
INTRODUCTION
Protein synthesis is a finely regulated process that begins with DNA replication, followed by transcription, and concluding with translation of the protein. Post-transcriptional modifications occur mainly between the transcription and translation processes, and ensure integrity and generate the diversity that characterizes the final protein products. Among post-transcriptional modifications, RNA splicing involves the removal of noncoding sequences (introns) from the primary transcript, or pre-mRNA (Figure 1). RNA splicing is orchestrated by small nuclear ribonucleoproteins (snRNPs), small nuclear RNAs, and protein factors that form spliceosomes. This process greatly expands the coding capacity of complex genomes, as it can generate protein products with distinct and even opposite properties from a single gene locus [1, 2]. It is therefore not surprising that dysfunction of alternative splicing (AS) can lead to various diseases, including cancers. Accordingly, abnormal AS has been demonstrated to contribute to many aspects of tumor initiation and addiction, including the control of cell proliferation and programmed cell death, metabolism of cancer cells, angiogenesis, metastasis, response to treatment and clinical outcome [3].
Acute myelogenous leukemia (AML) represents a heterogeneous spectrum of myeloid malignancies that harbor a constellation of chromosomal abnormalities, gene mutations, and epigenetic modifications. These genetic abnormalities have enabled understanding of the biology of the disease, helped generate the main diagnostic and prognostic tools, and represent key therapeutic targets. Recent reports have shown a significant deregulation of AS in AML, with approximately one-third of expressed genes being abnormally spliced in AML compared to normal CD34+ bone marrow cells [4]. Several reports have evidenced somatic mutations of splicing factors in AML, yet their frequency appears significantly lower than that observed in other myeloid malignancies such as in the myelodysplastic syndromes (MDS). In addition to spliceosome gene mutations, what other processes of factors can influence and alter AS in AML? Here we have concisely reviewed some of the main cellular processes known to influence AS, including those that are deregulated in AML such as deregulated transcription, epigenetic changes (histone modifications and CpG methylation), and WT1 gene overexpression.
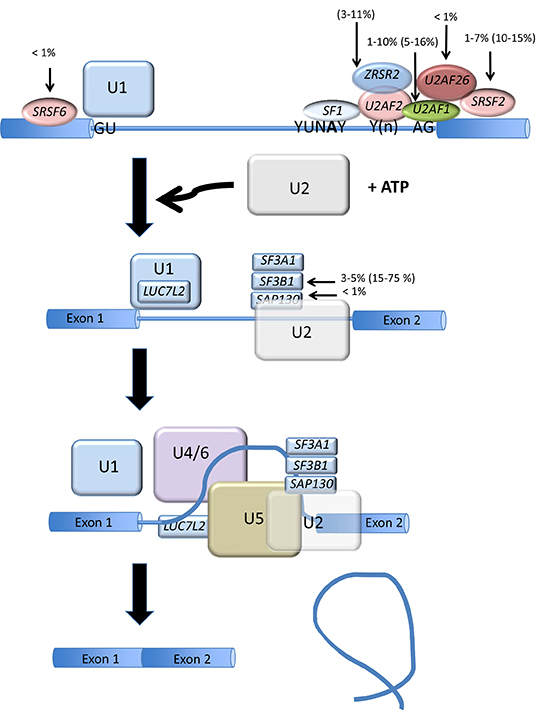
Figure 1: Splicing factor gene mutation in AML and MDS. The two main steps of mRNA splicing are represented. The steps are catalyzed by spliceosomes and specified by three RNA sequence elements called the 5’ splice site, the 3’ splice site and the branch site (YUNAY). The 5’ splice site is recognized by the U1 snRNA-protein particle (snRNP) while the branch site is recognized by the U2 snRNP complexed with proteins at the 3’ splice site. In a later step of the spliceosome formation pathway, a tri-snRNP complex composed of U4, U5 and U6 snRNPs joins the spliceosome. Further rearrangements of the spliceosome lead to catalysis of the splicing reaction and the production of the spliced product mRNA and the excised intron. The main splicing factor gene mutation is represented for AML and MDS (in parenthesis).
Abnormal mRNA splicing in AML and spliceosome gene mutations
Although abnormal RNA splicing has been previously demonstrated for several genes in AML, the connection between hematological malignancies and RNA splicing has recently emerged from studies based on next generation sequencing (NGS). Mutations in spliceosomal genes, in particular splicing factor 3 subunit b1 (SF3b1), were first identified in myelodysplastic syndromes (MDS) [5], myeloproliferative neoplasms (MPN) [6], MDS/MPN [7], and chronic lymphocytic leukemia (CLL) [8], as well as other hematological disorders [9]. Mutations in other spliceosomal genes such as U2 small nuclear RNA auxiliary factor 1 (U2AF1) [10] and serine arginine-rich splicing factor 2 (SRSF2) were subsequently identified [11]. The mutational landscape of the spliceosome is now available for hematological malignancies [12]. These findings have opened new avenues for understanding the underlying biology and for therapeutic intervention in this setting. Overall, the proportion of cases carrying spliceosome mutations ranges from <1–90% in MDS, MDS/MPN and secondary AML compared to <1–10.5% in de novo AML [5, 6] (Figure 1). In addition to this mutational spectrum, exon-array technologies and RNA sequencing (NGS) have enabled the assessment of the pattern of RNA splicing in hematological diseases. Certain spliceosome mutations have been found associated with missplicing in specific genes and might therefore help explain some aspects of the deregulated pattern of AS in AML. For example, in MDS, MPN, MDS/MPN, and secondary AML (sAML), U2AF1 (U2AF35) mutations, which are observed in 1–10% of de novo AML, have been found associated with abnormal splicing of genes involved in cell cycle progression and RNA processing that are somatically mutated or deleted in various cancers [13]. Yoshida et al. compared the effect of U2AF1 mutation on gene expression and splicing in HeLa cells and TF-1 myeloid cells [5]. The authors found that the S34F mutation quantitatively inhibited AS and triggered a significant enrichment of genes in nonsense-mediated mRNA decay (NMD), suggesting that the mutant U2AF35 triggered abnormal RNA splicing in HeLa and TF-1 cells, leading to the generation of unspliced RNA species carrying premature stop codons that induced NMD activity.
