
INTRODUCTION
Much of the effort directed to targeted anti-cancer drug development has focused on the inhibition of signaling regulators such as receptor tyrosine kinases [1–3]. Such approaches have shown limited effectiveness in treating metastatic disease or in patient subpopulations with cancers that do not express the targeted kinase [4]. In contrast to kinases, ion channels have received relatively little attention as potential cancer therapeutic targets. It is well established that ion channels play major roles in many cellular functions of cancerous cells. Potassium ion (K+) channels have been traditionally recognized for their role in controlling important cellular events including electrical transmission, contraction, and secretion. Recent research has revealed that K+ channels play fundamental roles in several other important cellular events including cell proliferation [5–7], which is important in cancer.
The human ether-a-go-go-related gene type 1 (hERG1) belongs to the ether a go-go (EAG) gene superfamily initially cloned in Drosophila and encodes for the voltage-gated (Kv) K+ channel Kv11.1. In adult humans, Kv11 channels are primarily found in cardiac myocytes and in neurons, where they contribute to cell excitability. However, breast cancers of different histogenesis exhibit overexpression of Kv11.1, while the corresponding non-cancerous cells do not exhibit significant levels of the channel. These data suggest that the Kv11.1 channel might be part of a general breast cancer oncogenic signature [8, 9].
Breast cancer is a highly heterogeneous disease that presents differences in morphology and responses to therapeutic options [10]. A fundamental study focusing on molecular characterization of different breast cancers has identified major gene signatures that include cells expressing estrogen receptors (ER+), cells lacking ERs (ER-) and expressing the human epidermal growth factor receptor 2 (HER2+), and cells lacking both ERs and HER2 (triple negative breast cancer; TNBC). Although this discovery has provided the opportunity to develop anti-cancer therapies that selectively aim specific targets and greatly improve the survival of breast cancer patients, particularly the ones bearing ER+ tumors, the same therapies have met limited success in patients with HER2+ and TNBC tumors. Unfortunately, the TNBC tumors also happen to be the most aggressive and present high frequency of recurrence and resistance to treatments [11–13].
In our previous studies, we have shown that pharmacologic stimulation of the Kv11.1 channel current activity with selective and chemically distinct small molecule activators NS1643 or PD115087 induces cell cycle arrest in cultured breast cancer cells that expressed Kv11.1. In contrast, these molecules did not produce any effect in non-transformed breast cells [9, 14, 15]. The antiproliferative effect of Kv11.1 activators coincided with activation of a cellular senescence program in cultured breast cancer cell lines independently of their molecular classification. Senescence is a distinct cellular phenotype that is characterized by permanent arrest of the cell cycle as a response to non-lethal stress such as the formation of reactive oxygen species (ROS) and/or DNA damage [16–20]. Traditionally, cellular senescence can be triggered by telomeric erosion during the normal process of aging or by telomere-independent factors such as oncogenic activation, ionizing radiations (UV or X-rays), and it is generally considered a physiological barrier against tumorigenesis, as senescent cells do not re-enter the cell cycle [21, 22]. Some drugs used in the management of breast cancer, such as the anthracycline doxorubicin, have been found to activate senescence in breast tumors, suggesting that at least in part, senescence mediates the multifactorial cytotoxic effects of doxorubicin on breast cancer cells [23]. Nevertheless, although the ability of doxorubicin to activate senescence appears to provide therapeutic benefit, the occurrence of severe cardiotoxicity [24–28] strongly hampers the use of anthracyclines for treatment of advanced breast cancer.
Several studies suggest an association between ion channels and oxidative stress. For instance, it has been demonstrated that activity of a variety of ion channels, including the Kv11.1 channel, can be directly or indirectly altered by ROS-dependent signaling in vitro [29–35] as well as in vivo [36, 37]. In several elegant studies, it has been demonstrated that superoxides can produce significant effects on K+ channels activity by direct oxidation of specific amino acid residues. Other studies have reported that activity of K+ channels can be directly related to the formation of ROS. Further, it is known that Ca2+ plays a central role in the process of ROS formation [38]. However, the contribution of ion channels in altering the cancer cell oxidative state is still unexplored. In this study, we aimed to characterize the antineoplastic effects of the Kv11.1 activator NS1643 on TNBC in vivo and in vitro. The present data demonstrate that NS1643 inhibits TNBC-derived tumor growth in vivo in association with increased expression of senescence markers. Acute and chronic application of NS1643 did not produce any significant effect on heart performance.
In addition, we found that NS1643 leads to the production of ROS which results in DNA damage through a Ca2+-dependent mechanism. Therefore, we propose that targeting Kv11.1 channels with activators could be considered as a potential pharmacological strategy against breast cancer.
RESULTS
Kv11.1 potassium channel gene is overexpressed in human breast carcinomas
In this study, we extracted microarray data from the publically available gene expression database Oncomine to examine relative hERG1 (alias KCNH2) gene expression levels in different histological subtypes of breast cancers compared to normal breast tissue and performed biomarker assessment by using Kaplan-Meier Plotter global portal [39]. We found that at least 7 independent studies showed a significant difference in the value of the expression (p ≤ 0.05) in breast cancers compared with normal breast tissue (Figure 1A).
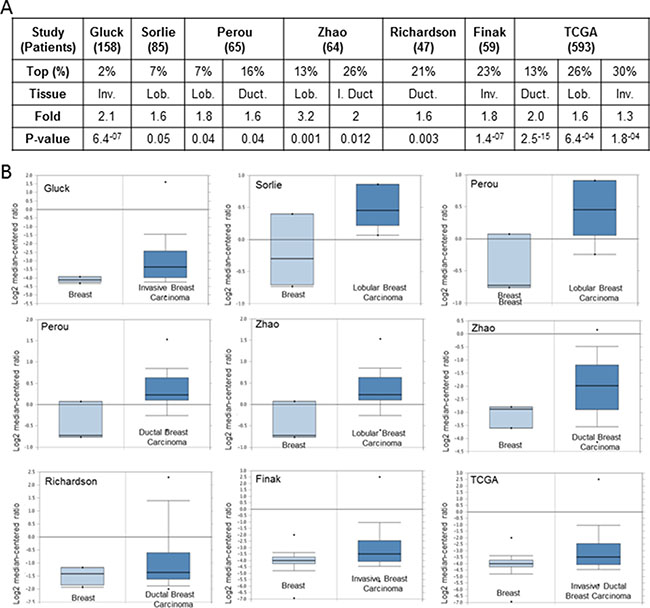
Figure 1: Analysis of KCNH2 mRNA levels in human breast cancer tissues. (A) Fold = fold change. P-value is reported only when < 0.05. Rank = Over-expression gene rank. Type = type of breast cancer: Inv = invasive; I Duct = invasive ductal; Lob = lobular; Duct = ductal.TCGA; http://tcga-data.nci.nih.gov/tcga/). (B) Box plot indicating KCNH2 expression level in different studies as indicated. Oncomine dataset; threshold by: P-Value: 1-4; Fold Change: 2; Gene rank: Top 10%.
