year old Caucasian girl (XP65BE) presented with a mild phenotype displaying sun sensitivity, no skin cancer, and no neurological symptoms [24]. This implicates that minor repair activities of splice variants may have a huge influence on the cell’s repair capabilities and therefore could be accounted for mild clinical features and strong influence on the severity of disease progression as well as skin cancer free survival time.
Unfortunately, the mechanism of mammalian ICL repair is not completely understood, yet. As unhooking is a key step during ICL repair, roles of the two endonucleases XPG and XPF/ERCC1 are of special interest. It depends on the position of an ICL whether XPF/ERCC1 is able to cleave on either side [25], but also XPG may be involved, cutting 3′ to a junction between duplex and single-stranded DNA [26]. Notably, XPG does not seem to be essential for ICL repair in contrast to XPF (see Figure 3B–3C) [27]. As previously shown, human cells deficient in XPG presented unhooking kinetics of psoralen + UVA induced ICLs close to WT levels, while XPF/ERCC1 deficient cells were not able to unhook these lesions [4, 28, 29].
This is in concurrence with our results that propose a fundamental role of the XPF/ERCC1 complex in ICL repair, as we could nearly detect no residual repair capability in our XPF KO cells (see Figure 3E–3F). XPF/ERCC1 is known to interact with the scaffold protein SLX4 and two other structure specific endonucleases Mus81/Eme1 and SLX1, and SLX4 is thought to enhance XPF’s endonuclease activity [30–32]. The interaction between SLX4 and XPF/ERCC1 seems to be important outside NER, as an siRNA knockdown of SLX4 rendered cells sensitive to crosslinking agents, but not UV irradiation [32]. Our results suggest an important protein interaction domain between the very N-terminal part of the XPF protein and SLX4, as XPF-003 showed residual repair capability in contrast to XPF-202 (see Figure 3D–3F). In regard thereto, a double knockout of XPF and SLX4 is of special importance to test the complementation ability of the XPF-003 splice variant. This double knockout should eradicate XPF-003’s complementation ability most importantly in regard to CP and TMP + UVA induced lesions, indicating dependence on the presence of SLX4. Moreover, XPF-202 showed a diffuse cellular localization and no clear nuclear accumulation (see Figure 2D). A disrupted interaction with SLX4, an important platform and scaffold protein to organize DNA repair factors, could also be an explanation for this. It is supposed that XPF/ERCC1 and SLX4 simultaneously load onto the ICL [33] and SLX4 also functions as a SUMO E3 ligase sumoylating itself, as well as XPF [34]. Furthermore, the XPF/ERCC1 endonuclease is also important for recombination and DSB repair [35–38] meriting further investigations to discover whether the interaction between XPF and SLX4 is important for unhooking or other crosslink repair associated pathways. Another important interaction factor is the single stranded DNA binding protein RPA. It is essential for ICL unhooking, by permitting XPF/ERCC1 to overcome inhibitory structures at ICLs, like the nascent leading strand [27]. So far only one essential residue for interaction between XPF/ERCC1 and RPA has been identified in the afore mentioned XFE patient [39], but there might also be other important residues, possibly in the first 12aa of the protein.
Commonly, all functionally active splice variants of XPF and XPG have evolved from alternative splicing and intron retention [11]. Intron gain or loss has been an important evolutionary engine over time [40]. In the literature, it is postulated that intron-retaining splice variants of transcription factors could be risk factors for various cancer entities, e.g. breast cancer, lymphoma or melanoma [41–43].
Most importantly, it remains to be elucidated how splice variants exert residual repair capabilities as XPG IsoV, VI, and XPF-003 lack at least parts of the endonuclease domains (see Figure 1). In addition to protein-protein interactions with other repair factors the existence of a cellular backup mechanism for the XPG endonuclease has been suggested demonstrating that endonuclease defective XPG was able to perform accurate NER in living cells [44]. It can be proposed that severely truncated XPG splice variants can structurally complement XPG-defective cells. This would result in functional NER by recruitment of other structure-specific endonucleases like e.g. Exo1 or Fen1, cleaving the DNA allowing the repair to proceed [45, 46]. It merits further investigations to prove this hypothesis. For example, recruitment of the XPF/ERCC1 complex to DNA damage in XPG-deficient cells expressing the truncated splice variants would be of special interest.
XPG and XPF splice variants exert a dominant negative effect on wild type NER capacities
We characterized the interference of splice variants with endogenous DNA repair to investigate dominant negative inhibition. Thereby, we identified two XPG splice variants (XPG IsoVI and XPG-201), as well as WT XPF and all three splice variants (XPF-201, XPF-003, XPF-202), to exhibit a dominant negative effect on MRC5Vi WT NER capabilities (see Figure 4C–4D). A larger part of protein-encoding genes is subject to physiological splicing, but pathological splicing has been reported in cancer tissue [47, 48]. Alternative splicing could function as a mechanism for repression of WT protein function, e.g. Chk2 splice variants have an effect on downstream substrates like p53, and Cdc25A/C involved in cell cycle arrest and apoptosis [49]. It has already been shown that a polymorphism of the splice acceptor site in the XPC gene leads to the expression of a functionally inhibitory splice variant reducing NER by about 50% [16]. It occurs as a haplotype with a second polymorphism in the XPC gene [50] that is associated with an increased risk for skin cancer development, SCCs and melanoma [17, 51]. Splice variants of the telomerase catalytic subunit (hTERT) lacking telomerase activity, due to missing parts of the catalytic core, can inhibit endogenous telomerase activity in developing tissues, resulting in telomere shortening and chromosome end-to-end fusions [52, 53]. In addition, dominant negative effects are often observed for splice variants of multisubunit proteins, e.g. isoleucyl-tRNA synthetase. Co-expression of the variant and the endogenous WT protein may result in competition for functional complex formation due to dysfunctional complexes in need of the same binding partners [54]. We would expect a similar mechanism behind the inhibitory effects of XPF and its splice variants in view of heterodimer formation with ERCC1. However, the interaction between ERCC1 and XPF is conferred by the (HhH)2 motif, which is only present in XPF-201, as XPF-202 and XPF-003 are severely C-terminally truncated.
