
INTRODUCTION
Salivary duct carcinoma (SDC) is an uncommon entity, and histologically resembles high-grade mammary ductal carcinoma [1]. It occurs as a de novo carcinoma (de novo SDC) or a carcinoma ex-pleomorphic adenoma (SDC ex-PA) [1]. SDCs are known to be one of the most aggressive types of salivary gland carcinoma, since patients frequently develop distant metastasis, even after curative resection [2, 3]. Surgical resection is the standard therapy for SDC; in many cases, postoperative radiotherapy is also performed [4]. Although there is no established regimen, systemic chemotherapy is often carried out in cases with distant metastasis [4, 5].
In the past decade, there have been remarkable advances in the research on multiple therapeutically-relevant genetic alterations in various cancers, and appropriate targeted therapies have been generated [6, 7]. In SDCs, however, there is little data in relation to personalized therapy, with the exception of therapies targeting HER2 and the androgen receptor (AR); evidence for which has gradually accumulated [4, 5]. On the other hand, recent studies in which limited comprehensive molecular profiling analyses were performed—especially those using next-generation sequencing—have revealed that several genes are altered in SDCs, including TP53 and genes related to cell signaling by receptor-tyrosine kinases, the PI3K/Akt pathway, and the RAS-MAPK cascade, which play integral roles in tumor growth and the survival of various types of carcinoma [8–17]. These studies have reported that the percentages of genetic alteration of TP53, ERBB2, PIK3CA, and H-RAS, are characteristically high in SDC. Although mutations of other key genes, such as CDKN2A, PTEN, KIT, and APC also have been described in the literature, those frequencies are low, being ≤ 10% for any of them [8–17]. Due to the rarity of the SDCs, the differences in the genetic mechanisms underlying carcinogenesis according to the histologic origin (i.e. de novo SDC or SDC ex-PA) [13, 14, 18] and the relationship between the aberrant expression of molecular markers and the clinical outcome have not been fully explored [8, 9, 14, 18]. However, it is essential to clarify these issues as an initial step in designing effective therapeutic options and—in the future—for selecting SDC patients who are candidates for targeted therapy.
The purpose of the present study, the largest cohort of SDCs to date, was to determine the gene alteration and expression status of selected potentially actionable cancer-related targets, and to evaluate their clinicopathological and prognostic significance in SDC patients.
RESULTS
Patient characteristics
The distribution of the patient characteristics is shown in Table 1. The median follow-up period of the survivors was 3.7 years (range, 0.4–18.7 years). The 3-year overall survival (OS) and progression-free survival (PFS) rates of the whole study population were 68.5% (95% confidence interval [CI]: 60.1–75.5) and 34.3% (95% CI: 26.7–42.1), respectively.
Table 1: Patient characteristics (n = 151)
Variables |
n |
% |
|---|---|---|
Age (years) |
||
< 65 |
84 |
56 |
≥ 65 |
67 |
44 |
Sex |
||
Male |
127 |
84 |
Female |
24 |
16 |
T classification |
||
1 |
13 |
8 |
2 |
39 |
26 |
3 |
30 |
20 |
4 |
69 |
46 |
N classification |
||
0 |
71 |
47 |
1 |
9 |
6 |
2 |
71 |
47 |
M classification |
||
0 |
142 |
94 |
1 |
9 |
6 |
Primary tumor site |
||
Parotid gland |
117 |
77 |
Submandibular gland |
30 |
20 |
Others |
4 |
3 |
Histologic origin |
||
De novo |
57 |
38 |
Ex-pleomorphic adenoma* |
89 |
59 |
Unknown |
5 |
3 |
*Salivary duct carcinomas ex-pleomorphic adenoma (SDCs ex-PA) include 13 intracapsular SDC ex-PA cases, 5 minimally invasive SDC ex-PA cases, and 71 widely invasive SDC ex-PA cases.
Gene alterations and the immunohistochemical findings
TP53 mutations were the most frequent genetic mutation among all patients with SDC (68%, 85/125 cases), followed by the PIK3CA (18%, 25/137 cases), H-RAS (16%, 23/140 cases), BRAF (3.7%, 5/136 cases), and AKT1 (1.5%, 2/133 cases) mutations. No K-RAS or N-RAS mutations were detected in any of the cases. Twenty-four of the patients showed no genetic mutations among the analyzed genes.
TP53 mutations were distributed throughout exons 4–10, and a subset of TP53-mutated cases simultaneously harbored missense, nonsense, and/or frameshift mutations in the same or different exons; 23 tumors (18% of the mutated tumors) carried more than one TP53 mutation (Supplementary Table 1). The percentages of missense (Figure 1A), nonsense, and frameshift TP53 mutations (Figure 1B) were 61%, 9.4%, and 27%, respectively, among the total mutations. The frameshift mutations (23 cases) included insertions (4 cases), deletions (19 cases) (Figure 1B), and in-frame mutations (2 cases). Consequently, the rates of each TP53 status were as follows: wild-type, 32%; missense mutation, 42%; and truncating mutation, 26%. PIK3CA mutations were detected in either exons 9 (14 cases) or 20 (11 cases) (Figure 1C) (Supplementary Table 2). The majority of H-RAS mutations were identified in exon 2 (21/23 cases) (Figure 1D) (Supplementary Table 2). All of the BRAF mutations were represented as a V600E mutation (Figure 1E). In addition, an AKT1 mutation was identified as an E17K mutation.
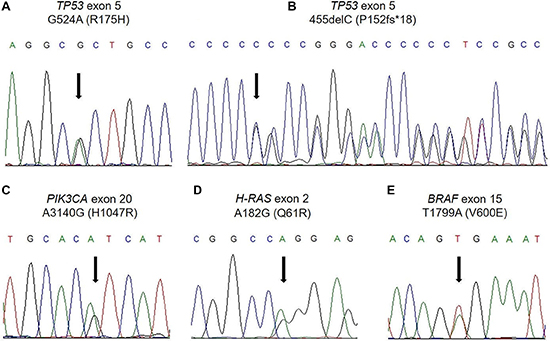
Figure 1: Examples of the direct DNA sequencing analysis. (A) TP53 missense mutation (R175H). (B) TP53 frameshift mutation (P152fs*18). (C) PIK3CA mutation (H1047R). (D) H-RAS mutation (Q61R). (E) BRAF mutation (V600E).
A dual color in situ hybridization (DISH) analysis revealed no EGFR gene amplification in any of the cases, whereas 14% (19/132 cases) of the cases demonstrated chromosome 7 polysomy (Figure 2).
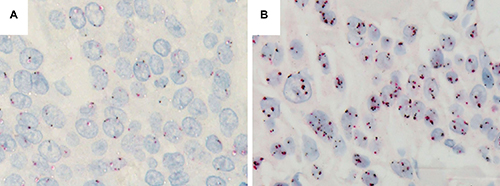
Figure 2: Dual color in situ hybridization for the EGFR gene. (A) No abnormalities. (B) The presence of chromosome 7 polysomy but no EGFR gene amplification (HER2 genes: black signal, CEN 17: red signal).