In the Gluck’s [40] and Finak’s [41] studies, KCNH2 was significantly upregulated in invasive breast carcinoma and it was ranked among the top 2% and 23% overexpressed genes respectively. While in Zhao’s [42] study, KCNH2 was overexpressed in invasive (top 13%) and invasive ductal (top 26%) vs normal tissue. In Sorlie’s [43] investigation, KCNH2 was upregulated in lobular carcinoma (top 7%) while in the study by Richardson et al. [44], KCNH2 was found to be overexpressed in ductal breast carcinoma (top 21%). Furthermore, in Perou’s study KCNH2 was upregulated in both lobular (top 7%) and ductal (top 16%) breast carcinomas. Finally, in the Cancer Genome Atlas KCNH2 was found to be overexpressed in ductal (top 13%), lobular (top 26%) and invasive (top 30%) breast carcinomas.
Stimulation of Kv11.1 activity inhibits tumor growth of RasV12 cells
The fruit fly Drosophila melanogaster is a well-established animal model that has significantly contributed toward elucidating the molecular basis of cancer [45, 46], and more recently has been demonstrated in multiple studies to be an effective preclinical assay for a broad range of cancers in drug discovery [47]. Several recent significant findings have begun to implicate K+ channels as important components in morphogenetic signaling [7, 48] Drosophila has a Kv11.1/KCNH2 homolog called seizure (sei) [49–51]. As an initial test in an in vivo setting, we investigated the effects of NS1643 on tumor growth in genetically modified Drosophila harboring GFP expressing oncogenic RasV12 with knockdown of polarity gene discs large DlgRNAi (RasV12, DlgRNAi tumors) [52, 53]. Drosophila harboring these tumors when fed with Kv11.1 activator NS1643 (25 μM) in their diet developed smaller tumor growth compared to animals that were fed with a vehicle control (Figure 2). These data suggest that application of Kv11.1 channel activator NS1643 can arrest tumor growth and that this response is likely to be strongly conserved across many cancer cell contexts.
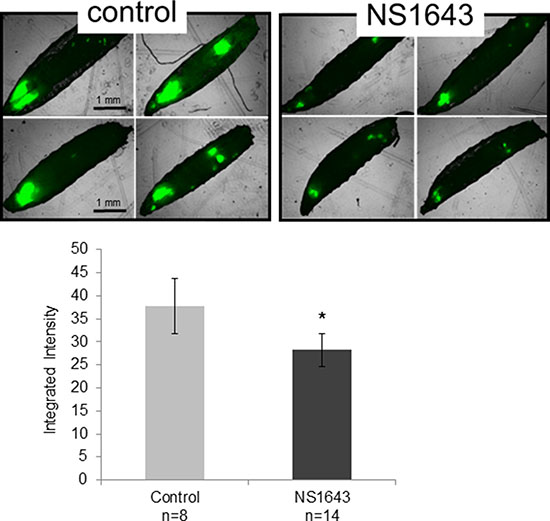
Figure 2: NS1643 arrests RasV12 tumor growth in Drosophila melanogaster. Inhibition of proliferation in Drosophila model by Kv11.1 stimulation (NS1634) in comparison to vehicle alone (control). On the seventh day after egg laying (3 days of drug treatment), larvae were harvested and imaged on an EVOS fluorescent microscope. Data is expressed as Mean ± SEM; *p < 0.05 (T-Test).
Kv11.1 activator NS1643 inhibits tumor growth in triple negative breast cancer xenograft model of breast cancer
In this study, we wanted to test the efficacy of stimulating the Kv11.1 K+ channel activity to in inhibiting TNBC tumor growth in vivo. MDA-MB-231 cell line was selected as a TNBC breast cancer cell line, which represents a cancer subtype that is aggressive and has few treatment options. In our previous studies, we showed that stimulation of Kv11.1 activity by the small molecule activator NS1643 inhibits proliferation independently of the molecular characterization of the breast cancer cell line including MDA-MB-231 but no effect of NS1643 was observed in normal cells [2, 11] (Supplementary Figure 1).
We, therefore, established a xenograft model of MDA-MB-231 cells in athymic nude mice. In this model, MDA-MB-231 cells developed measurable tumors of 50 mm3 as early as 6 days (Figure 3). Mice treated with NS1643 (6 mg/Kg) had significantly reduced tumor volumes in comparison to the vehicle-treated group as early as 6 days following injection (day 12). Smaller dose of NS1643 (1.5 mg/Kg) did not produce significant reduction of tumor growth in the time considered in this study. At the end of the study, we observed that the mean tumor areas of control mice were was1050 ± 200 mm3 (Mean ± SEM; Figure 3A, 3C) while that of the NS1643 treated group was significantly lower at 560 ± 120 mm3 (Mean ± SEM; Figure 3B, 3C).
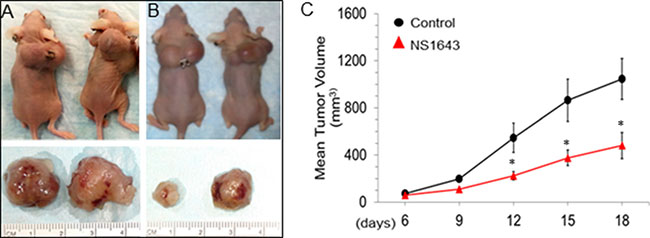
Figure 3: Kv11.1 stimulation inhibits primary tumor growth in xenograft model of breast cancer. MDA-MB-231 (ATCC®) cells were injected subcutaneously into the flanks of female athymic nude mice. When tumors were palpable the mice were injected intraperitoneally with vehicle alone or Kv11.1 activator NS1643 at 6 mg/kg every two days. (A) Representative mice with tumor burdens treated with vehicle (DMSO) alone. (B) Representative mice with tumor burdens treated with Kv11.1 activator NS1643. (C) Mean tumor volume in mice treated with vehicle alone (control N = 6) and in mice treated with Kv11.1 activator (NS1643 N = 6). Data is expressed as Mean ± SEM; *p < 0.05.