The development of natural compounds and synthetic analogues that target SFs is in progress [14]. The rationale for use of these spliceosome inhibitors in leukemia and MDS is that the majority of the splicing mutations found in hematological malignancies are heterozygous and considered gain of function or change of function/neomorphic mutations [5, 15]. Accordingly, it may be that treatment-triggered homozygous inactivation of spliceosomal genes may be more toxic to the heterozygous mutant cells than normal cells, and thus result in preferential killing of the mutant cells.
Recently Adamia et al. showed that approximately 29% of expressed genes are differentially and recurrently spliced in AML patients compared to normal bone marrow donors [4]. Alternative exon usages (AEUs) were found to involve oncogenes, tumor suppressor proteins, splicing factors and heterogeneous-nuclear-ribonucleoproteins, and proteins involved in apoptosis, cell proliferation, and spliceosome assembly [4]. In silico, these deregulations trigger important pathways involved in leukemogenesis. To date, their consequences on the physiopathology, diagnosis, response to treatment and disease outcome in AML remain unknown. In addition, little is understood regarding the causes and mechanisms that underlie abnormal AS in AML. Given such a proportion of abnormally spliced genes in AML, it could be proposed that additional factors other than spliceosomal mutations, which account for ≤10% of AML cases (Figure 1), would be involved in the deregulation of RNA splicing in AML. AS is influenced not only by signaling pathways that target the splicing machinery but also by transcription factors and chromatin structure. Accordingly, in addition to spliceosome gene mutations, numerous factors might influence RNA splicing in the context of AML. These include transcriptional deregulation, protein-protein interaction and epigenetic modifications.
Epigenetics in alternative mRNA splicing
In addition to RNA sequence elements and their associated splicing factors, chromatin structure and histone modifications have been found to interfere with AS regulation (reviewed in ref [16]). This became apparent when mRNA splicing was found to be closely coupled with DNA transcription in mammalian cells [17]. In fact, transcription and splicing influence one another. For example, the transcription machinery can recruit several splicing factors such as SRSF3 that binds to the carboxy-terminal domain of RNA polymerase II (Pol II) [18]. Alternatively, some factors interacting with DNA may create roadblocks to Pol II-induced elongation [19]. Such pausing during elongation has been evidenced with the DNA-binding protein CCCTC-binding factor (CTCF) that promotes inclusion of an alternative exon 5 in CD45 through a roadblock effect [20] (Figure 2A).
The link between chromatin structure and splicing was first observed through the demonstration that the more compact chromatin structure of a replicated reporter plasmid acted as a barrier to Pol II-mediated elongation and resulted in higher exon inclusion [19]. Nucleosome positioning, histone modification and DNA methylation have been found to interfere with AS. Nucleosomes are preferentially positioned at exons and are thereby presupposed to create transient pauses to Pol II-mediated elongation, providing extra time for the recognition of 3’ splice sites by splicing factors [21]. Histone post-translational modifications represent a main regulator of AS (reviewed in ref [16, 19]) and acts through two distinct mechanisms. First, as illustrated in Figure 2B, some histone modifications, such as acetylation of histone 3 at Lys9 (H3K9ac), increase Pol II-mediated elongation and thereby favor exon skipping. Schor et al. demonstrated this for the neural cell adhesion molecule (NCAM) exon 18 upon neuron depolarization [19, 22]. Alternatively, the same study showed that neuron differentiation promoted inclusion of exon 18 in NCAM through H3K9 methylation (H3K9me), causing a reduction in Pol II-mediated elongation [19, 22]. Second, histone modifications can trigger the recruitment of factors influencing splicing, such as in the case of H3K36me3 at the fibroblast growth factor receptor 2 (FGFR2) locus that recruits the negative splicing factor PTB through the adaptor protein MRG15, resulting in exclusion of an alternative exon [23]. Abnormal DNA methylation represents a key pathogenic pathway in AML, yet its pathogenicity has mainly been linked to the global transcriptional deregulation of key genes involved in tumor development [24]. However recent studies have suggested that CpG methylation and transcriptional silencing are not synonymous [25]. Figure 2A gives a schematic example of how CpG differential methylation might influence AS [20]. Indeed, in the above example of CD45 splicing, Shukla et al. showed that DNA methylation of the intronic site prevented CTCF binding, releasing Pol II and thereby facilitating the skipping of exon 5 [20]. Epigenetic modifications are the hallmark of AML and several recent studies have highlighted the misregulation of DNA and histone methylation in this disease [25]. For example the role of abnormal DNA methylation in leukemia has recently been reinforced by the discovery of DNMT3A [26] and TET2 mutations [27] in about 20% of AML. DNMT3A is an active DNA methyltransferase [25]. Its gene mutations are regularly heterozygous and are predicted to disrupt the catalytic activity of the enzyme. TET2 converts 5mC into several oxidative intermediates, including 5-hydroxymethylcytosine (5hmC), which are likely involved in the process of active DNA demethylation. Similarly to DNMT3a substitutions in AML, patient-associated TET2 mutations are largely loss-of-function mutations that consequently result in decreased 5hmC levels and a reciprocal increase in 5mC. Regarding the deregulation of histone methylation, several histone lysine methyltransferases (KMT) have been found mutated in cancers. For example inactivation of EZH2 by loss or mutation are present in in MDS and, to a lesser extent in AML [25]. EZH2 is the catalytic component of the PRC2 complex, which is primarily responsible for the methylation of H3K27. ASXL1 is an additional epigenetic modifier found mutated in AML. ASXL1 mutations result in loss of polycomb repressive complex 2 (PRC2)-mediated histone H3K27 tri-methylation. Mutations in ASXL1 exon 12 are present in 5% - 30% of de novo AML where its frequency is 5 times higher in older patients [28], whereas some studies suggest a higher prevalence in secondary AML. Given the aforementioned link between missplicing and epigenetic changes, it will be therefore an important task to assess whether or not; missplicing is involved in the functional consequences of the mutation of epigenetic modifiers such as TET2, EZH2 and ASXL1 in AML.