Positive immunoreactivity for PI3K and p-mTOR was exclusively observed in the cytoplasm, while p-Akt was sometimes detected in the nuclei and cytoplasm by immunostaining. The positive rate—in accordance with the sequential order of the signaling pathway axis—was as follows: PI3K, 34% (51/149 cases) (Figure 3A), p-Akt, 22% (32/147 cases), nuclear p-Akt, 14% (21/147 cases) (Figure 3B), and p-mTOR, 66% (98/149 cases) (Figure 3C). The loss of PTEN (Figure 3D) was observed in 49% of the cases (73/149 cases).
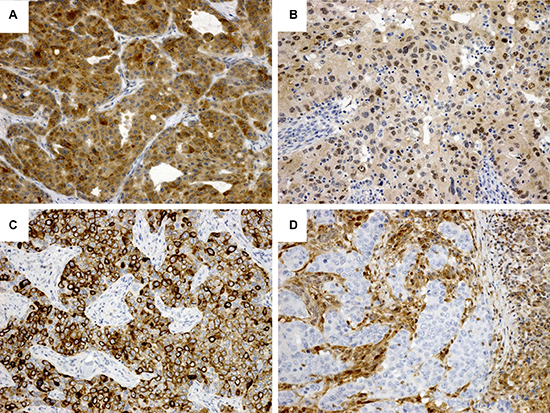
Figure 3: Immunohistochemical staining of the PI3K/Akt signaling pathway. (A) PI3K. Diffuse cytoplasmic staining. (B) p-Akt. Half of the tumor cells show both nuclear and cytoplasmic staining. (C) p-mTOR. Many tumor cells are positive with cytoplasmic and membranous staining pattern. (D) PTEN. Expression loss. Note that the expression of stromal and adjacent salivary gland parenchymal cells is retained.
Mutual relationship between the gene alterations and immunohistochemical findings
PIK3CA and H-RAS mutations, which were frequently co-mutated (P < 0.001) (Supplementary Table 2), had a mutually exclusive relationship with the HER2-positive status (P = 0.004 and < 0.001, respectively) (Table 2). In contrast, the HER2-positive cases commonly carried TP53 mutations (P = 0.044). Meanwhile, TP53 wild-type was associated with H-RAS mutations (P = 0.013). Cases harboring H-RAS mutations and chromosome 7 polysomy showed significantly lower and higher Ki-67 labeling index (LI) values, respectively (P = 0.001 and < 0.001, respectively). In addition, TP53 and PIK3CA mutations tended to have higher and lower Ki-67 LI values, respectively (P = 0.057 and 0.069, respectively).
Table 2: Mutual correlation between gene alterations and immunohistochemical findings in salivary duct carcinomas
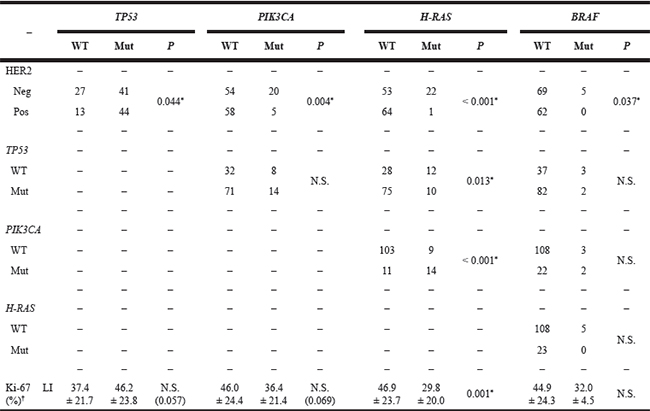
*Statistically significant (P < 0.05); †mean ± SD
Abbreviations: LI = labeling index; WT = wild type; Mut = mutation; Neg = negative; Pos = positive; N.S. = not significant.
With regard to the correlation between the TP53-mutation status and the p53-expression phenotype (Table 3) (Supplementary Table 1), TP53 mutations were more frequently identified in p53-extreme negative/positive cases than in p53-non-extreme cases (P = 0.002). Moreover, significant correlations were found between p53-extreme negative and TP53 truncating mutation and between p53-extreme positive and TP53 missense mutations (P < 0.001).
Table 3: Correlation between p53-expression phenotype and TP53 status
p53-expression phenotype |
n |
TP53 status |
||||
|---|---|---|---|---|---|---|
Wild type |
Mutation |
|||||
Missense |
Truncating |
|||||
Non-extreme |
71 |
31* |
40* |
|||
21 |
19 |
|||||
EN + EP |
53 |
9* |
44* |
|||
EN |
18 |
3 |
5† |
10† |
||
EP |
35 |
6 |
26† |
3† |
||
Total |
124 |
40 |
84 |
|||
*P = 0.002; †P < 0.001
Abbreviations: EN = extreme negative; EP = extreme positive.
The presence of chromosome 7 polysomy tended (with marginal significance [P = 0.065]) to be correlated with EGFR overexpression.
The interrelationship between the mutation and expression of molecules related to the PI3K/Akt signaling pathway is shown in Table 4. PTEN loss was found to be connected to PI3K negativity and nuclear p-Akt positivity (P = 0.006 and 0.007, respectively). Furthermore, both the overall and nuclear p-Akt expression were positively correlated with p-mTOR expression (P = 0.032 and 0.034, respectively). On the other hand, the PIK3CA status was not associated with the expression of PI3K/Akt signaling pathway proteins.
Table 4: Interrelation between the mutation and expression of molecules associated with the PI3K/Akt signaling pathway in salivary duct carcinomas
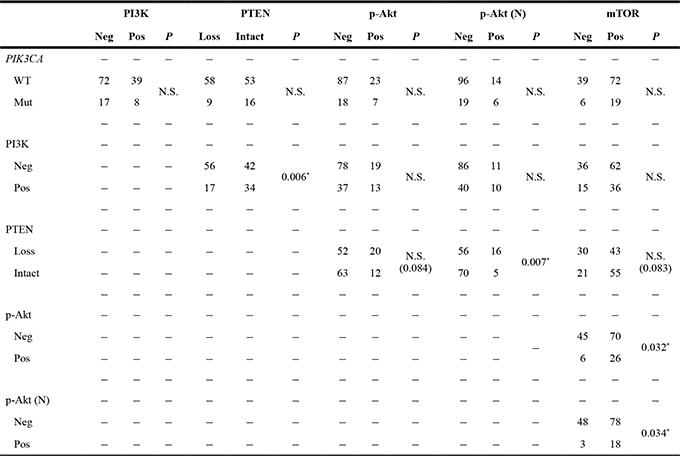
*Statistically significant (P < 0.05) Abbreviations: p-Akt (N) = p-Akt nuclear expression; WT = wild type; Mut = mutation; Neg = negative; Pos = positive; N.S. = not significant.