NS1643 suppresses proliferation and promotes senescence in breast tumors
We have shown that NS1643 activates a cellular senescence program [51] in breast cancer cells in culture, a phenotype that is characterized by suppressed proliferation without apoptosis. To confirm that this same mechanism was functional in vivo we analyzed the effect of NS1643 in regulating proliferation and senescence markers in tumors in of the MDA-MB-231 xenograft model.
High expression of the proliferation marker Ki67 and/or the extracellular signal-related kinase (ERK) levels [52–54] are associated with more aggressive clinical features of TNBC [55–60]. Our immunohistochemistry investigation showed that tumors extracted from mice that received NS1643 presented a significantly lower expression of Ki67 relative to the control group (Figure 4A, 4B). Similarly, we found that the activity of ERK, as indicated by its phosphorylation status, was strongly downregulated in tumors from mice treated with NS1643 compared to control (Figure 4C, 4D).
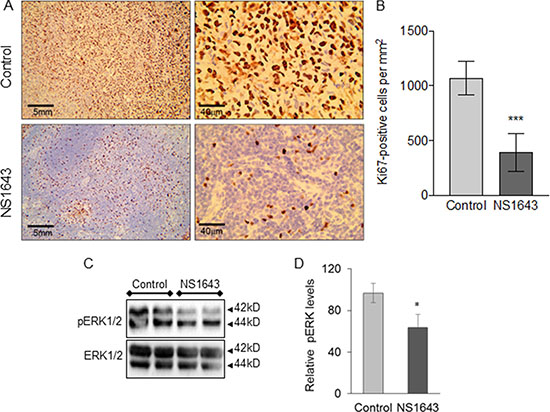
Figure 4: NS1643 treatment decreases tumor proliferation markers. (A) Detection of Ki67 (clone Mib-1, Glostrup, Denmark) protein expression in mice treated with DMSO (control) or NS1643 and (B) quantification of Ki67-positive cells per mm2. Data presented as mean ± S.D. Unpaired t-test. ***p < 0.001; n = 5 per group. (C) Representative immunoblots demonstrating the effects of NS1643 on phosphorylated ERK or total ERK in tumors extracted from mice receiving DMSO (control) or NS1643. (D) Quantification of the effect of NS1643 on activated ERK. Data is expressed as mean ± SEM; Unpaired t-test. *p < 0.05 (n = 6 per treatment).
In contrast, protein levels of the cellular senescence markers p21waf/cip and p16INK4A were increased in tumors from NS1643 treated mice compared to control (Figure 5A, 5B). The high mobility group A2 (HMGA2) is a DNA binding protein non-transcription factor that has been associated with a cellular senescent phenotype [54] and is highly associated with DNA damage [55]. Remarkably, we found that breast cancer cells treated with NS1643 presented a strong increase of the HMGA2 nuclear foci (Figure 5C, 5D), suggesting that HMGA2 can contribute to NS1643-dependent senescence and DNA damage.
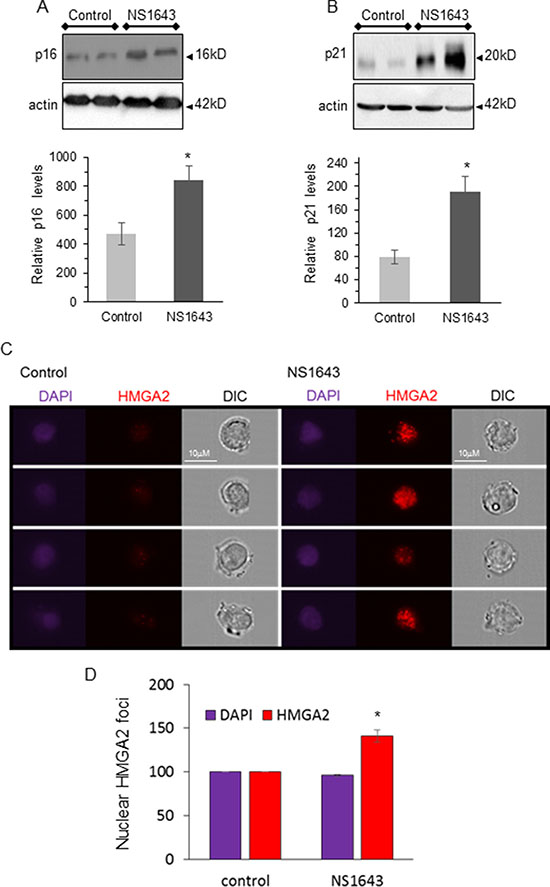
Figure 5: NS1643 treatment increases senescence markers in vivo. (A) Representative immunoblots and quantification of the effects of NS1643 on senescence markers p16INK4A and (B) p21waf/cip in tumors extracted from mice receiving DMSO (control) or NS1643. Data is expressed as mean ± SEM; Unpaired t-test. *p < 0.05 (n = 6 per treatment). (C) Representative images of HMGA2 nuclear foci in human breast cancer cells treated with NS1643 or DMSO (Control) for 24 hr. Genomic DNA was stained with DAPI. (D) Quantification of nuclear HMGA2 foci in cells treated with NS1643 or (DMSO) control. Shown are 4 representative cells per treatment from one experiment. Two independent experiments were quantified. Data is expressed as mean ± SEM; *p < 0.001.