Another possible explanation might be protein overload in the cell, due to the strong and artificial overexpression, resulting in unproductive protein interactions of splice variants with residual repair capability. Moreover, protein ratios of XPG/XPF could be disturbed. The dual incision process is highly coordinated between XPF/ERCC1 and XPG in subsequent binding and cutting steps and might be sensitive to changes in molecular ratios.
Implication of XPG isoforms for personalized medicine and further perspectives
We were able to detect differential patterns of three functional XPG transcripts between individuals as well as different tissues (see Figure 5A–5B). As mentioned before, expression levels of splice variants have been shown to be more suitable to distinguish between oncogene and non-oncogene samples than the primary gene transcript itself [12]. This could be an indicator for the existence of an individualized overall repair capacity that might even differ between a person’s tissues. Therefore, the expression of XP splice variants could determine a patient’s resistance to different therapies, e.g. in chemotherapy against cancer. A weak chemotherapeutic response could be explained by different levels of DNA repair and has already been shown to be a target for small molecule inhibitors [55]. Additionally, it is known that resistances to platinum-based therapy correlate with high expression of ERCC1 [56] and can be reversed by blocking the interaction between ERCC1 and XPA, which is essential for NER, sensitizing cancer cells to NER substrates. Furthermore, inhibitors disrupting the XPF-ERCC1 interaction have been identified in in silico drug screens [57]. High expression of XP genes, like ERCC1, has also been correlated with poor therapy response in different cancer entities like e.g. non-small cell lung cancer, esophageal cancer, breast cancer, colorectal cancer, as well as head and neck cancer [58–61]. Testes tumors with reduced levels of NER proteins could be cured after application of a cisplatin-based therapy, even though they were already in an advanced metastasizing state. Meaning that low expression of XPF and XPG is beneficial for patients with advanced testes tumors in regard to therapeutic responses [62]. Interestingly, in our expression analyses of 20 different tissues we found that both XPG isoforms, V and VI, were surpassingly high in testes compared to WT XPG levels.
Anyhow, tumor-specific splice variants often tend to be overexpressed [63–65]. In order to apply splice variants as prognostic marker, residual repair capabilities and duration of skin-cancer free survival time in patients missing the primary transcript [15] as well as dominant negative functions in healthy individuals have to be balanced and evaluated in larger collectives. The HCR assay has already been used in molecular epidemiology studies to correlate low NER function in peripheral blood lymphocytes with an increased risk of cancer [66]. Furthermore, Wei et al. showed decades ago that a reduced NER capacity as assessed with HCR and peripheral blood lymphocytes is an independent melanoma and basal cell carcinoma risk factor [67, 68].
Our small collective of 20 healthy donors as well as expression differences in 20 tissues have been assessed by qRT-PCR only for XPG and its two splice variants with residual repair function (IsoV and VI), whereas IsoVI also displayed dominant negative effects in WT cells (see Figures 4C and 5C). Notably, we could observe that individuals with a high expression of isoform VI showed a low expression level of the primary transcript, implicating a dominant negative effect of this variant on flXPG expression. As a future perspective, the quantification of functional splice variants of NER components, e.g. XPG or XPF, in certain high-risk cancer populations (even for different cancer types and tumor entities) could be applied to investigate this variants as prognostic marker for beneficial or negative influences of cancer development, disease outcome, therapeutic responses, and side effects. In summary, we present functionally active, alternatively spliced mRNA variants of XP genes as possible useful tools to estimate cancer susceptibility, responses to chemotherapeutics and therapeutic success [6–8, 69].
MATERIALS AND METHODS
Cell culture
WT immortalized MRC5Vi cells were kindly provided by Sarah Sertic (University of Milan, Department of Life Sciences, Milan, Italy). Cells were cultivated and passaged as previously described [13, 14]. For antibiotic selection cells were treated with 600 μg/ml G418 (neomycine) (Sigma-Aldrich, Munich, DE). Primary human fibroblasts from healthy donors were established from skin punch biopsies (WT = male, age 53) in our laboratory. XP20BE cells were purchased from the Coriell Cell Repository (Camden, NJ, USA), while HeLa cells were obtained from ATCC (Manassas, VA, USA). Experiments were conducted in accordance with the Declaration of Helsinki principles, as approved by the University’s ethics committee. Written informed consent was obtained from the donor.