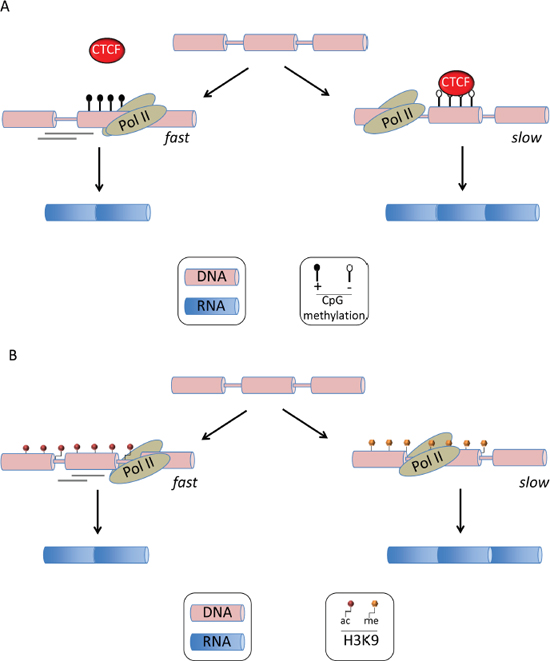
Figure 2: Epigenetic alterations in alternative mRNA splicing. (A) DNA CpG methylation influences mRNA splicing. The figure illustrates the role of differential CpG methylation in the recruitment of cellular factors that create a roadblock to Pol-II-mediated elongation, resulting in stalling of transcription and favoring exon inclusion. This has been demonstrated by Shukla et al. with CTCF that binds unmethylated CG-rich DNA sequences located downstream of an alternative exon [20]. (B) Histone post-translational modifications influence mRNA splicing. Acetylation of histone 3 at Lys9 has been demonstrated by Schor et al. to increase Pol II-mediated elongation and thereby favoring exon skipping.
WT1 possesses pleiotropic effects on the splicing machinery
WT1 expression in AML
The Wilms’ tumor gene (WT1) was among the first tumor suppressor genes to be cloned [29]. Originally named for its role in the pediatric kidney malignancy, Wilms’ tumor, it has since been implicated in many other cancers including hematologic malignancies. In AML ex vivo, reduction of WT1 expression levels leads to decrease of proliferation and apoptosis of leukemic cells [30], indicating that WT1 acts as an oncogene in these diseases. Although the diagnostic level of WT1 expression does not seem to possess a significant prognostic impact, its decreasing level upon induction of chemotherapy has been found to correlate with subsequent favorable outcome. WT1 mutations occur in about 10% of AML with normal karyotype, whereas their prognostic impact remains unclear. In addition to its prognostic usefulness, WT1 has become a broadly used marker for minimal residual diseases and a promising therapeutic target for anti-sense molecules, antibodies, and vaccine strategies [31].
More than 30 different WT1 isoforms are generated by a combination of alternative RNA splicing, the usage of different start codons and RNA editing [32]. The most widely studied isoforms are the inclusion or exclusion of exon 5 and an alternative splice donor site in exon 9, which encodes three amino acids, KTS (Figure 3). Thus, WT1 can be exon 5+ or exon 5- and KTS+ or KTS-, and all four isoforms are expressed in several tissues [33]. Interestingly, some of these isoforms exhibit specific biological properties, and their expressions depend on the age of patients and disease phenotype (i.e. MDS vs. AML) [34, 35].
At the molecular level, WT1 is a zinc-finger DNA-binding protein that can act as a transcriptional activator or repressor depending on the cellular or chromosomal context [36] (Figure 3). As a zinc finger transcription factor, WT1 can bind DNA, RNA, and protein, affecting the flow of eukaryotic genetic information from transcription to translation. WT1 isoforms possess distinct biochemical properties. The -KTS isoform colocalizes in the nucleus with regions of active transcription and binds DNA, RNA, and protein, whereas the +KTS isoform is capable of binding to RNA and proteins and colocalizes with splicing machinery in nuclear speckles [37]
Direct and indirect WT1-RNA interactions
The WT1-RNA interaction was first suggested by Larsson et al. who found that the +KTS WT1 isoform colocalizes with small nuclear RNA-protein particles (snRPs) in COS7 cells {Larsson, 1995 #148}. The mouse WT1 +KTS counterpart was further observed to accumulate on nascent transcripts when transfected in Xenopus oocyes [38], while Ladomery et al. [39] and Morrison et al. [40] identified the presence of WT1 in messenger ribonucleoprotein particles.