NS1643 generates ROS-dependent DNA damage in breast cancer cells
To understand the effect of NS1643 on tumor growth we observed that tumors extracted from NS1643 treated mice exhibited complex DNA fragmentation compared to tumors from untreated mice (Figure 6A, 6B), suggesting that NS1643 could cause DNA damage. Therefore, to better understand the effect of NS1643 on tumor growth, we tested the hypothesis that NS1643 could affect DNA integrity in breast cancer cell lines. A comet assay revealed that application of NS1643 produced significant DNA damage as early as 1h after treatment (Figure 6C, 6D). In addition, NS1643-treated cells showed upregulation of γ-H2AX protein levels, a measure of double-strand DNA damage [56, 57] (Figure 6E) and an increased poly ADP-ribosylation (PARylation), a measure of DNA damage response [58] (Supplementary Figure 2).
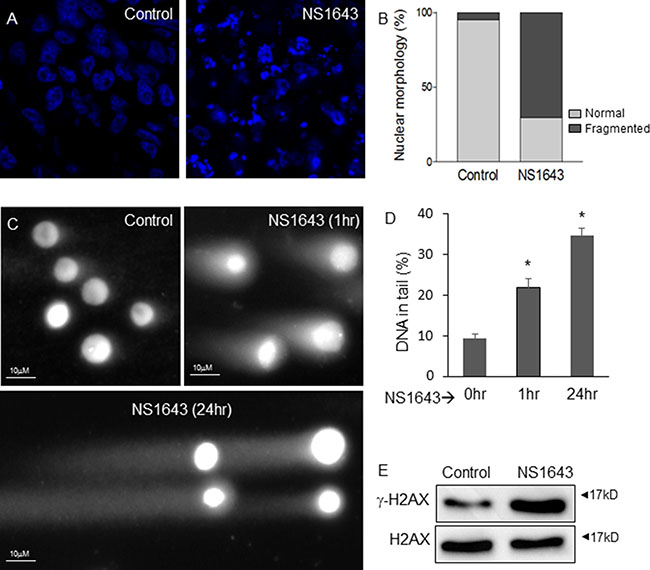
Figure 6: NS1643 generates DNA damage in breast cancer. (A) Single confocal sections of DAPI-stained tumors from control and NS1643-treated mice show fragmentation of nuclei indicative of DNA damage induced by NS1643 treatment. The number of normal (light grey) vs. fragmented (dark grey) nuclei was quantified by manual counting (n = 4, p < 0.001, (B) Light vs Dark represents normal vs fragmented nuclei. (C) Representative images of human MDA-MB-231 cells subjected to neutral comet assay after treatment with NS1643 (50 μM) for 0 hr, 1 hr or 24 hr. (D) Quantification of DNA in tail (percent) of cells treated as indicated. Data is expressed as mean ± SEM; Unpaired t-test. *p < 0.05 (E) Western blot analyses of phosphorylated γH2AX in MDA-MB-231 cells treated with NS1643 (50 μM) for 30 min compared to control (DMSO).
As mentioned before, since DNA damage is strongly associated with oxidative stress [6, 59, 60].This led us to we hypothesized that stimulation of Kv11.1 activity could produce ROS formation in breast cancer cells. Results showed that application of NS1643 produced a progressive and significant increase in ROS formation within 2h (Figure 7A) in both MDA-MB-231 (Figure 7A) and in HER2+ breast cancer cell lines (Supplementary Figure 3). Of note, this effect of NS1643 was completely inhibited by the Kv11.1 blocker E4031 (Figure 7A).
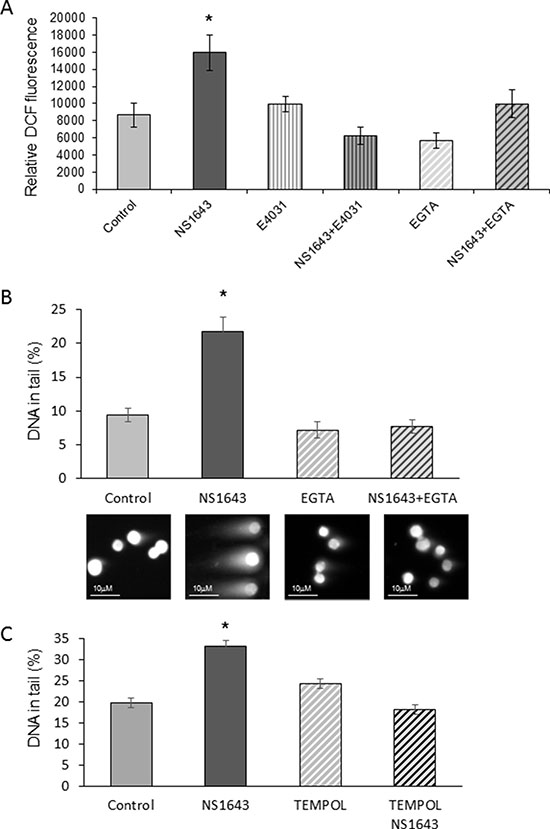
Figure 7: NS1643 generates reactive oxygen species (ROS) and DNA damage in a Ca2+-dependent manner. (A) Effect of NS1643 alone (50 μM), in association with the Kv11.1 blocker E-4031 (10 μM), or in the presence of the Ca2+ ion chelator EGTA (2.4 mM) on cellular ROS formation in human MDA-MB-231 cells (DCFH-DA to 2′,7′-dichlorofluorescein DCF, Thermo Fisher Sci; Fluorescence was analyzed in a plate reader (PHERAstar FS, BMG LABTECH) with excitation at 485 nm and emission at 520 nm). Data is expressed as mean ± SEM; *p < 0.001. (B) Quantification of DNA in tail (percent) via comet assay of cells treated with NS1643 alone or in the presence of EGTA. Representative images of cells subjected to neutral comet assay are presented below the bars corresponding to specific treatments. (C) Effect of the antioxidant TEMPOL (5 mM) on NS1643-dependent DNA damage in MDA-MB-231 cells. Data is expressed as mean ± SEM; *p < 0.05.
In our previous experiments, we have demonstrated that stimulation of Kv11.1 activity correlates with an increase of intracellular Ca2+ via passive Ca2+ entry [14]. Alteration of cytoplasmic Ca2+ can be a major cause of ROS formation [61]. Our experiments showed that generation of both ROS and DNA damage was prevented by treating the cells with the Ca2+ ion chelator EGTA (Figure 7A, 7B), suggesting that NS1643-dependent ROS formation and DNA damage were regulated by the Ca2+ entry. The same results were obtained in the HER2+ breast cancer cell line SKBR3 (Supplementary Figure 3). In addition, we found that treatment of the MDA-MB-231 cells with the antioxidant TEMPOL rescued the NS1643-dependent DNA damage (Figure 7C). Our data suggest that stimulation of Kv11.1 channel alters the cellular oxidative state which and produces DNA damage.