Cloning
All until now, physiologically occurring spontaneous mRNA splice variants (see Figure 1) of the XPF (XPF-201, XPF-003, XPF-202) and XPG (XPG IsoII – VI, XPG-201 and XPG-202) genes were amplified from WT MRC5Vi mRNA and cloned into different expression plasmids: pcDNA3.1(+) (for functional testing), pcDNA3.1(-)mycHisA2 (for protein level analyses), and pcDNA3.1 (+)eGFP (C-terminal, for subcellular localization studies). mRNA was isolated from WT MRC5Vi using the RNeasy Kit from Qiagen. Obligatory DNase digestion of RNA samples was performed for 30 minutes at room temperature using the RNase free DNase Set (Qiagen, Hilden, DE) and 1μg of total RNA was used for cDNA synthesis (Revert Aid H Minus Kit, Thermo Fisher Scientific, Waltham, MA, USA) according to manufacturer’s instructions. Splice variants as well as full-length sequences were amplified from cDNA via PCR performed in the Arktik Thermal Cycler (Thermo Fisher Scientific, Waltham, MA, USA). Phusion polymerase (Thermo Fisher Scientific, Waltham, MA, USA) was used according to manufacturer’s instructions. Specifically designed primer pairs were utilized for amplification of the different splice variants and are listed in Table 1. Amplification products of the right size were purified by gel-extraction (Wizard Gel extraction and PCR Clean Up kit, Promega, Mannheim, DE). Restriction sites for specific restriction enzymes were added by a second PCR reaction of the purified fragments and afterwards precipitated with ethanol under high salt conditions. Amplification fragments as well as the target vector were digested with appropriate restriction enzymes (listed in Table 2) according to manufacturer’s protocol (NEB, Ipswich, MA, USA). Ligation was performed in a 3:1 (fragment : vector) ratio using a T4 Ligase (Thermo Fisher Scientific, Waltham, MA, USA). Constructs were transformed in chemically competent DH5α (E.coli) cells and isolated from single clones grown on LB Agar plates using the NucleoBond Mini plasmid kit (Machery + Nagel, Düren, DE). Sequences were verified by Sanger sequencing using the BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) as described in Schäfer et al. [70] (primer see Table 1).
Table 1: Oligonucleotides used for cloning, sequencing, and qRT-PCR
Gene |
Oligonucleotide |
|---|---|
Amplification primer XPF/XPG and splice variants |
|
XPF_fwd |
5′- ATGGAGTCAGGGCAGCC -3’ |
XPF_rev |
5′- TCACTTTTTCCCTTTTCCTTTTGA -3’ |
XPF_rev_w/oStop |
5′- CTTTTTCCCTTTTCCTTTTGATAC -3’ |
XPF201_fwd |
5′- ATGGCGCCGCTGCTGGA -3’ |
XPF003_rev |
5′- TTAACCCCACAAGATACCTTCCC -3’ |
XPF003_rev_w/oStop |
5′- ACCCCACAAGATACCTTCCCCT -3’ |
XPG_fwd |
5′- GTGCAGTCCGTCGTAGAAG -3’ |
XPG_rev |
5′- CATTACAAATGGCTGTCATAAC -3’ |
XP20BE allele_fwd |
5′- GTGCAGTCCGTCGTAGAAG -3’ |
XP20BE allele_rev |
5′- AACTTGGGTAAGACTGGGTAG -3’ |
XPG IsoII_fwd |
5′- GTGCAGTCCGTCGTAGAAG -3’ |
XPG IsoII_rev |
5′- CTTTTTTAAAACTTCATCTCTAACACG -3’ |
XPG IsoIII_fwd |
5′- GTGCAGTCCGTCGTAGAAG -3’ |
XPG IsoIII_rev |
5′- CTAGGGCTGCAGCAGAG -3’ |
XPG IsoIV_fwd |
5′- GTGCAGTCCGTCGTAGAAG -3’ |
XPG IsoIV_rev |
5′- GTTACGGTATTACCAAATTAATATC -3’ |
XPG IsoV_fwd |
5′- GTGCAGTCCGTCGTAGAAG -3’ |
XPG IsoV_rev |
5′- GGAAATCCTACCGTTCCAG -3’ |
XPG IsoVI_fwd |
5′- GTGCAGTCCGTCGTAGAAG -3’ |
XPG IsoVI_rev |
5′- GTTACGGTATTACCAAATTAATATC -3’ |
XPG201_fwd |
5′- GAAAGAATGAATCAAAAACAAGC -3’ |
XPG201_rev |
5′- CATTACAAATGGCTGTCATAAC -3’ |
XPG202_fwd |
5′- GAAACAGATAGAGTTGCAACTTG -3’ |
XPG202_rev |
5′- CATTACAAATGGCTGTCATAAC -3’ |
Sequencing primer |
|
M13 |
5′- TAGAAGGCACAGTCGAG -3’ |
T7 |
5′- TAATACGACTCACTATAGGG -3’ |
XPFseq_exon1_fwd |
5′- ATGGAGTCAGGGCAGC -3’ |
XPFseq_exon4_fwd |
5′- GTTCCATGTAGCAGTAAACT -3’ |
XPFseq_exon7_fwd |
5′- CAAGTGATGACCGAACATGT -3’ |
XPFseq_exon9_rev |
5′- TCACTTTTTCCCTTTTCCT -3’ |
XPFseq_exon3_rev |
5′- CTTGGCCACAGATACAGTT -3’ |
XPFseq_exon8_rev |
5′- GCTTGGCCACAGATACAGT -3’ |
XPGseq1_fwd |
5′- ACCTCTATGTTTTGCCTCCTT -3’ |
XPGseq2_fwd |
5′- CAACATTCAGGACACATCCG -3’ |
XPGseq3_fwd |
5′- CACTTCTGCAACTGTCTTAGC -3 |
XPGseq4_fwd |
5′- CCACAGACTCAGTTCCAAA -3’ |
XPGseq5_fwd |
5′- GAAGCCTTTCCGATAAGTGAT -3’ |
XPGseq6_fwd |
5′- CTCGGAAGAAAGTGAATCTG -3’ |
XPGseq7_fwd |
5′- ACAACTCTGAGAGGGACGAC -3 |
XPGseq8_fwd |
5′- GGATGTAGGGAATGCCGAAC -3’ |
XPGseq9_fwd |
5′- CTGTCTTCGTTCTGTTCCAG -3 |
CMVpromoter _fwd |
5′- CTGCTTAGGGTTAGGCGTTTTGCGCT -3’ |
Oligonucleotides for quantitative real-time PCR purchased from Qiagen, Hilden GER |
|
Gene |
Order number |
GAPDH |
QT00079247 |