As reported previously, AR immunoreactivity was found in 144 of 150 cases (96%) [19]. Besides, PI3K (Figure 4A) and p-mTOR negativity, and the loss of PTEN (Figure 4B) were associated with a lower AR LI (P = 0.001, 0.098 [marginal significance], and < 0.001, respectively).
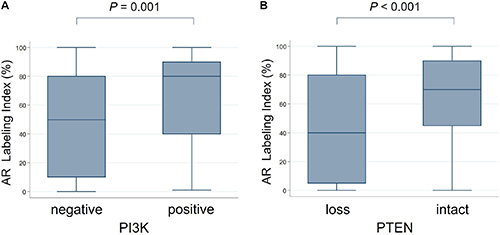
Figure 4: The comparison of androgen receptor labeling index (AR LI) with PI3K/PTEN expression. PI3K negativity (A) and PTEN loss (B) are significantly associated with lower AR LI (PI3K: mean [%] ± SD, 47.6 ± 34.5 versus 66.2 ± 29.6; PTEN: 42.7 ± 34.8 versus 64.7 ± 29.6).
Correlation between the molecular profile and clinicopathologic factors
The significant clinicopathological correlations included the presence of PIK3CA gene mutations with higher age (P = 0.025), PI3K and p-mTOR positivity with an early N classification (P = 0.039 and 0.028, respectively), and the loss of PTEN with an advanced T classification (P = 0.009).
Of note, the PIK3CA and H-RAS status showed significant differences according to the histological origin of SDC (Table 5). PIK3CA and H-RAS mutations were more commonly identified in de novo SDC than in SDC ex-PA (P < 0.001 for each). Similarly, the frequency of simultaneous PIK3CA and H-RAS mutations was also higher in de novo SDCs (P < 0.001). Even though the incidence was low, BRAF mutations were only found in de novo SDC. AKT1 mutations were identified in one case each of de novo SDC and SDC ex-PA. The histological origin was not significantly associated with the TP53 mutation status, chromosome 7 polysomy, or the expression of PI3K, p-Akt, p-mTOR, and PTEN.
Table 5: Association between the histologic origin and therapeutically-relevant genetic alterations in salivary duct carcinomas
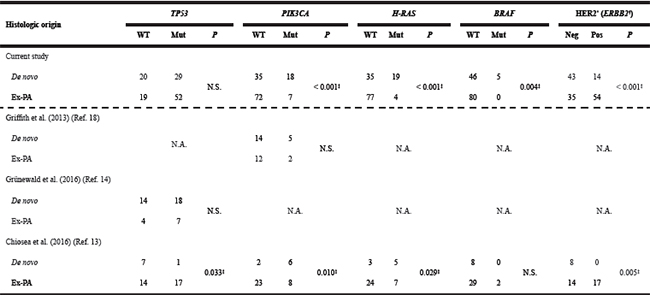
*HER2 status was assessed by a combination of immunohistochemistry and FISH analysis in accordance with the 2013 ASCO/CAP guideline. (Data from our previous report [Ref. 19])
†ERBB2 positive was defined as > 8 genes gain by next-generation sequencing (Ref. 13).
‡Statistically significant (P < 0.05)
Abbreviations: PA = pleomorphic adenoma; WT = wild type; Mut = mutation; Neg = negative; Pos = positive; N.S. = not significant; N.A. = not available.
Prognostic impact
The data of the univariate and multivariate Cox proportional hazard analyses of the molecular and immunohistochemical variables associated with survival are summarized in Table 6. The univariate analysis revealed that patients with TP53 mutated SDCs showed poorer PFS in comparison to those with carried wild-type TP53 (P = 0.017); however, the difference was not statistically significant in the multivariate analysis. Furthermore, the presence of a TP53 truncating mutation was a significant independent predictor of poor PFS in both the univariate and multivariate analyses (P = 0.008 and 0.004, respectively). While, p-Akt-positive expression was associated with a favorable clinical outcome indicated by better OS and PFS in a univariate analysis (P = 0.017 and 0.023, respectively); these associations showed marginal trend in the multivariate analysis (P = 0.064 and 0.106, respectively).
Table 6: Univariate and multivariate analysis of clinical outcomes in patients with salivary duct carcinoma
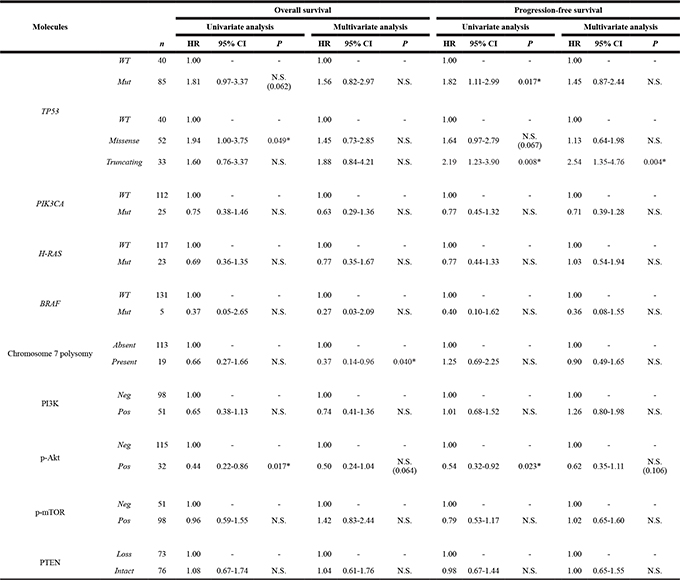
Adjusted by age, sex, primary site, TNM clasification, definitive treatment, and histologic origin.
*Statistically significant (P < 0.05)
Abbreviations: HR = hazard ratio; CI = confidence interval; WT = wild type; Mut = mutation; Neg = negative; Pos = positive; N.S. = not significant.
Kaplan-Meier survival curves indicated that patients with TP53 mutated SDC showed worse OS and PFS (P = 0.060 [marginal significance] and 0.015, respectively) than SDC patients with wild-type TP53; furthermore, TP53 truncating mutations were significantly associated with poorest PFS (P = 0.026) (Figure 5). In the HER2-negative subgroup, a TP53 mutation was an indicator of worse OS and PFS (P = 0.041 and 0.013, respectively). In contrast, p-Akt positivity was associated with higher OS and PFS (P = 0.013 and 0.018, respectively) (Figure 6). The other molecular and immunohistochemical factors that were examined showed no association with the survival rates.