Effects of NS1643 on heart function
Cardiac toxicity is one of the most serious adverse effects of anticancer drugs. Cardiac contractility was assessed by transthoracic echocardiograms (TTE) performed on mice with tumors within 1–2h after NS1643 injection [to assess acute effects of the drug] and on mice with tumors that were treated with NS1643 for 25 days (to assess the effect of chronic exposure).
We found that in both acute and chronic exposure, NS1643 did not produce a significant change in any of the measured parameters compared to control (Figure 8; Supplementary Figure 4). To assess whether chronic or acute exposure to NS1643 would generate myocardial damage, we monitored biomarkers for myocardial injuries such as creatine kinase (CK) and CK myocardial band (CK-M) [62]. CK was measured in blood extracted from mice with tumors that were treated with NS1643 and controls for 25 days, while CK-M activity was measured in animals that were treated within 1–2 h before blood extraction. We found that the level of both CK and CK-M activity in blood samples from NS1643 treated animals were not different from controls (Figure 8B, 8C). Overall, these data indicate that exposure to the Kv11.1 activator NS1643 did not significantly alter cardiac function or damage in mice.
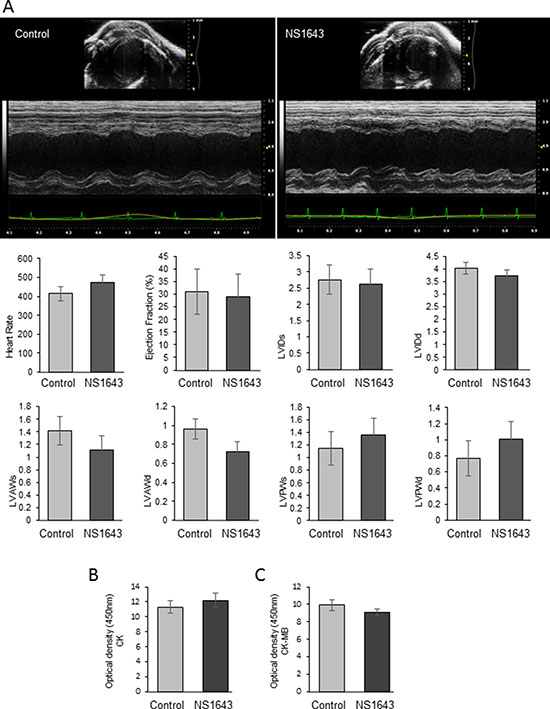
Figure 8: Effect of NS1643 on in vivo Cardiac function. Transthoracic M-mode echocardiograms echocardiography (Vevo 2100 with MS400 series transducer, Visualsonics Inc.) of (A) control (n = 3) and NS1643-treated mice (n = 3). Figure represents a motion mode of the left ventricle, which is obtained with a single ultrasound beam transmitted through the heart with the resulting image displayed over time. Light Gray bar=control; Dark gray bar=NS1643. HR=heart rate (beats per min); EF=ejection fraction (%); LVIDs & LVIDd=left ventricular internal diameter systole and diastole; LVAWs & LVAWd=left ventricular posterior wall systole and diastole (mm). (B) Colorimetric assay detecting serum level of creatine phosphokinase and (C) serum level of creatine phosphokinase-Myocardial band (CK-MB) assay in blood extracted from DMSO (control) vs NS1643 treated mice. Beat-to-beat detection was achieved using preset detection and analysis settings for mice (typical QRS width 10 ms, R waves > 60 ms apart, Pre-P baseline 10 ms, Maximum PR 50 ms, Maximum RT 40 ms, ST height at 10 ms, averaging 4 beats). Averaged ECG measurements were taken from a minimum of 20 beats per mouse.
DISCUSSION
The concerted activity of a multitude of ion channels plays a critical role in maintaining homeostasis [7, 63–66] of every living cell by generating and controlling oscillation of ion fluxes across cellular membranes [67–69]. In recent years, the role of ion channels in cancer biology has received significant attention [9, 70]. Particularly, aberrant expression of K+ ion channels in cancer tissues, including breast cancers, has been implicated in cancer pathogenesis [71, 72]. It is increasingly recognized that specific changes in ionic concentration across membranes can selectively affect intricate intracellular signaling mechanisms regulating fundamental processes such as cell proliferation, migration, and differentiation [66, 73]. For instance, in our initial study on the effect of NS1643 on breast cancer, we showed that loss of K+ ions from the cytoplasm suppresses growth in breast cancer cells as a result of upregulation of tumor suppressors [14, 15, 74, 75]. Accordingly, a recent study demonstrated that accumulation of K+ ions in the cytoplasm can stimulate proliferation by amplification of oncogenic signaling such as K-Ras [76]. This indicates that activity of a K+channel such as Kv11.1 can play a critical role in cancer. Although pharmacologic manipulation of K+ channels with both activators and blockers strongly affects ionic oscillations and therefore alters cancer cell biology [6, 8, 63, 70, 76–78], still very little is known about the role of K+ channels in cancer biology or if they can be considered as potential targets for anticancer therapy.
Our bioinformatics investigation indicates that KCNH2 gene presents a differential expression profile independent of breast cancer histological subtypes, but there is a higher degree of expression in the metastatic phenotype. Although we believe that it is still premature to withdraw a definitive conclusion about the possible role of KCNH2 in oncogenesis, we believe that these data suggest that the KCNH2 gene presents tumor suppressor characteristics at the least in breast cancer. This hypothesis is supported by previous reports showing that the KCNH2 promoter can be found hypermethylated in a variety of cancers, including breast carcinomas, suggesting that in these cancers suppression of Kv11.1 expression might contribute to a hyperproliferative phenotype.