Oligonucleotides for quantitative real-time PCR |
|
XPG_fwd |
5’- GGATCTTCAAGTGAACATGCTGAA -3’ |
XPG_rev |
5’- TGCGAATCTGAAGCACTGGT -3’ |
XPGIsoV_fwd |
5‘- GAATCTGCAGGCCAGGATTT -3‘ |
XPGIsoV_rev |
5’- CCTACCGTTCCAGATTTCTACAAAA -3 |
XPGIsoVI_fwd |
5’- GAGCCACAGGAAGCTGAGAAA -3‘ |
XPGIsoVI_rev |
5’- CAGCAAGAAGTCGAAACACAATG-3‘ |
For cloning into pcDNA3.1(+) (KpnI (5’- ttaGGTACC -3’), XbaI (5’- ttaTCTAGA -3’), pcDNA3.1(-)mycHisA2 (XbaI, KpnI), and pcDNA3.1(+)eGFP (KpnI, XbaI) respective restriction recognition sequences were added.
Table 2: Restriction enzymes
Description |
Manufacturer |
|---|---|
KpnI 10u/μl |
Thermo Fisher Scientific, Waltham, MA, USA |
PvuI |
Thermo Fisher Scientific, Waltham, MA, USA |
XbaI 10u/μll |
Thermo Fisher Scientific, Waltham, MA, USA |
Generation of stable cell lines
Stable cell lines overexpressing the full-length protein or respective splice variants were generated via spontaneous integration of a linearized expression plasmid into MRC5Vi WT cells. An expression plasmid coding for the full-length XPG/XPF protein or one of the splice variants, e.g. pcDNA3.1 (+)XPF, was linearized using the PvuI restriction enzyme (Thermo Fisher Scientific, Waltham, MA, USA) according to manufacturer’s instructions. Successful linearization was analyzed on an agarose gel and the linearized plasmids were purified by ethanol precipitation. Afterwards, the linearized plasmids were transiently transfected into MRC5Vi WT cells seeded on 10mm cell culture dishes and positive clones were selected using G418. Single cell clones were established by serial dilution and checked for plasmid integration via PCR using specific primer pairs for the CMV (cytomegalo virus)-promoter and the integrated gene (see Table 1), as well as immunoblot analyses.
Immunoblot analyses
Whole protein lysates were extracted from cell pellets, washed twice using 10ml PBS, resuspended in PBS containing PMSF and complete proteinase stop. Cells were disrupted by rotational freezing in liquid nitrogen followed by thawing on ice for three times and sedimented by centrifugation (10 min, 14 000 rpm, 4°C). The supernatant was transferred into a new reaction tube to get rid of cell debris. Lysates were adjusted to the desired concentration by mixing with 3% SDS splitting buffer and boiling at 95°C for 10min. Equal amounts of protein extracts (50–100 μg) were loaded onto the gels. SDS-PAGE and immunoblotting were performed as described before [14] using an anti-XPF mouse monoclonal antibody clone 3F2/3 (Santa Cruz, Dallas, TX, USA), an anti-β-actin mouse monoclonal clone AC-74 (Sigma-Aldrich, Munich, DE) or two different XPG antibodies (Abcam, Cambridge, UK; Bethyl, Montgomery, TX, USA) diluted 1:250, 1:5000, 1:250, and 1:1000 in blocking solution, respectively.
Reporter gene assays
Analyses of NER, as well as ICL repair capacity and complementation with WT full-length XPF or XPG, as well as the respective splice variants was assessed via HCR performed according to the descriptions in Lehmann et al. [14] after 48 h for XPF in immortalized MRC5Vi cells and 72 h for XPG in primary patients or WT fibroblasts. Liposomal transient transfections were carried out in a 24-well format (Greiner Bio-One, Kremsmünster, AUT) using Attractene transfection reagent (Qiagen, Hilden, DE) according to the manufacturer’s instructions. Unirradiated Renilla luciferase was used for normalization. Luminescence was measured 48 h or 72 h after transfection with the GloMax® Discover Multimode Detection System (Promega). The relative repair capability [%] was calculated as the quotient of treated cells to untreated firefly/Renilla ratios. Cells were treated with either UVC (750–1000J/m2), with diammineplatinum(II) dichloride (CP) (Sigma-Aldrich, Munich, DE) in a molecular ratio of 1:20 or 1:40 (vector:CP) or with 4,5’,8-trimethylpsoralen (TMP) (Sigma Aldrich, Munich, DE) (1:25 (primary cells) or 1:50 (immortalized cells) (vector:TMP)) followed by irradiation with 1 J/cm² UVA. A batch treatment of plasmids was done and used for all respective experiments. The assay was repeated at least four times in triplicates.
qRT PCR
Specific primer pairs for isoforms as well as for WT XPG were utilized to analyze RNA expression in 20 different healthy human tissues obtained from Ambion® (product discontinued). cDNA was prepared from RNA samples and quantified as described in [14]. Specific primer pairs binding to the intronic regions of the isoforms were applied (see Table 1).