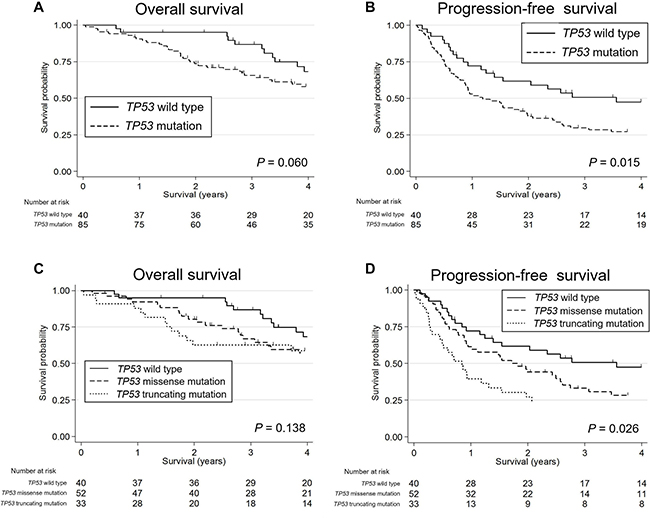
Figure 5: Kaplan-Meier survival curves of patients with salivary duct carcinoma stratified by their TP53 status. (A) TP53 mutated patients tended to show poorer overall survival (OS) than TP53 wild-type patients, with marginal significance. (B) The progression-free survival (PFS) of the TP53 mutated patients was significantly worse than that of the TP53 wild-type patients. (C) The OS of patients with TP53 wild-type, missense mutations, and truncating mutations tended to decrease in this order. (D) The PFS of patients with TP53 truncating mutations was significantly worst, followed in order by missense mutations and wild-type.
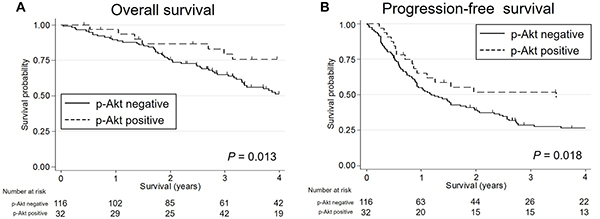
Figure 6: The Kaplan-Meier survival curves for patients with salivary duct carcinoma stratified by the p-Akt expression status. Both overall survival (A) and progression-free survival (B) were significantly worse in p-Akt-negative patients than in p-Akt-positive patients.
DISCUSSION
SDC has a very poor prognosis and the treatment options are limited [1–5]. We performed extensive molecular and immunohistochemical analyses of genes and biological markers either related to the driving tumorigenesis and/or relevant treatment targets in 151 cases of SDC. In patients with SDC, the most frequent gene alterations were observed in TP53, followed by PIK3CA, H-RAS, BRAF, and AKT1; this finding was in agreement with previous comprehensive genetic studies [9–17]. However, there is little in the way of convincing data in relation to the clinicopathological and prognostic implications of these molecular changes, mostly due to the rarity of SDC. Thus, the present study, which includes a large cohort of SDC patients, provides results that can more reliably interpret the associations between the mutation/expression status and clinicopathological features, including histogenesis and the clinical outcome. First, we found that PIK3CA/H-RAS/BRAF mutations were preferentially identified in de novo SDCs but not in SDCs ex-PA. These mutations and HER2 overexpression/amplification were mutually exclusive. Second, TP53 mutations were common events in SDC, and the truncating form of the given gene was independently associated with a worse prognosis. Additionally, TP53 mutations were frequently detected in cases with an extreme p53-negative/positive immunophenotype, and TP53 missense and truncating mutations were associated with extreme p53 positivity and negativity, respectively. Finally, we demonstrated that at least one-fifth of SDC cases aberrantly expressed PI3K/Akt signaling pathway proteins, along with a mutual correlation with the expression profile according to the axis. Furthermore, we revealed that p-Akt positivity indicates a favorable clinical outcome. Moreover, PI3K negativity and PTEN loss were associated with low AR expression. We believe that in the future these data will be important in the implementation of personalized therapy for SDC.
SDC can occur as a de novo tumor, and can develop through malignant transformation from PA. It has been shown that the survival of patients with SDC ex-PA and those with de novo SDC does not differ to a statistically significant extent [20, 21]. Recently, studies analyzing small cohorts have shown that the underlying molecular mechanism and therapeutically-relevant genetic alterations in SDC might differ between de novo and ex-PA (Table 5) [13, 14, 18, 19]. However, conflicting results have been reported [13, 14, 18]. Chiosea et al. examined 39 cases and found that PIK3CA and H-RAS mutations were predominantly detected in de novo SDCs, whereas TP53 mutations and ERBB2 amplification were more commonly observed in SDCs ex-PA [13]. They investigated the presence of pre-existing PA based on a combination of histomorphology and the PLAG1 and HMGA2 rearrangement status. On the other hand, Griffith et al. failed to find an association between the PIK3CA status and the histologic origin in their analysis of 34 cases [18]. In the study of 43 cases, Grünewald et al. demonstrated that the TP53 mutation rate was not influenced—to a statistically significant extent—by the histologic origin [14]. Our current analysis of much larger series of patients with SDC revealed hot spot mutations of PIK3CA and H-RAS in 18.2% and 16.2% of the cases, respectively, and showed the intimate relationship between the presence of PIK3CA mutations, frequently accompanied by simultaneous H-RAS mutations, and de novo occurrence. Although these findings confirm the data presented by Chiosea et al. [13], we identified cases of SDC ex-PA by a histomorphological evaluation using multi-step sections from the whole tumor. We found no significant association between the histological origin and the expression of PI3K, p-Akt, p-mTOR, and PTEN. Moreover, in agreement with the results presented by Grünewald et al., the TP53 mutation rate did not differ according to the histologic origin [14]. Conversely, although the mutation rate was low, BRAF mutations (V600E)—one of the major therapeutic targets [22]—were only detected in de novo SDCs. Furthermore, our data showed that PIK3CA/H-RAS/BRAF mutations and HER2 positivity are mutually exclusive, and that SDCs ex-PA more frequently overexpressed HER family members, including HER2 (and/or HER2 amplification), EGFR, and HER3 [19]. These findings indicate that PIK3CA and H-RAS mutations in de novo SDCs and the disruption of the HER family members in SDCs ex-PA may play crucial roles in their carcinogenesis. These insights should be considered when designing clinical trials and when planning potential targeted therapeutic strategies against SDCs based on the histological origin. Instead, consistent with previous studies, these molecular changes were not correlated with the patient outcome [8, 9, 18, 19]. The further clarification of the mechanisms associated with the development of SDCs is expected to lead to novel targeted therapies.