Based on the hypothesis that an increased activity of Kv11.1 can be a limiting factor for cancer cell proliferation, we tested the effects of the small molecule Kv11.1 K+ channel activator NS1643 on tumor growth. We demonstrate that NS1643 significantly arrested tumor growth in TNBC MDA-MB-231-induced xenografts and in a Ras-dependent tumor model of Drosophila. Interestingly, none of the mice presented any significant stress signs during the whole duration of the study which might indicate that NS1643 might inhibit the metastatic process in MDA-MB-231. However, our analysis of tissue distribution of metastasis at sacrifice revealed that only 2 control animals presented few and small metastasis (lungs) which indicate the limitation of the current animal model for studying metastatic processes.
Our data provide evidence that chronic stimulation of Kv11.1 produces strong limitation on proliferative potential of breast cancer cells, which overrides oncogenic signals. We found that tumors isolated from NS1643-treated mice presented a strong reduction of proliferation markers, with a concomitant upregulation of cellular senescence markers. Senescence is a form of normal aging resulting from replicative exhaustion and it is generally characterized by a permanent arrest of the cell cycle and loss of cell’s ability to respond to mitotic and/or apoptotic stimuli [17]. We have previously demonstrated that stimulation of Kv11.1 activity produces an increased net negative intracellular electric charge (hyperpolarization) in breast cancer cells [15]. This is important because several studies have demonstrated that a dynamic change in ionic gradients is necessary to promote cell proliferation [66, 79–83]. For example, it has been found that highly proliferative cells present a smaller negative intracellular charge (depolarized) compared to differentiated cells. Therefore, our approach of altering bioelectric controls [84] by “pharmacologically induced forced hyperpolarization” might be functional for inducing senescence and therefore, inhibition of proliferation. In addition, several epidemiological studies have shown that changes in ion intake (e.g. K+ or Na+) might be associated with carcinogenesis. Although it is not clear what the mechanism linking dietary ions to cancer is, we can speculate that the anticancer effects of targeting ion channels might be potentiated by an appropriate dietary control of slats intake. However, while of obvious etiologic interests, these data should be interpreted with cautions and more dedicated studies need to be developed. Furthermore, cellular senescence has also been detected in benign human tumors [85] and can be activated by several molecular factors, including oncogenes such as RAS, Ca2+ or ROS [86]. Interestingly, it has been shown that RAS can alter the cellular oxidation state of the cell, suggesting that ROS plays a critical role in senescence, at least when triggered by an oncogene. Furthermore, ion channels can be directly or indirectly targeted by ROS [29–34], suggesting that these proteins can play a critical role in cellular oxidation processes. Our data demonstrate that stimulation of Kv11.1 activity produced a rapid increase of ROS and a high degree of DNA damage in both in vivo and in vitro. These data indicate that Kv11.1 K+ channel activity controls the production of ROS and it is not only a downstream effector or mediator of oxidative stress.
Furthermore, in our previous work, we have demonstrated that stimulation of Kv11.1 activity can result in a hyperpolarization-dependent Ca2+ entry in breast cancer cells [15]. This phenomenon can be explained by the increasing attractive force that hyperpolarization exerts on extracellular Ca2+ ions [87, 88]. It has also been shown that Ca2+ plays a critical role in the ROS formation process. Remarkably, application of EGTA [a membrane impermeable Ca2+ ion chelator] prevented NS1643-dependent ROS formation and DNA damage, suggesting that Kv11.1 controls ROS formation by regulating Ca2+ influx.
Overall, our data support the hypothesis that Kv11.1 activity can control Ca2+-dependent ROS formation and DNA damage which, contributes to a Kv11.1-induced senescence process. Nevertheless, at this time, we don’t know whether Ca2+ entry alone can activate NS1643-dependent ROS production, whether this event requires the intervention of other [e.g. intracellular] Ca2+ sources and/or whether mitochondria is necessarily involved in mediating the effects of NS1643. To answer these questions, more mechanistic experiments need to be developed.
Several studies have shown that senescence markers were upregulated in breast cancer samples obtained from patients that were treated with anthracyclines such as doxorubicin. This is an important finding because it suggests that, at least in part, senescence is associated with the antineoplastic effects of doxorubicin and thus finding drugs that can trigger senescence could have a significant impact on cancer therapy. Unfortunately, cardiac toxicity associated with these drugs can result in the irreversible and lethal condition of congestive heart failure and it is often the cause for discontinuing the treatment. Cardiac toxicity is also an important limiting factor for potential therapeutic molecules that act on Kv11.1. It is well known that Kv11.1 blockers can be associated with prolongation of the QT (Long QT; LQT) interval in the heart’s electrical cycle which is a marker for ventricular fibrillation and a risk factor for sudden death [89–91]. For example, several studies have routinely tested the effects of Kv11.1 inhibitors on different cancer cells but, despite their success in impeding cancer cell proliferation, use of these Kv11.1 blockers as anticancer therapeutics is discouraged due to the associated, often lethal severe cardiac arrhythmias [73, 77, 78, 92, 93]. In contrast, cardiac side effects related to Kv11.1 activators (which were initially tested to correct LQT) might include tachycardia (increased heart rate) [94].
Remarkably, our echocardiogram experiments demonstrate that mice treated with acute application of the Kv11.1 activator NS1643 did not present significant alteration of cardiac performance or changes in biochemical parameters that are typically associated with drugs acting on Kv11.1 channel. Our experiments revealed a slight increase in the heart rate in mice treated with NS1643 compared to untreated mice; however, these changes were not statistically significant. In addition, no alteration of the LQT was observed in NS1643-treated mice. Nevertheless, studies on the effect of NS1643 or other Kv11.1 channel activators in humans are still lacking. However, at this time we can speculate that eventual acute effects produced by these drugs on heart performance could be corrected by Kv11.1 blockers which are routinely used in clinic. In addition, chronic exposure to NS1643 did not reveal any significant side effects on the heart caused by current chemotherapeutic or targeting agents (e.g. doxorubicin). Therefore, our data suggest that at the effective dose for arresting tumor growth, NS1643 does not elicit severe cardiac side effects. We would like to point out that tachycardia is easily corrected by the routine measure of intervention [e.g. by beta-blockers] during treatment of patients with breast cancer. Therefore, in comparison to the severe often fatal side effect associated with the use of current antimetastatic agents [24, 25] [e.g. doxorubicin-dependent congestive heart failure], the potential benefits of Kv11.1 activators as anticancer drugs outweigh their potential side effects.