Abbreviations
6–4PPs : 6–4photoproducts, aa: amino acids, CPDs: cyclobutane pyrimidine dimers, CP: cis-diammineplatinum(II) dichloride, CS: Cockayne syndrome, fl: full-length, GGR: global genome repair, HCR: host cell reactivation, (HhH)2 Helix-hairpin-Helix motif, ICL: interstrand crosslink, KO: knockout, NER: nucleotide excision repair, NLS: nuclear localization signal, TCR: transcription coupled repair, TFIIH: transcription factor IIH, TMP: 4,5’,8-trimethylpsoralen, WT: wildtype, XP: xeroderma pigmentosum.
ACKNOWLEDGMENTS
The authors wish to acknowledge Sarah Sertic (University of Milan) for the supply of the MRC5Vi cells, Sebastian Monecke (UMG, Goettingen, GER) for providing eGFP for subcloning, and Petra Rieper (Clinic for Psychiatry and Psychotherapy, University Medical Center Goettingen) for additional technical support.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
FUNDING
This work was supported by the German Cancer Aid (Deutsche Krebshilfe e.V.) [111377], the Claudia von Schilling Foundation for Breast Cancer Research, and the Heinz and Heide Duerr foundation [2014–2.2.1/02].
REFERENCES
1. Kraemer KH, DiGiovanna JJ. Nucleotide excision repair in eukaryotes. Cold Spring Harb Perspect Biol. 2013; 5:a012609.
2. Ahmad A, Enzlin JH, Bhagwat NR, Wijgers N, Raams A, Appledoorn E, Theil AF, J Hoeijmakers JH, Vermeulen W, J Jaspers NG, Schärer OD, Niedernhofer LJ. Forty years of research on xeroderma pigmentosum at the US National Institutes of Health. Photochem Photobiol. 2015; 91:452–459.
3. Lehmann J, Schubert S, Emmert S. Xeroderma pigmentosum: diagnostic procedures, interdisciplinary patient care, and novel therapeutic approaches. J Dtsch Dermatol Ges. 2014; 12:867–872.
4. De Silva IU, McHugh PJ, Clingen PH, Hartley JA. Defects in interstrand cross-link uncoupling do not account for the extreme sensitivity of ERCC1 and XPF cells to cisplatin. Nucleic Acids Res. 2002; 30:3848–3856.
5. Khan SG, Oh KS, Shahlavi T, Ueda T, Busch DB, Inui H, Emmert S, Imoto K, Muniz-Medina V, Baker CC, DiGiovanna JJ, Schmidt D, Khadavi A, et al. Reduced XPC DNA repair gene mRNA levels in clinically normal parents of xeroderma pigmentosum patients. Carcinogenesis. 2006; 27:84–94.
6. Li C, Yin M, Wang LE, Amos CI, Zhu D, Lee JE, Gershenwald JE, Grimm EA, Wei Q. Polymorphisms of nucleotide excision repair genes predict melanoma survival. J Invest Dermatol. 2013; 133:1813–1821.
7. Hu W, Pan J, Zhao P, Yang G, Yang S. Genetic polymorphisms in XPG could predict clinical outcome of platinum-based chemotherapy for advanced non-small cell lung cancer. Tumour Biol. 2014; 35:5561–5567.
8. Sun K, Gong A, Liang P. Predictive impact of genetic polymorphisms in DNA repair genes on susceptibility and therapeutic outcomes to colorectal cancer patients. Tumour Biol. 2015; 36:1549–1559.
9. Brookman KW, Lamerdin JE, Thelen MP, Hwang M, Reardon JT, Sancar A, Zhou ZQ, Walter CA, Parris CN, Thompson LH. ERCC4 (XPF) encodes a human nucleotide excision repair protein with eukaryotic recombination homologs. Mol Cell Biol. 1996; 16:6553–6562.
10. Scharer OD. XPG: its products and biological roles. Adv Exp Med Biol. 2008; 637:83–92.
11. Emmert S, Schneider TD, Khan SG, Kraemer KH. The human XPG gene: gene architecture, alternative splicing and single nucleotide polymorphisms. Nucleic Acids Res. 2001; 29:1443–1452.
12. Zhang Z, Pal S, Bi Y, Tchou J, Davuluri RV. Isoform level expression profiles provide better cancer signatures than gene level expression profiles. Genome Med. 2013; 5: 33.
13. Moriwaki S, Stefanini M, Lehmann AR, Hoeijmakers JH, Robbins JH, Rapin I, Botta E, Tanganelli B, Vermeulen W, Broughton BC, Kraemer KH. DNA repair and ultraviolet mutagenesis in cells from a new patient with xeroderma pigmentosum group G and cockayne syndrome resemble xeroderma pigmentosum cells. J Invest Dermatol. 1996; 107:647–653.
14. Lehmann J, Seebode C, Smolorz S, Schubert S, Emmert S. XPF knockout via CRISPR/Cas9 reveals that ERCC1 is retained in the cytoplasm without its heterodimer partner XPF. Cell Mol Life Sci. 2017; 74:2081–2094.
15. Thorel F, Constantinou A, Dunand-Sauthier I, Nouspikel T, Lalle P, Raams A, Jaspers NG, Vermeulen W, Shivji MK, Wood RD, Clarkson SG. Definition of a short region of XPG necessary for TFIIH interaction and stable recruitment to sites of UV damage. Mol Cell Biol. 2004; 24:10670–10680.