TP53 is well known to be the most frequently mutated gene in human malignancies; mutations are found in at least 50% of human cancers [23–25]. In the current study, TP53 mutations were found in 68% of the SDC cases, which is in line with previous reports [9, 13, 15, 16]. Pathogenic mutations result in the loss or abrogation of wild-type p53 activity and the dominant-negative effect on the remaining wild-type p53, in addition some p53 mutants often gain oncogenic functions to promote tumorigenesis and progression [26]. Given the broad role of p53 in malignancy, there is an intense focus on its potential as a therapeutic target [27, 28]. Furthermore, TP53 mutations have been shown to be an independent marker of a poor prognosis in cancers of several organs [23–25]. Meanwhile, it should be noted that the prognostic significance of TP53 mutations shows extreme variation according to the type of tumor [23–25]. In SDCs, it is not clear whether TP53 mutation independently predicts a poor prognosis. Most recently, Grünewald et al. reported that TP53 mutated SDCs tended to be associated with a worse OS; however, the result did not reach statistical significance due to the relatively small number of patients [14]. In the present study, TP53 mutations were significantly associated with a poor PFS in a univariate analysis, but not in the multivariate analysis. A subgroup analysis using Kaplan-Meier survival curves showed that TP53 mutations were associated with worse OS and PFS in HER2-negative patients. In general (not limited to SDCs) major TP53 mutations are missense and cause single amino-acid changes at many different positions [23–25]. The mutations, however, are diverse in their type, sequence context, and structural impact—these differences influence the biological behavior of various cancers [23–25]. Missense substitutions, particularly certain “hot spots” mutations, have been shown to induce loss of DNA binding activity, transactivation capacity, and dominant-negative effects, as well as gain-of-function (GOF) properties, including altered cancer spectrum, deregulated metabolic pathways, increased metastasis and enhanced chemotherapy resistance [28–30]. Conversely, truncating TP53 mutations, such as frameshift insertions and deletions and nonsense mutations, correspond to loss-of-function (LOF) TP53 mutations and are thought to give rise to p53-null alleles [31–33]. In ovarian carcinomas, it has been suggested that distinguishing between GOF and LOF TP53 mutations is clinically important as LOF mutations have been associated with worst prognosis in comparison to GOF mutations and wild-type [34, 35]. We found that in SDCs the rates of wild-type, missense mutation, and truncating mutation were 32%, 42%, and 26% of the cases, respectively. Thus, by dividing TP53 status into 3 types (wild-type, missense mutations, and truncating mutations), we—for the first time—demonstrated that truncating mutations were a strongly independent adverse prognostic factor in SDC. Although mutations out of ‘‘hot spots’’ or large deletions/insertions might have been missed in our study, the molecular signatures of TP53 are very useful for determining therapeutic strategies.
The p53 expression (which is determined by immunohistochemistry) is widely used as a surrogate marker of TP53 mutation status; however, its reliability has not been established in SDCs. It has been generally accepted that wild-type p53 protein is relatively unstable and has a short half-life, which makes it undetectable by immunohistochemistry, whereas mutant p53 has a much longer half-life, and therefore accumulates in the nucleus creating a stable target for immunohistochemical detection [30]. However, the overexpression of wild-type p53 can sometimes occur due to impaired degradation under cellular stress [30]. On the other hand, truncating mutations invariably lead to the disruption of protein synthesis, and the biological consequences might differ from those of missense mutations [33]. In the present study of SDCs—as has been demostrated in breast [36] and ovarian carcinomas [37–39]—we proved that extreme p53 positivity and negativity was associated with TP53 missense and truncating mutations, respectively; this possibility has been suggested previously [11, 14]. This could provide a reasonable explanation of why extreme p53 positivity/negativity was associated with a worse prognosis in SDCs, as was shown our recent study [19]. It should be noted that the absence of protein expression tends to confound the interpretation of immunostaining, leading to false-negative results, especially in the case of truncating mutations.
The PI3K/Akt pathway is central in the regulation of multiple cancer-relevant regulatory processes that affect cell survival, cell growth, and cell cycle progression [40–42]. PI3K and its downstream components (including Akt and mTOR) in turn cross-talk with a number of other pathways, thereby leading to a complex network of signals; the disruption of which may have dramatic consequences [40–42]. In addition, PTEN is a major negative regulator of the PI3K activity [40, 43, 44]. The deregulation of the PI3K/Akt pathway—including activation and somatic mutations—in this pathway is frequently found in various cancers; thus it represents an attractive target for therapy [40–42, 45, 46]. Aberrations in this pathway have also recently been implicated in salivary gland tumorigenesis [13, 18, 47–49]. We disclosed that the overexpression of PI3K, activated Akt (p-Akt), and activated mTOR (p-mTOR), and the loss of PTEN are common in SDC. Furthermore, PTEN loss was negatively and positively correlated to PI3K expression and its downstream p-Akt nuclear expression, respectively. Moreover, a positive correlation was found between the expression of p-Akt and the downstream expression of p-mTOR. These observations indicate that activation of the PI3K/Akt pathway through the axis plays a crucial role in the pathogenesis of a subset of SDCs. On the other hand, we have shown that PIK3CA mutations are not associated with the high expression of downstream-activated proteins; this finding is in line with other cancer studies [50, 51].
The correlation between the expression of the PI3K/Akt signaling pathway proteins and the clinical outcome of salivary gland carcinomas has not been fully clarified. To the best of our knowledge, this is the first study to report that p-Akt negativity is associated with an unfavorable prognosis in patients with SDC; however, similar findings have been described in relation to salivary adenoid cystic carcinoma [48] and “high-grade tumors,” without focusing on sub-entities such as SDC [47]. In the current study, the expression of PI3K, PTEN, and p-mTOR had no impact on the survival of SDC patients. Furthermore, we could not demonstrate any significant association between the PIK3CA status and the clinical outcome, even though wild-type PIK3CA tumors showed higher proliferation index values. Similar findings have been reported in breast cancer patients [51, 52].
It is well known that SDC demonstrates the high expression of AR, as was shown in our results [19]. Thus, AR has been the target of androgen deprivation therapy (ADT) for SDC in recent years [4, 5], similarly to prostate cancer [53]. The PI3K/Akt pathway is important in both androgen-sensitive and hormone-refractory prostate cancer, with functional cross-talk between the pathway and the AR activity involved in both the initiation and progression of prostate cancer [54–57]. Interestingly, we found that PI3K negativity and PTEN loss were significantly correlated with the lower expression of AR in SDC. This study may provide novel evidence demonstrating interaction between the PI3K/Akt and AR pathways in SDC, and may be the first step toward elucidating the mechanisms of ADT therapy.
In conclusion, the present study, which included the largest series of patients with SDC, confirms the molecular variability and heterogeneity of treatment-relevant targets in this tumor. Our data shows that the genetic mechanisms underlying carcinogenesis differ according to the histogenesis and highlight the importance of subclassifying SDC based on specific molecular abnormalities. Almost all gene mutations in TP53, PIK3CA, H-RAS, BRAF, and AKT1 that were identified in this study were covered by the Catalogue of Somatic Mutations in Cancer (COSMIC), and most of these mutations are oncogenic, for which a functional analysis has already been performed [58–62]. However, an integrated molecular evaluation, including whole-exome sequencing, transcriptome and proteome analyses, and in vitro cell culture and patient-derived xenograft experiments, is warranted for gaining a greater understanding of the pathogenesis of this high-grade salivary gland carcinoma and the activity of targeted therapies. Besides, our analysis suggests that TP53 truncating mutations, which are correlated with p53-extreme negative expression, is an adverse prognostic factor in SDC. Additionally, the activation of the PI3K/Akt pathway with interaction with AR, which shows some prognostic impact in SDC, implies that it has the potential to be a biomarker of tumor aggressiveness, which may be useful for selecting hormonal and targeted therapy. Further validation in clinical trials would be expected for the development of therapy selection and treatment strategy in patients with SDC.