We have previously shown that while chemically distinct, Kv11.1 activators arrest proliferation in breast cancer cell lines without producing any effects on normal cells. Therefore, our experiments are significant and important because they indicate that pharmacologic stimulation of Kv11.1 can selectively force breast cancer cells to undergo cellular senescence and arrest tumor growth without causing major cardiac toxicity.
Although much more research is needed to understand the full benefit of targeting ion channels as an anticancer therapy, we propose that stimulation of the activity of K+ channels such as Kv11.1 could be considered as a potential approach to design a novel anti-breast cancer therapy. Overall, these studies demonstrate that manipulation of ion channels activity could be considered as an important and novel tool to better understand cancer biology and to develop potential anticancer approaches.
MATERIALS AND METHODS
Cells, antibodies, and reagents
All cell lines were purchased from ATCC and maintained at 37°C and 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM, 4.5 g/L glucose), supplemented with 10% fetal bovine serum, non-essential amino acids, 1% Penicillin (100 mg/ml) and streptomycin (100 mg/mL) antibiotics. Mouse anti-Ki-67, rabbit anti-phospho-ERK1/2, ERK1/2, p21waf/cip, and HMGA2 antibodies were purchased from Cell Signaling Technologies, Inc (Boston, MA, USA). Mouse anti-p16INK4A antibody was purchased from Sigma (St. Louis, MO, USA), Mouse anti-phospho H2AX antibody was purchased from EMD Millipore (Darmstadt, Germany), and rabbit anti-H2AX antibody was purchased from Abcam (Cambridge, MA, USA). Horse anti-mouse and goat anti-rabbit secondary antibodies were purchased from Cell Signaling Technologies, Inc. (Boston, MA, USA). NS1643 was purchased from Alomone Labs (Jerusalem, Israel), EGTA was purchased from Calbiochem (San Diego, CA, USA), TEMPOL was purchased from Sigma (St. Louis, MO, USA) and E-4031 was purchased from Abcam (Cambridge, MA, USA).
Drosophila tumor metastasis
The “tumor tester” line with genotype eyFLP; UAS-RasV12, UAS-dlgRNai, UAS-GFP/CyO, GAL80ts [44] was outcrossed to yw to eliminate Gal80ts, which inhibits GAL4 driven transgene expression. After four days of tumor progression at 25°C, GFP-positive [tumor-bearing] larvae were harvested and relocated to new vials with Drosophila food treated with either: NS1643 (25 μM, in DMSO) or vehicle control (DMSO). Three days after drug treatment, larvae were harvested and imaged on an EVOS fluorescent microscope. Data are expressed as Mean±SEM of the integrated GFP intensity in the larvae; *p < 0.05, one-tail t-test.
Tumor xenograft models
Female athymic nude mice (n = 12), aged 6 weeks, received a subcutaneous injection of 4 × 106 MDA-MB-231 cells into the flanks – (100 mL 1:1 PBS). Mice were randomly divided into two groups (n = 6): control (A) and NS1643 treatment (B) Mice in the NS1643 treatment group received NS1643 (6 mg/Kg/2days interval) while the control group received DMSO by intraperitoneal (IP) injections. Tumor size was measured every 3 days using a digital caliper. All animal study protocols were approved by Loyola University’s Institutional Animal Care and Use Committee
Western blot analysis
Tumors were grinded with a pestle and lysed with a buffer containing 50 mM HEPES, 1% Triton X-100, 150 mM NaCl, 5 mM EDTA, 1mM Na3VO4, 1 mM NaF, 1 mM PMSF, and 1x Protease inhibitor cocktail (Cell Signaling Technologies Inc, Boston, MA, USA). Cultured cells were harvested by trypsinization with 0.25% Trypsin-EDTA, washed in PBS, and lysed with cold radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris HCl (pH 8.0), 150 mM NaCl, 1% Tergitol (Sigma-Aldrich, St. Louis, MO, USA), 0.5% Na-deoxycholate, 0.1% SDS, 1 mM PMSF, 1 mM NaF, 1 mM Na3VO4, and 1 × Protease Inhibitor Cocktail (Cell Signaling Technologies Inc, Boston, MA, USA). Protein concentration was determined by BCA assay (Thermo Scientific). An equal amount of protein samples were incubated with 2X or 4X Laemmli buffer at 95°C for 5min. The samples (32 μg) were subjected to SDS-polyacrylamide gel electrophoresis on 4–15% gradient mini-gels and transferred onto a nitrocellulose membrane. Membranes were blocked with 5% nonfat milk in Tris-Buffered Saline containing 0.1% Tween 20 (Sigma-Aldrich) (TBST). Following washes with TBST, the membranes were incubated overnight at 4°C with primary antibodies diluted 1:1000 in 5% bovine serum albumin in TBST. The membranes were then washed with TBST and incubated with anti-rabbit or anti-mouse secondary antibody (1:2000) in TBST with 5% nonfat milk for 1h at room temperature. Following secondary antibody incubation, the membranes were thoroughly washed with TBST and visualized using Luminata Forte Western HRP Substrate [EMD Millipore, Darmstadt, Germany].
Immunohistochemistry of Ki67 protein expression in xenograft breast tumors
Paraffin sections of four tumors from mice that received DMSO or NS1643 were incubated with the Ki-67 antibody at a dilution of 1:200. A polyclonal biotinylated F’ab antibody to mouse immunoglobulin G reactive with all mouse isotypes and a streptavidin–biotinylated peroxidase complex or the Envision kit (Dako, Carpinteria, CA, USA) served as detection systems for the primary antibody. The sections were faintly counterstained with Harris’ hematoxylin. Negative controls were performed without primary antibody.
HMGA2 foci
Human MDA-MB-231 cells were treated with NS1643 or DMSO (control) and stained with DAPI (purple) and immunostained with an antibody recognizing HMGA2 (red). For each sample, a minimum of 75 cells was analyzed using LSM710 ZEN software and the mean percentage value+SE was determined.