Yeast two-hybrid screens identified the splice factors U2AF2 (U2AF65) [41], WTAP [42], and RBM4 [42] as WT1-interacting proteins (Figure 4A). U2 auxiliary factor 2 (U2AF2/65) interacts with the zinc finger domain of both + and –KTS WT1 isoforms via a serine-arginine domain within its N terminus. WT1 associated protein (WTAP) bounds the +KTS but not -KTS WT1 isoform (Figure 2A), and is involved in 3’ splice site selection [41]. Interestingly WTAP has been recently found to play an important role in abnormal proliferation and arrested differentiation of leukemia cells [44]. RNA-binding motif protein 4 (RBM4) is a splice factor that promotes skeletal muscle-specific exons [45] and is involved in stem-cell differentiation in the central nervous system [46] and the pancreas [47]. It is also involved in embryonic development in Drosophila and translational control in mouse and human [48, 49]. Markus et al. identified RBM4 as a binding partner of WT1 through yeast two-hybrid assays [43 d]. In contrast to U2AF65 and similar to WTAP, RBM4 is specific to the +KTS WT1 isoform [43] (Figure 2A). Mechanistically, minigene experiments have shown that WT1 counteracted the splicing effect of RBM4. Morrison et al. found that WT1 cofractionated and co-immunoprecipitated with the splice factor PSF in nuclear extracts prepared from mouse mesonephric fetal kidney M15 cells [50].
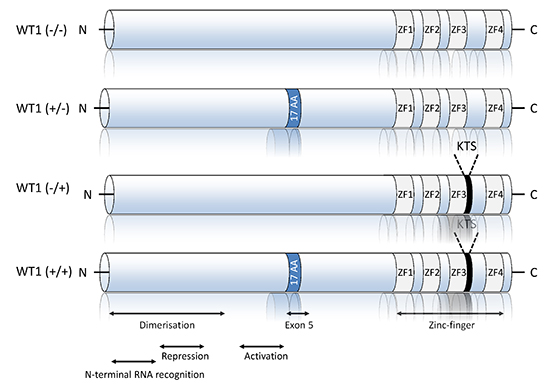
Figure 3: Schematic representation of the four main Wilms tumor suppressor gene (WT1) isoforms. The best studied splice variants of WT1 include mammalian-specific 17 amino acids encoded by exon 5 and three amino acids (KTS) at the terminal end of zinc finger three, encoded by an alternative splice donor site in exon 9. These generate the four main WT1 isoforms, with molecular masses of 52-54 kDa. The N-terminal domain includes dimerization, transcriptional activation, RNA recognition [41, 66, 67] and repression domains. DNA and RNA binding activities are both mediated by the zinc-finger domain.
Besides its interaction with splice factors, WT1 interacts with RNA processing through RNA binding via its zinc finger domain [38, 51]. Accordingly WT1 is not restricted to the nucleus but shuttles between the nucleus and cytoplasm, where it is involved in the regulation of mRNA export, localization, and translation. In vitro binding assays have shown that a hairpin loop is critically required for RNA binding by WT1 zinc fingers [38]. Interestingly Morrison et al. found through immunoprecipitation coupled with PCR differential-display that WT1 interacted with the RNA of p54nrb, which encodes a splice factor related to PSF [50].
Functional and pathogenic consequences of WT1-RNA interactions
The above-summarized interactions between WT1 and the splicing machinery are presupposed to alter the alternative exon usage that characterizes AML cells [4]. Two recent studies have addressed the consequences of WT1-RNA interactions and unmasked the effects of WT1 mutants on the splicing of the vascular endothelial growth factor (VEGF) and its consequences on angiogenesis, hematopoiesis, and tumor development [51, 52].
Angiogenesis is a key pathogenic mechanism in cancer and leukemia [54]. It is positively and negatively regulated by VEGF165 and VEGF165b splice isoforms, respectively [55]. VEGF splicing is controlled at least in part by serine-arginine-rich proteins (SRSFs), which include SRSF1, also called ASF, SF2, SF2/ASF, or ASF/SF2 [56]. SRSF1 promotes the expression of the angiogenic VEGF-A165 isoform via the selection of a specific 3’ proximal splicing site (3’ PSS) whereas the anti-angiogenic VEGF-A165b is expressed through the selection of a 3’ distal splicing site (3’ DSS, Figure 4B). Its nuclear localization is brought about by phosphorylation by a number of splicing factors, including SRPK1 [57], and Amin et al. demonstrated that WT1 isoforms lacking the KTS domain bind to the SRPK1 promoter and repress expression through a specific WT1 binding site [52] (Figure 4B). This repression results in SRSF1 hypophosphorylation that results in the selection of the 3’ DSS, thereby inhibiting angiogenesis through the expression of the VEGF-A165b isoform. In contrast, certain WT1 mutants carrying a substitution in the zinc-finger domain were unable to repress SRPK1 expression, leading to a SRPK1-mediated phosphorylation of SRSF1 that triggers the expression of the pro-angiogenic VEGF-A165 isoform [52].
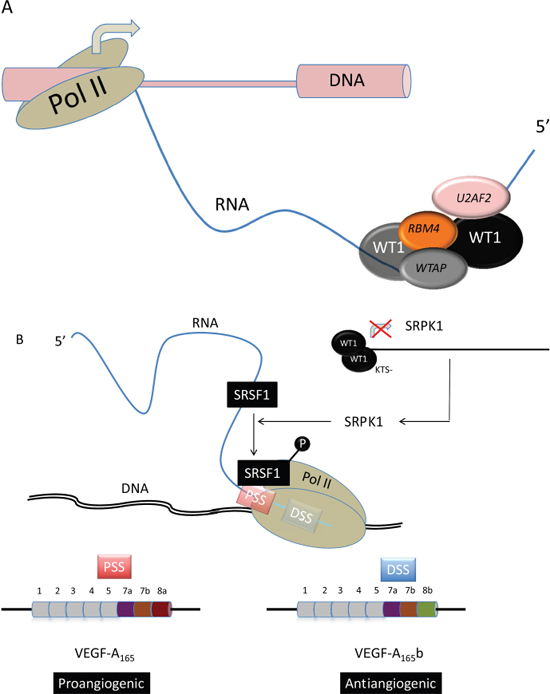
Figure 4: WT1 in alternative mRNA splicing. (A) WT1 interacts with U2AF2, RBM4, and WTAP splicing factors. (B) -KTS (Figure 3) WT1 represses SRPK1 transcription, leading to SRSF1 hypophosphorylation and thereby for the selection of a 3' proximal splicing site (PSS) during VEGF-A synthesis. This PSS usage results in the expression of the VEGF-A165 pro-angiogenic factor [52, 57].