MATERIALS AND METHODS
Patients and tissue specimens
The present multicenter collaborative retrospective study was approved by the Institutional Review Board of each facility.
In total, 151 SDC patients were diagnosed and treated at 7 institutions between 1992 and 2014. Patients who underwent anti-HER2 or anti-AR therapy as an initial treatment were excluded. These patients included cases that were previously reported by our collaborative facilities [2, 19, 63]. All of the cases in the central review system were correctly diagnosed by an expert pathologist (T.N.) based on strict histological criteria in accordance with the 2017 WHO Classification of Tumors [1]. In principle, multi-step sections from the whole tumor were subjected to a histological review. Cases were considered to be SDC ex-PA when a pre-existing PA component was histologically detected in the tumor, even if it was represented by a markedly hyalinized nodule. On the other hand, in cases where no PA component was identified in any of the sections examined, the tumor was diagnosed as de novo SDC. When only biopsy samples were available for examination, the histologic origin was categorized as “unknown” (n = 5). Tumor stage was determined according to the UICC TNM classification and staging system (2009, 7th edition). Clinical information was collected from the patients’ medical records by the person-in-charge in each facility.
Mutational analysis
With microdissection methods, DNA of the carcinoma cell-rich part in each case was carefully extracted from the paraffin-embedded thin-sliced sections using a QIAmp DNA FFPE Tissue Kit (QIAGEN GmgH, Hilden, Germany), purified using QIAquick Spin Kit (QIAGEN GmgH), and subjected to a purity check using Nano Drop (Thermo Fisher Scientific, MA). A gene alteration analysis was performed for TP53 (exons 4–10), PIK3CA (exons 9 and 20), AKT1 (exon 2), K-RAS (exons 1-2), N-RAS (exons 1-2), H-RAS (exons 1-2), and BRAF (exon 15) by Sanger sequencing. The primer sequences are described in Supplementary Table 3.
DISH
The EGFR gene copy number was assessed by a DISH analysis using Benchmark ULTRA (Ventana Medical Systems, CA); staining was performed according to the manufacturer’s protocol (Roche, Basel Switzerland). Briefly, 20 tumor cells in which both EGFR signaling (black signal) and chromosome 7 centromere (CEN7) (red signal) were depicted and counted to calculate the ratio of the total number of EGFR signals to the total number of CEN7 signals. An EGFR/CEN7 ratio of ≥2.0 was considered to indicate EGFR gene amplification, whereas a mean CEN7 signal number of ≥3.0/cell was considered to indicate the presence of chromosome 7 polysomy.
Immunohistochemistry
Immunohistochemical staining was performed for PI3K, p-Akt, p-mTOR, and PTEN using 4-μm-thick formalin-fixed, paraffin-embedded tissue sections. The antibodies used in the assay are shown in Supplementary Table 4. The cases were regarded as positive for PI3K, p-Akt, and p-mTOR when the percentage of cytoplasmic and/or nuclear staining cells was >10%. With regard to p-Akt, nuclear positivity was indicated as “p-Akt (N)”. The PTEN expression level was scored semiquantitatively, as described previously [64]. The normal surrounding epithelium served as an internal control. Tumor tissues were scored as follows: score 2, same staining intensity as that of the normal epithelium; score 1, staining intensity weaker than normal; score 0, no staining. We considered scores of 0 and 1 to indicate a loss of PTEN.
Statistical analysis
The chi-squared test and Mann-Whitney U test were used to compare non-continuous and continuous variables, respectively. Univariate and multivariate Cox proportional hazards models and the Kaplan-Meier product-limit method were used to investigate the associations between the molecular/immunohistochemical results and OS and PFS. The potential confounders in the multivariate analysis included age, gender, TNM classification, primary site, first-line treatment, and histologic origin (de novo vs. SDC ex-PA). The strength of an association was determined by the hazard ratio and the 95% CI. The statistical analyses were performed using the STATA software program (version 13, StataCorp., College Station, TX). All of the tests were two-sided, and P values of < 0.05 were considered to indicate statistical significance.
ACKNOWLEDGMENTS AND FUNDING
This work was supported by the JSPS Grant-in-Aid for Scientific Research (C) to Dr. Yuichiro Tada (No. 15K10823) and Dr. Toshitaka Nagao (No. 17K08705), Grant-in-Aid for Young Scientists (B) to Dr. Daisuke Kawakita (No. 17K18006), the Tokyo Medical University Research Grant to Dr. Soichiro Takase (2015), and the Tokyo Medical University Cancer Research Foundation Grant to Dr. Toshitaka Nagao (2015). The authors thank Mr. Hiroaki Iobe, Ms. Mayumi Yokotsuka, and Ms. Hitomi Shimayama, Tokyo Medical University, for their technical assistance.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest in association with the present study.
REFERENCES
1. Nagao T, Licitra L, Leoning T, Vielh P, Williams MD. Salivary duct carcinoma. In: El-Naggar AK, Chan JKC, Grandis JR, Takata T, Slootweg PJ, editors. WHO Classification of Head and Neck Tumours. 4th ed. Lyon, IARC. 2017; p. 173–174.
2. Otsuka K, Imanishi Y, Tada Y, Kawakita D, Kano S, Tsukahara K, Shimizu A, Ozawa H, Okami K, Sakai A, Sato Y, Ueki Y, Sato Y, et al. Clinical outcomes and prognostic factors for salivary duct carcinoma: a multi-institutional analysis of 141 patients. Ann Surg Oncol. 2016; 23:2038–2045.
3. Osborn V, Givi B, Lee A, Sheth N, Roden D, Schwartz D, Schreiber D. Characterization, treatment and outcomes of salivary ductal carcinoma using the National Cancer Database. Oral Oncol. 2017; 71:41–46.
4. Lewis AG, Tong T, Maghami E. Diagnosis and management of malignant salivary gland tumors of the parotid gland. Otolaryngol Clin North Am. 2016; 49:343–380.
5. Alfieri S, Granata R, Bergamini C, Resteghini C, Bossi P, Licitra LF, Locati LD. Systemic therapy in metastatic salivary gland carcinomas: A pathology-driven paradigm? Oral Oncol. 2017; 66:58–63.
6. Amirouchene-Angelozzi N, Swanton C, Bardelli A. Tumor evolution as a therapeutic target. Cancer Discov. 2017 Jul 20. [Epub ahead of print].
7. Kamel HFM, Al-Amodi HSAB. Exploitation of gene expression and cancer biomarkers in paving the path to era of personalized medicine. Genomics Proteomics Bioinformatics. 2017.
8. Nardi V, Sadow PM, Juric D, Zhao D, Cosper AK, Bergethon K, Scialabba VL, Batten JM, Borger DR, Iafrate AJ, Heist RS, Lawrence DP, Flaherty KT, et al. Detection of novel actionable genetic changes in salivary duct carcinoma helps direct patient treatment. Clin Cancer Res. 2013; 19:480–490.