16. Khan SG, Muniz-Medina V, Shahlavi T, Baker CC, Inui H, Ueda T, Emmert S, Schneider TD, Kraemer KH. The human XPC DNA repair gene: arrangement, splice site information content and influence of a single nucleotide polymorphism in a splice acceptor site on alternative splicing and function. Nucleic Acids Res. 2002; 30:3624–3631.
17. Blankenburg S, Konig IR, Moessner R, Laspe P, Thoms KM, Krueger U, Khan SG, Westphal G, Berking C, Volkenandt M, Reich K, Neumann C, Ziegler A, et al. Assessment of 3 xeroderma pigmentosum group C gene polymorphisms and risk of cutaneous melanoma: a case-control study. Carcinogenesis. 2005; 26:1085–1090.
18. Aracil M, Dauffenbach LM, Diez MM, Richeh R, Moneo V, Leal JF, Fernandez LF, Kerfoot CA, Galmarini CM. Expression of XPG protein in human normal and tumor tissues. Int J Clin Exp Pathol. 2013; 6:199–211.
19. Dunand-Sauthier I, Hohl M, Thorel F, Jaquier-Gubler P, Clarkson SG, Scharer OD. The spacer region of XPG mediates recruitment to nucleotide excision repair complexes and determines substrate specificity. J Biol Chem. 2005; 280:7030–7037.
20. Tillhon M, Cazzalini O, Nardo T, Necchi D, Sommatis S, Stivala LA, Scovassi AI, Prosperi E. p300/CBP acetyl transferases interact with and acetylate the nucleotide excision repair factor XPG. DNA Repair (Amst). 2012; 11:844–852.
21. Fischer JA, Muller-Weeks S, Caradonna S. Proteolytic degradation of the nuclear isoform of uracil-DNA glycosylase occurs during the S phase of the cell cycle. DNA Repair (Amst). 2004; 3:505–513.
22. Ahmad A, Enzlin JH, Bhagwat NR, Wijgers N, Raams A, Appledoorn E, Theil AF, Hoeijmakers JHJ, Vermeulen W, Jaspers NGJ, Schärer OD, Niedernhofer LJ. Mislocalization of XPF-ERCC1 nuclease contributes to reduced DNA repair in XP-F patients. PLoS Genet. 2010; 6: e1000871.
23. Kashiyama K, Nakazawa Y, Pilz DT, Guo C, Shimada M, Sasaki K, Fawcett H, Wing JF, Lewin SO, Carr L, Li TS, Yoshiura K, Utani A, et al. Malfunction of nuclease ERCC1-XPF results in diverse clinical manifestations and causes Cockayne syndrome, xeroderma pigmentosum, and Fanconi anemia. Am J Hum Genet. 2013; 92:807–819.
24. Emmert S, Slor H, Busch DB, Batko S, Albert RB, Coleman D, Khan SG, Abu-Libdeh B, DiGiovanna JJ, Cunningham BB, Lee MM, Crollick J, Inui H, et al. Relationship of neurologic degeneration to genotype in three xeroderma pigmentosum group G patients. J Invest Dermatol. 2002; 118:972–982.
25. Kuraoka I, Kobertz WR, Ariza RR, Biggerstaff M, Essigmann JM, Wood RD. Repair of an interstrand DNA cross-link initiated by ERCC1-XPF repair/recombination nuclease. J Biol Chem. 2000; 275:26632–26636.
26. Evans E, Fellows J, Coffer A, Wood RD. Open complex formation around a lesion during nucleotide excision repair provides a structure for cleavage by human XPG protein. Embo j. 1997; 16:625–638.
27. Abdullah UB, McGouran JF, Brolih S, Ptchelkine D, El-Sagheer AH, Brown T, McHugh PJ. RPA activates the XPF-ERCC1 endonuclease to initiate processing of DNA interstrand crosslinks. Embo j. 2017; 36:2047–2060.
28. De Silva IU, McHugh PJ, Clingen PH, Hartley JA. Defining the roles of nucleotide excision repair and recombination in the repair of DNA interstrand cross-links in mammalian cells. Mol Cell Biol. 2000; 20:7980–7990.
29. Rothfuss A, Grompe M. Repair kinetics of genomic interstrand DNA cross-links: evidence for DNA double-strand break-dependent activation of the Fanconi anemia/BRCA pathway. Mol Cell Biol. 2004; 24:123–134.
30. Andersen SL, Bergstralh DT, Kohl KP, LaRocque JR, Moore CB, Sekelsky J. Drosophila MUS312 and the vertebrate ortholog BTBD12 interact with DNA structure-specific endonucleases in DNA repair and recombination. Mol Cell. 2009; 35:128–135.
31. Fekairi S, Scaglione S, Chahwan C, Taylor ER, Tissier A, Coulon S, Dong MQ, Ruse C, Yates JR 3rd, Russell P, Fuchs RP, McGowan CH, Gaillard PH. Human SLX4 is a Holliday junction resolvase subunit that binds multiple DNA repair/recombination endonucleases. Cell. 2009; 138:78–89.
32. Munoz IM, Hain K, Declais AC, Gardiner M, Toh GW, Sanchez-Pulido L, Heuckmann JM, Toth R, Macartney T, Eppink B, Kanaar R, Ponting CP, Lilley DM, et al. Coordination of structure-specific nucleases by human SLX4/BTBD12 is required for DNA repair. Mol Cell. 2009; 35:116–127.