In addition to their differential roles in angiogenesis, VEGF isoforms have been found to possess distinct effect on hematopoiesis. Cunningham et al. showed that WT1-deficient mouse embryonic stem cells exhibit reduced hematopoietic potential [53]. This diminished hematopoiesis was caused by a VEGF-A-dependent apoptosis of hematopoietic progenitor cells associated with a shift in VEGF-A isoforms toward VEGF-A 120. Interestingly, high levels of VEGF-121 (the human counterpart of murine VEGF-A120) have recently been identified as an independent prognostic factor associated with poor survival in AML [58].
Conclusion and perspective
In addition to acquired and/or selected somatic mutations, AS offers AML cells a rapid, dynamic, and reversible means to deal with their environment, which depends on numerous factors influencing tumor development, maintenance and recurrence. These influencing factors include the bone marrow stroma, immune system and treatments. Given the huge excess of abnormally spliced genes in AML [4], it is reasonable to propose that alteration of splicing regulation participates in the phenotypic plasticity of AML cells. Accordingly, validating the in silico evidenced pathways deregulated in AML upon RNA missplicing will allow better assessment of the pathogenic implications of AS in this disease [4]. The example of S34F U2AF1 mutation, which has been demonstrated to trigger a specific pattern of AS [5], suggests that specific patterns of AS possessing specific functional consequences might be related to specific defects in the splicing process.
Numerous questions remain to be addressed (Figure 5). For example, with the present reviewed data in mind, it becomes important to determine whether and how histone modifications, DNA methylation and WT1 expression modify AS and participate in the AML AS landscape. The main biological prognostic factors in AML have consisted of cytogenetic, gene mutations and transcriptional deregulations. Some patterns of AS have been found to correlate with tumor aggressiveness in solid tumors [58] while in AML, the expression of certain isoforms, such as for WT1 itself, TP53 [60], HOXA9 [61], BAALC [62], VEGF [58], or BCL-X [63], have been found to exhibit distinct effects on disease outcome. This encourages the investigation of the prognostic implication of the newly evidenced myriad of AML-associated alternative exon events [4]. Histone deacetylase inhibitors, DNA hypomethylating agents, and WT1 vaccination are currently used in many AML patients. Given that epigenetic and WT1 interfere with AS, it is possible that the antileukemic effect of these drugs relies at least in part on modification of AS. A recent meta analysis of published cancer vaccine trials has shown that objective clinical responses (including stable disease) are observed in 64% of evaluable WT1-vaccinated patients with hematological malignancies while immunogenicity of WT1-based cancer vaccines was demonstrated by the detection of a specific immunological response in 68% of cases [64]. Some drugs directly target the spliceosome, such as spliceostatin that targets SF3B1, have been found to possess antitumor effect ex vivo [14]. Whether these specific splicing factor inhibitors act in non-mutated AML remains to be assessed. In addition to these approachs, steric-blocking oligonucleotides have been designed to specifically redirect alternative splicing, repair defective RNA, and restore protein production. Although these product were designed to treat genetic disorders such as Duchenne muscular dystrophy [65], it will be interesting to develop such strategy to target the specific splicing defects observed in AML.
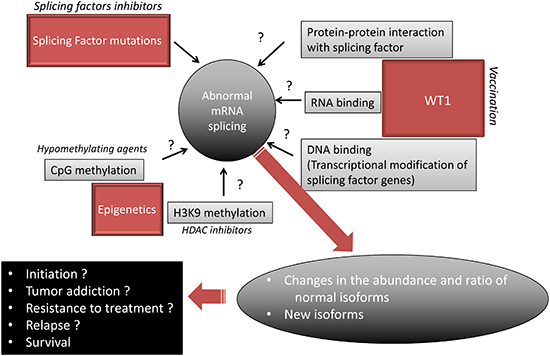
Figure 5: Proposed model of mRNA missplicing in AML. Hypothetical mechanisms are denoted by a question mark. A targeted treatment is represented for each proposed mechanism.
Conflict of interest
The authors declare no conflict of interest
REFERENCES
1. Blencowe BJ. Alternative splicing: new insights from global analyses. Cell. 2006; 126:37–47.
2. Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010; 463:457–463.
3. David CJ, Manley JL. Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes & development. 2010; 24:2343–2364.
4. Adamia S, Haibe-Kains B, Pilarski PM, Bar-Natan M, Pevzner S, Avet-Loiseau H, Lode L, Verselis S, Fox EA, Burke J, Galinsky I, Dagogo-Jack I, Wadleigh M, Steensma DP, Motyckova G, Deangelo DJ, et al. A genome-wide aberrant RNA splicing in patients with acute myeloid leukemia identifies novel potential disease markers and therapeutic targets. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014; 20:1135–1145.
5. Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, Chalkidis G, Suzuki Y, Shiosaka M, Kawahata R, Yamaguchi T, Otsu M, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011; 478:64–69.
6. Lasho TL, Finke CM, Hanson CA, Jimma T, Knudson RA, Ketterling RP, Pardanani A, Tefferi A. SF3B1 mutations in primary myelofibrosis: clinical, histopathology and genetic correlates among 155 patients. Leukemia. 2012; 26:1135–1137.