9. Ku BM, Jung HA, Sun JM, Ko YH, Jeong HS, Son YI, Baek CH, Park K, Ahn MJ. High-throughput profiling identifies clinically actionable mutations in salivary duct carcinoma. J Transl Med. 2014; 12:299.
10. Grünewald I, Vollbrecht C, Meinrath J, Meyer MF, Heukamp LC, Drebber U, Quaas A, Beutner D, Hüttenbrink KB, Wardelmann E, Hartmann W, Büttner R, Odenthal M, et al. Targeted next generation sequencing of parotid gland cancer uncovers genetic heterogeneity. Oncotarget. 2015; 6:18224–18237. https://doi.org/10.18632/oncotarget.4015.
11. Chiosea SI, Williams L, Griffith CC, Thompson LD, Weinreb I, Bauman JE, Luvison A, Roy S, Seethala RR, Nikiforova MN. Molecular characterization of apocrine salivary duct carcinoma. Am J Surg Pathol. 2015; 39:744–752.
12. Luk PP, Weston JD, Yu B, Selinger CI, Ekmejian R, Eviston TJ, Lum T, Gao K, Boyer M, O’Toole SA, Clark JR, Gupta R. Salivary duct carcinoma: Clinicopathologic features, morphologic spectrum, and somatic mutations. Head Neck. 2016; 38:E1838–1847.
13. Chiosea SI, Thompson LD, Weinreb I, Bauman JE, Mahaffey AM, Miller C, Ferris RL, Gooding WE. Subsets of salivary duct carcinoma defined by morphologic evidence of pleomorphic adenoma, PLAG1 or HMGA2 rearrangements, and common genetic alterations. Cancer. 2016; 122:3136–3144.
14. Grünewald I, Trautmann M, Busch A, Bauer L, Huss S, Schweinshaupt P, Vollbrecht C, Odenthal M, Quaas A, Büttner R, Meyer MF, Beutner D, Hüttenbrink KB, et al. MDM2 and CDK4 amplifications are rare events in salivary duct carcinomas. Oncotarget. 2016; 7:75261–75272. https://doi.org/10.18632/oncotarget.12127.
15. Dalin MG, Desrichard A, Katabi N, Makarov V, Walsh LA, Lee KW, Wang Q, Armenia J, West L, Dogan S, Wang L, Ramaswami D, Ho AL, et al. Comprehensive molecular characterization of salivary duct carcinoma reveals actionable targets and similarity to apocrine breast cancer. Clin Cancer Res. 2016; 22:4623–4633.
16. Wang K, Russell JS, McDermott JD, Elvin JA, Khaira D, Johnson A, Jennings TA, Ali SM, Murray M, Marshall C, Oldham DS, Washburn D, Wong SJ, et al. Profiling of 149 salivary duct carcinomas, carcinoma ex pleomorphic adenomas, and adenocarcinomas, not otherwise specified reveals actionable genomic alterations. Clin Cancer Res. 2016; 22:6061–6068.
17. Khoo TK, Yu B, Smith JA, Clarke AJ, Luk PP, Selinger CI, Mahon KL, Kraitsek S, Palme C, Boyer MJ, Dinger ME, Cowley MJ, O’Toole SA, et al. Somatic mutations in salivary duct carcinoma and potential therapeutic targets. Oncotarget. 2017; 8:75893–75903. https://doi.org/10.18632/oncotarget.18173.
18. Griffith CC, Seethala RR, Luvison A, Miller M, Chiosea SI. PIK3CA mutations and PTEN loss in salivary duct carcinomas. Am J Surg Pathol. 2013; 37:1201–1207.
19. Takase S, Kano S, Tada Y, Kawakita D, Shimura T, Hirai H, Tsukahara K, Shimizu A, Imanishi Y, Ozawa H, Okami K, Sato Y, Sato Y, et al. Biomarker immunoprofile in salivary duct carcinomas: Clinicopathological and prognostic implications with evaluation of the revised classification. Oncotarget. 2017; 8:59023-59035. https://doi.org/10.18632/oncotarget.19812
20. Lim CM, Hobson C, Kim S, Johnson JT. Clinical outcome of patients with carcinoma ex pleomorphic adenoma of the parotid gland: a comparative study from a single tertiary center. Head Neck. 2015; 37:543–547.
21. Gilbert MR, Sharma A, Schmitt NC, Johnson JT, Ferris RL, Duvvuri U, Kim S. A 20-year review of 75 cases of salivary duct carcinoma. JAMA Otolaryngol Head Neck Surg. 2016; 142:489–495.
22. Hyman DM, Puzanov I, Subbiah V, Faris JE, Chau I, Blay JY, Wolf J, Raje NS, Diamond EL, Hollebecque A, Gervais R, Elez-Fernandez ME, Italiano A, et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med. 2015; 373:726–736.
23. Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harb Perspect Biol. 2010; 2.
24. Robles AI, Harris CC. Clinical outcomes and correlates of TP53 mutations and cancer. Cold Spring Harb Perspect Biol. 2010; 2.
25. Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010; 2.
26. Muller PA, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell. 2014; 25:304–317.
27. Mantovani F, Walerych D, Sal GD. Targeting mutant p53 in cancer: a long road to precision therapy. FEBS J. 2017; 284:837–850.
28. Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nat Rev Cancer. 2014; 14:359–370.
29. Zhu J, Sammons MA, Donahue G, Dou Z, Vedadi M, Getlik M, Barsyte-Lovejoy D, Al-awar R, Katona BW, Shilatifard A, Huang J, Hua X, Arrowsmith CH, et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature. 2015; 525:206–211.
30. Hientz K, Mohr A, Bhakta-Guha D, Efferth T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget. 2017; 8:8921–8946. https://doi.org/10.18632/oncotarget.13475.
31. Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D, Jacks T. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004; 119:847–860.
32. Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, Valentin-Vega YA, Terzian T, Caldwell LC, Strong LC, El-Naggar AK, Lozano G. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 2004; 119:861–872.
33. Hainaut P, Pfeifer GP. Somatic TP53 mutations in the era of genome sequencing. Cold Spring Harb Perspect Med. 2016; 6.
34. Shahin MS, Hughes JH, Sood AK, Buller RE. The prognostic significance of p53 tumor suppressor gene alterations in ovarian carcinoma. Cancer. 2000; 89:2006–2017.
35. Wojnarowicz PM, Oros KK, Quinn MC, Arcand SL, Gambaro K, Madore J, Birch AH, de Ladurantaye M, Rahimi K, Provencher DM, Mes-Masson AM, Greenwood CM, Tonin PN. The genomic landscape of TP53 and p53 annotated high grade ovarian serous carcinomas from a defined founder population associated with patient outcome. PLoS One. 2012; 7:e45484.
36. Boyle DP, McArt DG, Irwin G, Wilhelm-Benartzi CS, Lioe TF, Sebastian E, McQuaid S, Hamilton PW, James JA, Mullan PB, Catherwood MA, Harkin DP, Salto-Tellez M. The prognostic significance of the aberrant extremes of p53 immunophenotypes in breast cancer. Histopathology. 2014; 65:340–352.