33. Klein Douwel D, Boonen RA, Long DT, Szypowska AA, Raschle M, Walter JC, Knipscheer P. XPF-ERCC1 acts in Unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol Cell. 2014; 54:460–471.
34. Guervilly JH, Takedachi A, Naim V, Scaglione S, Chawhan C, Lovera Y, Despras E, Kuraoka I, Kannouche P, Rosselli F, Gaillard PH. The SLX4 complex is a SUMO E3 ligase that impacts on replication stress outcome and genome stability. Mol Cell. 2015; 57:123–137.
35. Adair GM, Rolig RL, Moore-Faver D, Zabelshansky M, Wilson JH, Nairn RS. Role of ERCC1 in removal of long non-homologous tails during targeted homologous recombination. Embo j. 2000; 19:5552–5561.
36. Ahmad A, Robinson AR, Duensing A, van Drunen E, Beverloo HB, Weisberg DB, Hasty P, Hoeijmakers JH, Niedernhofer LJ. ERCC1-XPF endonuclease facilitates DNA double-strand break repair. Mol Cell Biol. 2008; 28:5082–5092.
37. Niedernhofer LJ, Essers J, Weeda G, Beverloo B, de Wit J, Muijtjens M, Odijk H, Hoeijmakers JH, Kanaar R. The structure-specific endonuclease Ercc1-Xpf is required for targeted gene replacement in embryonic stem cells. Embo j. 2001; 20:6540–6549.
38. Sargent RG, Rolig RL, Kilburn AE, Adair GM, Wilson JH, Nairn RS. Recombination-dependent deletion formation in mammalian cells deficient in the nucleotide excision repair gene ERCC1. Proc Natl Acad Sci U S A. 1997; 94:13122–13127.
39. Niedernhofer LJ, Garinis GA, Raams A, Lalai AS, Robinson AR, Appeldoorn E, Odijk H, Oostendorp R, Ahmad A, van Leeuwen W, Theil AF, Vermeulen W, van der Horst GT, et al. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature. 2006; 444:1038–1043.
40. Rogers J. Exon shuffling and intron insertion in serine protease genes. Nature. 1985; 315:458–459.
41. Busse A, Rietz A, Schwartz S, Thiel E, Keilholz U. An intron 9 containing splice variant of PAX2. J Transl Med. 2009; 7: 36.
42. Honda A, Valogne Y, Bou Nader M, Brechot C, Faivre J. An intron-retaining splice variant of human cyclin A2, expressed in adult differentiated tissues, induces a G1/S cell cycle arrest in vitro. PLoS One. 2012; 7: e39249.
43. Whiley PJ, Guidugli L, Walker LC, Healey S, Thompson BA, Lakhani SR, Da Silva LM, Tavtigian SV, Goldgar DE, Brown MA, Couch FJ, Spurdle AB. Splicing and multifactorial analysis of intronic BRCA1 and BRCA2 sequence variants identifies clinically significant splicing aberrations up to 12 nucleotides from the intron/exon boundary. Hum Mutat. 2011; 32:678–687.
44. Schubert S. Characterization of the multifunctional XPG protein during Nucleotide-excision-repair. [Doctoral thesis]; Department of Dermatology, Venereology and Allergology, Georg-August-University Goettingen. 2014.
45. Giannattasio M, Follonier C, Tourriere H, Puddu F, Lazzaro F, Pasero P, Lopes M, Plevani P, Muzi-Falconi M. Exo1 competes with repair synthesis, converts NER intermediates to long ssDNA gaps, and promotes checkpoint activation. Mol Cell. 2010; 40:50–62.
46. Godon C, Mourgues S, Nonnekens J, Mourcet A, Coin F, Vermeulen W, Mari PO, Giglia-Mari G. Generation of DNA single-strand displacement by compromised nucleotide excision repair. Embo j. 2012; 31:3550–3563.
47. Srebrow A, Kornblihtt AR. The connection between splicing and cancer. J Cell Sci. 2006; 119:2635–2641.
48. Zhao S, Liu W, Li Y, Liu P, Li S, Dou D, Wang Y, Yang R, Xiang R, Liu F. Alternative Splice Variants Modulates Dominant-Negative Function of Helios in T-Cell Leukemia. PLoS One. 2016; 11: e0163328.
49. Berge EO, Staalesen V, Straume AH, Lillehaug JR, Lonning PE. Chk2 splice variants express a dominant-negative effect on the wild-type Chk2 kinase activity. Biochim Biophys Acta. 2010; 1803:386–395.
50. Khan SG, Metter EJ, Tarone RE, Bohr VA, Grossman L, Hedayati M, Bale SJ, Emmert S, Kraemer KH. A new xeroderma pigmentosum group C poly(AT) insertion/deletion polymorphism. Carcinogenesis. 2000; 21:1821–1825.
51. Shen H, Sturgis EM, Khan SG, Qiao Y, Shahlavi T, Eicher SA, Xu Y, Wang X, Strom SS, Spitz MR, Kraemer KH, Wei Q. An intronic poly (AT) polymorphism of the DNA repair gene XPC and risk of squamous cell carcinoma of the head and neck: a case-control study. Cancer Res. 2001; 61:3321–3325.
52. Colgin LM, Wilkinson C, Englezou A, Kilian A, Robinson MO, Reddel RR. The hTERTalpha splice variant is a dominant negative inhibitor of telomerase activity. Neoplasia. 2000; 2:426–432.