7. Malcovati L, Papaemmanuil E, Bowen DT, Boultwood J, Della Porta MG, Pascutto C, Travaglino E, Groves MJ, Godfrey AL, Ambaglio I, Galli A, Da Via MC, Conte S, Tauro S, Keenan N, Hyslop A, et al. Clinical significance of SF3B1 mutations in myelodysplastic syndromes and myelodysplastic/myeloproliferative neoplasms. Blood. 2011; 118:6239–6246.
8. Wan Y, Wu CJ. SF3B1 mutations in chronic lymphocytic leukemia. Blood. 2013; 121:4627–4634.
9. Je EM, Yoo NJ, Kim YJ, Kim MS, Lee SH. Mutational analysis of splicing machinery genes SF3B1, U2AF1 and SRSF2 in myelodysplasia and other common tumors. International journal of cancer Journal international du cancer. 2013; 133:260–265.
10. Graubert TA, Shen D, Ding L, Okeyo-Owuor T, Lunn CL, Shao J, Krysiak K, Harris CC, Koboldt DC, Larson DE, McLellan MD, Dooling DJ, Abbott RM, Fulton RS, Schmidt H, Kalicki-Veizer J, et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nature genetics. 2012; 44:53–57.
11. Thol F, Kade S, Schlarmann C, Loffeld P, Morgan M, Krauter J, Wlodarski MW, Kolking B, Wichmann M, Gorlich K, Gohring G, Bug G, Ottmann O, Niemeyer CM, Hofmann WK, Schlegelberger B, et al. Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood. 2012; 119:3578–3584.
12. Visconte V, Makishima H, Maciejewski JP, Tiu RV. Emerging roles of the spliceosomal machinery in myelodysplastic syndromes and other hematological disorders. Leukemia. 2012; 26:2447–2454.
13. Przychodzen B, Jerez A, Guinta K, Sekeres MA, Padgett R, Maciejewski JP, Makishima H. Patterns of missplicing due to somatic U2AF1 mutations in myeloid neoplasms. Blood. 2013; 122:999–1006.
14. Bonnal S, Vigevani L, Valcarcel J. The spliceosome as a target of novel antitumour drugs. Nature reviews Drug discovery. 2012; 11:847–859.
15. Abdel-Wahab O, Levine R. The spliceosome as an indicted conspirator in myeloid malignancies. Cancer cell. 2011; 20:420–423.
16. Luco RF, Allo M, Schor IE, Kornblihtt AR, Misteli T. Epigenetics in alternative pre-mRNA splicing. Cell. 2011; 144:16–26.
17. Ameur A, Zaghlool A, Halvardson J, Wetterbom A, Gyllensten U, Cavelier L, Feuk L. Total RNA sequencing reveals nascent transcription and widespread co-transcriptional splicing in the human brain. Nature structural & molecular biology. 2011; 18:1435–1440.
18. Das R, Yu J, Zhang Z, Gygi MP, Krainer AR, Gygi SP, Reed R. SR proteins function in coupling RNAP II transcription to pre-mRNA splicing. Molecular cell. 2007; 26:867–881.
19. Kornblihtt AR, Schor IE, Allo M, Dujardin G, Petrillo E, Munoz MJ. Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nature reviews Molecular cell biology. 2013; 14:153–165.
20. Shukla S, Kavak E, Gregory M, Imashimizu M, Shutinoski B, Kashlev M, Oberdoerffer P, Sandberg R, Oberdoerffer S. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature. 2011; 479:74–79.
21. Andersson R, Enroth S, Rada-Iglesias A, Wadelius C, Komorowski J. Nucleosomes are well positioned in exons and carry characteristic histone modifications. Genome research. 2009; 19:1732–1741.
22. Schor IE, Rascovan N, Pelisch F, Allo M, Kornblihtt AR. Neuronal cell depolarization induces intragenic chromatin modifications affecting NCAM alternative splicing. Proceedings of the National Academy of Sciences of the United States of America. 2009; 106:4325–4330.
23. Luco RF, Pan Q, Tominaga K, Blencowe BJ, Pereira-Smith OM, Misteli T. Regulation of alternative splicing by histone modifications. Science. 2010; 327:996–1000.
24. Itzykson R, Fenaux P. Epigenetics of myelodysplastic syndromes. Leukemia. 2014; 28:497–506.
25. Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012; 150:12–27.
26. Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J, Harris CC, Lichti CF, Townsend RR, Fulton RS, Dooling DJ, Koboldt DC, et al. DNMT3A mutations in acute myeloid leukemia. The New England journal of medicine. 2010; 363:2424–2433.
27. Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, Kosmider O, Le Couedic JP, Robert F, Alberdi A, Lecluse Y, Plo I, Dreyfus FJ, Marzac C, Casadevall N, Lacombe C, et al. Mutation in TET2 in myeloid cancers. The New England journal of medicine. 2009; 360:2289–2301.
28. Metzeler KH, Becker H, Maharry K, Radmacher MD, Kohlschmidt J, Mrozek K, Nicolet D, Whitman SP, Wu YZ, Schwind S, Powell BL, Carter TH, Wetzler M, Moore JO, Kolitz JE, Baer MR, et al. ASXL1 mutations identify a high-risk subgroup of older patients with primary cytogenetically normal AML within the ELN Favorable genetic category. Blood. 2011; 118:6920–6929.
29. Call KM, Glaser T, Ito CY, Buckler AJ, Pelletier J, Haber DA, Rose EA, Kral A, Yeger H, Lewis WH, et al. Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms' tumor locus. Cell. 1990; 60:509–520.