37. Yemelyanova A, Vang R, Kshirsagar M, Lu D, Marks MA, Shih IeM, Kurman RJ. Immunohistochemical staining patterns of p53 can serve as a surrogate marker for TP53 mutations in ovarian carcinoma: an immunohistochemical and nucleotide sequencing analysis. Mod Pathol. 2011; 24:1248–1253.
38. Cole AJ, Dwight T, Gill AJ, Dickson KA, Zhu Y, Clarkson A, Gard GB, Maidens J, Valmadre S, Clifton-Bligh R, Marsh DJ. Assessing mutant p53 in primary high-grade serous ovarian cancer using immunohistochemistry and massively parallel sequencing. Sci Rep. 2016; 6:26191.
39. Köbel M, Piskorz AM, Lee S, Lui S, LePage C, Marass F, Rosenfeld N, Mes Masson AM, Brenton JD. Optimized p53 immunohistochemistry is an accurate predictor of TP53 mutation in ovarian carcinoma. J Pathol Clin Res. 2016; 2:247–258.
40. Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008; 27:5497–5510.
41. Chan CH, Jo U, Kohrman A, Rezaeian AH, Chou PC, Logothetis C, Lin HK. Posttranslational regulation of Akt in human cancer. Cell Biosci. 2014; 4:59.
42. Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K pathway in human disease. Cell. 2017; 170:605–635.
43. Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell. 2008; 133:403–414.
44. Lim HJ, Crowe P, Yang JL. Current clinical regulation of PI3K/PTEN/Akt/mTOR signalling in treatment of human cancer. J Cancer Res Clin Oncol. 2015; 141:671–689.
45. Keppler-Noreuil KM, Parker VE, Darling TN, Martinez-Agosto JA. Somatic overgrowth disorders of the PI3K/AKT/mTOR pathway & therapeutic strategies. Am J Med Genet C Semin Med Genet. 2016; 172:402–421.
46. Janku F. Phosphoinositide 3-kinase (PI3K) pathway inhibitors in solid tumors: From laboratory to patients. Cancer Treat Rev. 2017; 59:93–101.
47. Ettl T, Schwarz-Furlan S, Haubner F, Müller S, Zenk J, Gosau M, Reichert TE, Zeitler K. The PI3K/AKT/mTOR signalling pathway is active in salivary gland cancer and implies different functions and prognoses depending on cell localisation. Oral Oncol. 2012; 48:822–830.
48. Ouyang DQ, Liang LZ, Ke ZF, Zheng GS, Weng DS, Yang WF, Su YX, Liao GQ. Association between high expression of phosphorylated Akt and mammalian target of rapamycin and improved survival in salivary gland adenoid cystic carcinoma. Head Neck. 2017; 39:1145–1154
49. Wang K, McDermott JD, Schrock AB, Elvin JA, Gay L, Karam SD, Raben D, Somerset H, Ali SM, Ross JS, Bowles DW. Comprehensive genomic profiling of salivary mucoepidermoid carcinomas reveals frequent BAP1, PIK3CA, and other actionable genomic alterations. Ann Oncol. 2017; 28:748–753.
50. Trigka EA, Levidou G, Saetta AA, Chatziandreou I, Tomos P, Thalassinos N, Anastasiou N, Spartalis E, Kavantzas N, Patsouris E, Korkolopoulou P. A detailed immunohistochemical analysis of the PI3K/AKT/mTOR pathway in lung cancer: correlation with PIK3CA, AKT1, K-RAS or PTEN mutational status and clinicopathological features. Oncol Rep. 2013; 30:623–636.
51. Beelen K, Opdam M, Severson TM, Koornstra RH, Vincent AD, Wesseling J, Muris JJ, Berns EM, Vermorken JB, van Diest PJ, Linn SC. PIK3CA mutations, phosphatase and tensin homolog, human epidermal growth factor receptor 2, and insulin-like growth factor 1 receptor and adjuvant tamoxifen resistance in postmenopausal breast cancer patients. Breast Cancer Res. 2014; 16:R13.
52. Buttitta F, Felicioni L, Barassi F, Martella C, Paolizzi D, Fresu G, Salvatore S, Cuccurullo F, Mezzetti A, Campani D, Marchetti A. PIK3CA mutation and histological type in breast carcinoma: high frequency of mutations in lobular carcinoma. J Pathol. 2006; 208:350–355.
53. Prostate Cancer Trialists’ Collaborative Group. Maximum androgen blockade in advanced prostate cancer: an overview of the randomised trials. Lancet. 2000; 355:1491–1498.
54. Wang Y, Kreisberg JI, Ghosh PM. Cross-talk between the androgen receptor and the phosphatidylinositol 3-kinase/Akt pathway in prostate cancer. Curr Cancer Drug Targets. 2007; 7:591–604.
55. Sircar K, Yoshimoto M, Monzon FA, Koumakpayi IH, Katz RL, Khanna A, Alvarez K, Chen G, Darnel AD, Aprikian AG, Saad F, Bismar TA, Squire JA. PTEN genomic deletion is associated with p-Akt and AR signalling in poorer outcome, hormone refractory prostate cancer. J Pathol. 2009; 218:505–513.
56. Choucair K, Ejdelman J, Brimo F, Aprikian A, Chevalier S, Lapointe J. PTEN genomic deletion predicts prostate cancer recurrence and is associated with low AR expression and transcriptional activity. BMC Cancer. 2012; 12:543.
57. Edlind MP, Hsieh AC. PI3K-AKT-mTOR signaling in prostate cancer progression and androgen deprivation therapy resistance. Asian J Androl. 2014; 16:378–386.
58. Kato S, Han SY, Liu W, Otsuka K, Shibata H, Kanamaru R, Ishioka C. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc Natl Acad Sci U S A. 2003; 100:8424–8429.
59. Soussi T, Kato S, Levy PP, Ishioka C. Reassessment of the TP53 mutation database in human disease by data mining with a library of TP53 missense mutations. Hum Mutat. 2005; 25:6–17.
60. Gymnopoulos M, Elsliger MA, Vogt PK. Rare cancer-specific mutations in PIK3CA show gain of function. Proc Natl Acad Sci U S A. 2007; 104:5569–5574.
61. Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012; 72:2457–2467.
62. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, et al. Mutations of the BRAF gene in human cancer. Nature. 2002; 417:949–954.
63. Masubuchi T, Tada Y, Maruya S, Osamura Y, Kamata SE, Miura K, Fushimi C, Takahashi H, Kawakita D, Kishimoto S, Nagao T. Clinicopathological significance of androgen receptor, HER2, Ki-67 and EGFR expressions in salivary duct carcinoma. Int J Clin Oncol. 2015; 20:35–44.
64. Sakr RA, Barbashina V, Morrogh M, Chandarlapaty S, Andrade VP, Arroyo CD, Olvera N, King TA. Protocol for PTEN expression by immunohistochemistry in formalin-fixed paraffin-embedded human breast carcinoma. Appl Immunohistochem Mol Morphol. 2010; 18:371–374.