53. Kilian A, Bowtell DD, Abud HE, Hime GR, Venter DJ, Keese PK, Duncan EL, Reddel RR, Jefferson RA. Isolation of a candidate human telomerase catalytic subunit gene, which reveals complex splicing patterns in different cell types. Hum Mol Genet. 1997; 6:2011–2019.
54. Michaels JE, Schimmel P, Shiba K, Miller WT. Dominant negative inhibition by fragments of a monomeric enzyme. Proc Natl Acad Sci U S A. 1996; 93:14452–14455.
55. Gentile F, Tuszynski JA, Barakat KH. New design of nucleotide excision repair (NER) inhibitors for combination cancer therapy. J Mol Graph Model. 2016; 65:71–82.
56. Barakat KH, Jordheim LP, Perez-Pineiro R, Wishart D, Dumontet C, Tuszynski JA. Virtual screening and biological evaluation of inhibitors targeting the XPA-ERCC1 interaction. PLoS One. 2012; 7: e51329.
57. Jordheim LP, Barakat KH, Heinrich-Balard L, Matera EL, Cros-Perrial E, Bouledrak K, El Sabeh R, Perez-Pineiro R, Wishart DS, Cohen R, Tuszynski J, Dumontet C. Small molecule inhibitors of ERCC1-XPF protein-protein interaction synergize alkylating agents in cancer cells. Mol Pharmacol. 2013; 84:12–24.
58. Bilen N, Tekin SB, Topdagi O. ERCC1 Expression in Non-Small Cell Lung and Esophageal Cancer. Eurasian J Med. 2014; 46:84–88.
59. Choueiri MB, Shen JP, Gross AM, Huang JK, Ideker T, Fanta P. ERCC1 and TS Expression as Prognostic and Predictive Biomarkers in Metastatic Colon Cancer. PLoS One. 2015; 10: e0126898.
60. Gerhard R, Carvalho A, Carneiro V, Bento RS, Uemura G, Gomes M, Albergaria A, Schmitt F. Clinicopathological significance of ERCC1 expression in breast cancer. Pathol Res Pract. 2013; 209:331–336.
61. Hayes M, Lan C, Yan J, Xie Y, Gray T, Amirkhan RH, Dowell JE. ERCC1 expression and outcomes in head and neck cancer treated with concurrent cisplatin and radiation. Anticancer Res. 2011; 31:4135–4139.
62. Welsh C, Day R, McGurk C, Masters JR, Wood RD, Koberle B. Reduced levels of XPA, ERCC1 and XPF DNA repair proteins in testis tumor cell lines. Int J Cancer. 2004; 110:352–361.
63. Blair CA, Zi X. Potential molecular targeting of splice variants for cancer treatment. Indian J Exp Biol. 2011; 49:836–839.
64. Brinkman BM. Splice variants as cancer biomarkers. Clin Biochem. 2004; 37:584–594.
65. Yi Q, Tang L. Alternative spliced variants as biomarkers of colorectal cancer. Curr Drug Metab. 2011; 12:966–974.
66. Mendez P, Taron M, Moran T, Fernandez MA, Requena G, Rosell R. A modified host-cell reactivation assay to quantify DNA repair capacity in cryopreserved peripheral lymphocytes. DNA Repair (Amst). 2011; 10:603–610.
67. Wei Q, Lee JE, Gershenwald JE, Ross MI, Mansfield PF, Strom SS, Wang LE, Guo Z, Qiao Y, Amos CI, Spitz MR, Duvic M. Repair of UV light-induced DNA damage and risk of cutaneous malignant melanoma. J Natl Cancer Inst. 2003; 95:308–315.
68. Wei Q, Matanoski GM, Farmer ER, Hedayati MA, Grossman L. DNA repair capacity for ultraviolet light-induced damage is reduced in peripheral lymphocytes from patients with basal cell carcinoma. J Invest Dermatol. 1995; 104:933–936.
69. Colmegna B, Uboldi S, Frapolli R, Licandro SA, Panini N, Galmarini CM, Badri N, Spanswick VJ, Bingham JP, Kiakos K, Erba E, Hartley JA, D’Incalci M. Increased sensitivity to platinum drugs of cancer cells with acquired resistance to trabectedin. Br J Cancer. 2015; 113:1687–1693.
70. Schafer A, Schubert S, Gratchev A, Seebode C, Apel A, Laspe P, Hofmann L, Ohlenbusch A, Mori T, Kobayashi N, Schurer A, Schon MP, Emmert S. Characterization of three XPG-defective patients identifies three missense mutations that impair repair and transcription. J Invest Dermatol. 2013; 133:1841–1849.
71. de Laat WL, Sijbers AM, Odijk H, Jaspers NG, Hoeijmakers JH. Mapping of interaction domains between human repair proteins ERCC1 and XPF. Nucleic Acids Res. 1998; 26:4146–4152.
72. Fisher LA, Bessho M, Wakasugi M, Matsunaga T, Bessho T. Role of interaction of XPF with RPA in nucleotide excision repair. J Mol Biol. 2011; 413:337–346.
73. Li L, Peterson CA, Lu X, Legerski RJ. Mutations in XPA that prevent association with ERCC1 are defective in nucleotide excision repair. Mol Cell Biol. 1995; 15:1993–1998.
74. McNeil EM, Melton DW. DNA repair endonuclease ERCC1-XPF as a novel therapeutic target to overcome chemoresistance in cancer therapy. Nucleic Acids Res. 2012; 40:9990–10004.