30. Nishida S, Hosen N, Shirakata T, Kanato K, Yanagihara M, Nakatsuka S, Hoshida Y, Nakazawa T, Harada Y, Tatsumi N, Tsuboi A, Kawakami M, Oka Y, Oji Y, Aozasa K, Kawase I, et al. AML1-ETO rapidly induces acute myeloblastic leukemia in cooperation with the Wilms tumor gene, WT1. Blood. 2006; 107:3303–3312.
31. Ochsenreither S, Fusi A, Busse A, Bauer S, Scheibenbogen C, Stather D, Thiel E, Keilholz U, Letsch A. “Wilms Tumor Protein 1” (WT1) peptide vaccination-induced complete remission in a patient with acute myeloid leukemia is accompanied by the emergence of a predominant T-cell clone both in blood and bone marrow. Journal of immunotherapy. 2011; 34:85–91.
32. Rivera MN, Haber DA. Wilms' tumour: connecting tumorigenesis and organ development in the kidney. Nature reviews Cancer. 2005; 5:699–712.
33. Morrison AA, Viney RL, Ladomery MR. The post-transcriptional roles of WT1, a multifunctional zinc-finger protein. Biochimica et biophysica acta. 2008; 1785:55–62.
34. Kramarzova K, Stuchly J, Willasch A, Gruhn B, Schwarz J, Cermak J, Machova-Polakova K, Fuchs O, Stary J, Trka J, Boublikova L. Real-time PCR quantification of major Wilms' tumor gene 1 (WT1) isoforms in acute myeloid leukemia, their characteristic expression patterns and possible functional consequences. Leukemia. 2012; 26:2086–2095.
35. Luna I, Such E, Cervera J, Barragan E, Ibanez M, Gomez-Segui I, Lopez-Pavia M, Llop M, Fuster O, Dolz S, Oltra S, Alonso C, Vera B, Lorenzo I, Martinez-Cuadron D, Montesinos P, et al. WT1 isoform expression pattern in acute myeloid leukemia. Leukemia research. 2013; 37:1744–1749.
36. Rauscher FJ 3rd, Morris JF, Tournay OE, Cook DM, Curran T. Binding of the Wilms' tumor locus zinc finger protein to the EGR-1 consensus sequence. Science. 1990; 250:1259–1262.
37. Larsson SH, Charlieu JP, Miyagawa K, Engelkamp D, Rassoulzadegan M, Ross A, Cuzin F, van Heyningen V, Hastie ND. Subnuclear localization of WT1 in splicing or transcription factor domains is regulated by alternative splicing. Cell. 1995; 81:391–401.
38. Ladomery M, Sommerville J, Woolner S, Slight J, Hastie N. Expression in Xenopus oocytes shows that WT1 binds transcripts in vivo, with a central role for zinc finger one. Journal of cell science. 2003; 116:1539–1549.
39. Ladomery MR, Slight J, Mc Ghee S, Hastie ND. Presence of WT1, the Wilm's tumor suppressor gene product, in nuclear poly(A)(+) ribonucleoprotein. The Journal of biological chemistry. 1999; 274:36520–36526.
40. Morrison AA, Ladomery MR. Presence of WT1 in nuclear messenger RNP particles in the human acute myeloid leukemia cell lines HL60 and K562. Cancer letters. 2006; 244:136–141.
41. Davies RC, Calvio C, Bratt E, Larsson SH, Lamond AI, Hastie ND. WT1 interacts with the splicing factor U2AF65 in an isoform-dependent manner and can be incorporated into spliceosomes. Genes & development. 1998; 12:3217–3225.
42. Little NA, Hastie ND, Davies RC. Identification of WTAP, a novel Wilms' tumour 1-associating protein. Human molecular genetics. 2000; 9:2231–2239.
43. Markus MA, Heinrich B, Raitskin O, Adams DJ, Mangs H, Goy C, Ladomery M, Sperling R, Stamm S, Morris BJ. WT1 interacts with the splicing protein RBM4 and regulates its ability to modulate alternative splicing in vivo. Experimental cell research. 2006; 312:3379–3388.
44. Bansal H, Yihua Q, Iyer SP, Ganapathy S, Proia D, Penalva LO, Uren PJ, Suresh U, Carew JS, Karnad AB, Weitman S, Tomlinson GE, Rao MK, Kornblau SM, Bansal S. WTAP is a novel oncogenic protein in acute myeloid leukemia. Leukemia. 2014.
45. Kar A, Havlioglu N, Tarn WY, Wu JY. RBM4 interacts with an intronic element and stimulates tau exon 10 inclusion. The Journal of biological chemistry. 2006; 281:24479–24488.
46. Brooks YS, Wang G, Yang Z, Smith KK, Bieberich E, Ko L. Functional pre- mRNA trans-splicing of coactivator CoAA and corepressor RBM4 during stem/progenitor cell differentiation. The Journal of biological chemistry. 2009; 284:18033–18046.
47. Lin JC, Yan YT, Hsieh WK, Peng PJ, Su CH, Tarn WY. RBM4 promotes pancreas cell differentiation and insulin expression. Molecular and cellular biology. 2013; 33:319–327.
48. Markus MA, Morris BJ. Lark is the splicing factor RBM4 and exhibits unique subnuclear localization properties. DNA and cell biology. 2006; 25:457–464.
49. Kojima S, Matsumoto K, Hirose M, Shimada M, Nagano M, Shigeyoshi Y, Hoshino S, Ui-Tei K, Saigo K, Green CB, Sakaki Y, Tei H. LARK activates post transcriptional expression of an essential mammalian clock protein, PERIOD1. Proceedings of the National Academy of Sciences of the United States of America. 2007; 104:1859–1864.
50. Morrison AA, Venables JP, Dellaire G, Ladomery MR. The Wilms tumour suppressor protein WT1 (+KTS isoform) binds alpha-actinin 1 mRNA via its zinc-finger domain. Biochemistry and cell biology = Biochimie et biologie cellulaire. 2006; 84:789–798.
51. Caricasole A, Duarte A, Larsson SH, Hastie ND, Little M, Holmes G, Todorov I, Ward A. RNA binding by the Wilms tumor suppressor zinc finger proteins. Proceedings of the National Academy of Sciences of the United States of America. 1996; 93:7562–7566.
52. Amin EM, Oltean S, Hua J, Gammons MV, Hamdollah-Zadeh M, Welsh GI, Cheung MK, Ni L, Kase S, Rennel ES, Symonds KE, Nowak DG, Royer-Pokora B, Saleem MA, Hagiwara M, Schumacher VA, et al. WT1 mutants reveal SRPK1 to be a downstream angiogenesis target by altering VEGF splicing. Cancer cell. 2011; 20:768–780.
53. Cunningham TJ, Palumbo I, Grosso M, Slater N, Miles CG. WT1 regulates murine hematopoiesis via maintenance of VEGF isoform ratio. Blood. 2013; 122:188–192.
54. Shih TT, Hou HA, Liu CY, Chen BB, Tang JL, Chen HY, Wei SY, Yao M, Huang SY, Chou WC, Hsu SC, Tsay W, Yu CW, Hsu CY, Tien HF, Yang PC. Bone marrow angiogenesis magnetic resonance imaging in patients with acute myeloid leukemia: peak enhancement ratio is an independent predictor for overall survival. Blood. 2009; 113:3161–3167.
55. Cebe Suarez S, Pieren M, Cariolato L, Arn S, Hoffmann U, Bogucki A, Manlius C, Wood J, Ballmer-Hofer K. A VEGF-A splice variant defective for heparan sulfate and neuropilin-1 binding shows attenuated signaling through VEGFR-2. Cellular and molecular life sciences : CMLS. 2006; 63:2067–2077.
56. Nowak DG, Amin EM, Rennel ES, Hoareau-Aveilla C, Gammons M, Damodoran G, Hagiwara M, Harper SJ, Woolard J, Ladomery MR, Bates DO. Regulation of vascular endothelial growth factor (VEGF) splicing from pro-angiogenic to anti-angiogenic isoforms: a novel therapeutic strategy for angiogenesis. The Journal of biological chemistry. 2010; 285:5532–5540.
57. Zhong XY, Ding JH, Adams JA, Ghosh G, Fu XD. Regulation of SR protein phosphorylation and alternative splicing by modulating kinetic interactions of SRPK1 with molecular chaperones. Genes & development. 2009; 23:482–495.
58. Mourah S, Porcher R, Lescaille G, Rousselot P, Podgorniak MP, Labarchede G, Naimi B, Medioni J, Dombret H, Calvo F. Quantification of VEGF isoforms and VEGFR transcripts by qRT-PCR and their significance in acute myeloid leukemia. The International journal of biological markers. 2009; 24:22–31.
59. Dutertre M, Lacroix-Triki M, Driouch K, de la Grange P, Gratadou L, Beck S, Millevoi S, Tazi J, Lidereau R, Vagner S, Auboeuf D. Exon-based clustering of murine breast tumor transcriptomes reveals alternative exons whose expression is associated with metastasis. Cancer research. 2010; 70:896–905.
60. Anensen N, Hjelle SM, Van Belle W, Haaland I, Silden E, Bourdon JC, Hovland R, Tasken K, Knappskog S, Lonning PE, Bruserud O, Gjertsen BT. Correlation analysis of p53 protein isoforms with NPM1/FLT3 mutations and therapy response in acute myeloid leukemia. Oncogene. 2012; 31:1533–1545.
61. He M, Chen P, Arnovitz S, Li Y, Huang H, Neilly MB, Wei M, Rowley JD, Chen J, Li Z. Two isoforms of HOXA9 function differently but work synergistically in human MLL-rearranged leukemia. Blood cells, molecules & diseases. 2012; 49:102–106.
62. Mizushima Y, Taki T, Shimada A, Yui Y, Hiraumi Y, Matsubara H, Watanabe M, Watanabe K, Kamitsuji Y, Hayashi Y, Tsukimoto I, Kobayashi R, Horibe K, Tawa A, Nakahata T, Adachi S. Prognostic significance of the BAALC isoform pattern and CEBPA mutations in pediatric acute myeloid leukemia with normal karyotype: a study by the Japanese Childhood AML Cooperative Study Group. International journal of hematology. 2010; 91:831–837.
63. Yamaguchi H, Inokuchi K, Dan K. The study for loss of bcl-xs expression as a prognostic factor in acute myeloid leukemia. Leukemia research. 2002; 26:1119–1123.
64. Van Driessche A, Berneman ZN, Van Tendeloo VF. Active specific immunotherapy targeting the Wilms' tumor protein 1 (WT1) for patients with hematological malignancies and solid tumors: lessons from early clinical trials. The oncologist. 2012; 17:250–259.
65. Kole R, Krainer AR, Altman S. RNA therapeutics: beyond RNA interference and antisense oligonucleotides. Nature reviews Drug discovery. 2012; 11:125–140.
66. Davies R, Moore A, Schedl A, Bratt E, Miyahawa K, Ladomery M, Miles C, Menke A, van Heyningen V, Hastie N. Multiple roles for the Wilms' tumor suppressor, WT1. Cancer research. 1999; 59:1747s–1750s; discussion 1751s.
67. Kennedy D, Ramsdale T, Mattick J, Little M. An RNA recognition motif in Wilms' tumour protein (WT1) revealed by structural modelling. Nature genetics. 1996; 12:329–331.