PAR immunocytochemistry
Cells plated on coverslips (N = 2/group) were fixed (4% PFA in PBS, 20 min), and blocked (10% normal goat serum (NGS), 1% BSA, 0.5% TritonX-100 in PBS, for 1 hr). Coverslips were labeled (2 hrs) with an anti-PAR mouse monoclonal antibody (1:100; ALX-804-220; Enzo Life Sciences, Farmingdale, NY) in antibody diluent (3% NGS, 1% BSA, 0.5% TritonX-100). After PBS washes (3 × 5 min), coverslips were incubated (1 hr) with an Alexa 594-conjugated secondary antibody (1:1000; 115-585-166; Jackson ImmunoResearch Laboratories, West Grove, PA) in antibody diluent (3% NGS, 1% BSA, 0.5% TritonX-100). Coverslips were incubated with DAPI (1:11 in PBS; NucBlue; Molecular Probes, Eugene, OR; 10 min) and washed (PBS, 3 x 5 min), before mounting on microscope slides (Aqua-Poly/Mount; Polysciences, Inc, Warrington, PA) and drying. All procedures took place at room temperature.
Images were collected in DAPI and Alexa 594 channels using an inverted wide-field fluorescence microscope (IX81, Olympus, Center Valley, PA) equipped with a 12-bit CCD camera (QIClick; QImaging, Surrey, BC Canada) and a motorized stage (Proscan III; Prior, Rockland, MA). Constrained iterative deconvolution was performed on image stacks using cellSens software (Olympus, Center Valley, PA). Regions of interest (ROIs) around nuclei were created by thresholding DAPI images in ImageJ (NIH, Bethesda, MD), and PAR immunofluorescence within ROIs was then quantified.
DNA damage detection
Tumors from mice that received DMSO or NS1643 were excised and processed for cryosectioning. 12 mm thick sections were stained with DAPI (100 ng/μL) and imaged using a Leica SPE confocal microscope (Leica Microsystems). The number of nuclei exhibiting fragmented vs. normal morphology was counted manually in ImageJ software. Data are expressed as a fraction of the normalized total number of nuclei.
Neutral comet assay was performed for detecting DNA double-strand breaks in human MDA-MB-231 cells. The images were captured on a fluorescent microscope and quantified by using TriTek CometScore ™
Detection of reactive oxygen species (ROS)
Cells were seeded at 4 × 104 cells/ml in 96 well plates. Adhered cells were incubated in the dark with DCFH-DA to 2′,7′-dichlorofluorescein (1 h/37°C; 40 μM, ThermoFisher Sci). Cells were washed and subjected to different drug treatments for 2h. Fluorescence was analyzed in a plate reader (PHERAstar FS, BMG LABTECH) with excitation at 485 nm and emission at 520 nm
Echocardiogram and ECG analysis
Cardiac function was assessed using echocardiography (Vevo 2100 with MS400 series transducer, Visualsonics Inc.). Briefly, mice were anesthetized with 1.2-1.5% isoflurane and placed on a heating pad to maintain body temperature. Parasternal short-axis 2-D echocardiograms and M-mode cine loops were taken at the level of the papillary muscles. The total length of anesthesia was less than 10 mins. Evaluation of stored data was performed offline by a sonographer blind to treatment groups.
ECG analysis was performed in LabChart8 (ADInstruments)
CKs activity
Creatine kinase (Abcam) and myocardial creatine kinase (MyBioSource) activity were measured by ELISA method in blood samples extracted from NS1643-treated or untreated mice according to manufacturer instructions.
Abbreviations
K+: potassium; Ca2+: calcium; hERG : human ether-a-go-go-related gene type 1; Kv: voltage-gated potassium channel; ER+: estrogen receptor; HER2: human epidermal growth factor receptor 2; TNBC: triple negative breast cancer; ROS: reactive oxygen species; HMGA2: high mobility group A2; EGTA: ethylene glycol-bis(β-aminoethyl ether)-N,N,N’,N’-tetraacetic acid; TTE: transthoracic echocardiograms; CK: creatine kinase; CK-M: creatine kinase-myocardial band;
Author contributions
SG, DFF-L, VR designed the study. WKJ provided administrative support. VR, DW, AB, E-KB, CO, JJZ, MB, SK, DK, SB, collected, assembled, analyzed the data and interpreted results. SG, DFF-L, WKJ, VR, JJZ, SK participated in the manuscript writing. All authors read and approved the final manuscript.
ACKNOWLEDGMENTS
The authors would like to thank Alexander Rockwell for excellent technical support.
The Edward Hines Jr. VA Hospital, Hines, IL, Dr. John P. and Therese E. Mulcahy Endowed Professor in Ophthalmology and intramural support by the Cardiovascular Research Institute at Loyola University Chicago is gratefully acknowledged. The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
FUNDING
MB and JJZ were supported in part by American Cancer Research Grant #IRG-14-195-01.
REFERENCES
1. Barni S, Cabiddu M, Guarneri P, Lonati V, Petrelli F. The risk for anemia with targeted therapies for solid tumors. Oncologist. 2012; 17:715–24.
2. Mayer EL, Krop IE. Advances in targeting SRC in the treatment of breast cancer and other solid malignancies. Clin Cancer Res. 2010; 16:3526–32.
3. Villanueva A, Llovet JM. Targeted therapies for hepatocellular carcinoma. Gastroenterology. 2011; 140:410–26.
4. Tinoco G, Warsch S, Gluck S, Avancha K, Montero AJ. Treating breast cancer in the 21st century: emerging biological therapies. J Cancer. 2013; 4:117–32.
5. Urrego D, Tomczak AP, Zahed F, Stuhmer W, Pardo LA. Potassium channels in cell cycle and cell proliferation. Philos Trans R Soc Lond B Biol Sci. 2014; 369:20130094.
6. Pardo LA, Stuhmer W. The roles of K(+) channels in cancer. Nat Rev Cancer. 2014; 14:39–48.
7. Bates E. Ion channels in development and cancer. Annu Rev Cell Dev Biol. 2015; 31:231–47.
8. Jehle J, Schweizer PA, Katus HA, Thomas D. Novel roles for hERG K(+) channels in cell proliferation and apoptosis. Cell Death Dis. 2011; 2:e193.
9. Rao VR, Perez-Neut M, Kaja S, Gentile S. Voltage-gated ion channels in cancer cell proliferation. Cancers (Basel). 2015; 7:849–75.
10. Dai X, Xiang L, Li T, Bai Z. Cancer Hallmarks, Biomarkers and Breast Cancer Molecular Subtypes. J Cancer. 2016; 7:1281–94.
11. Ismail-Khan R, Bui MM. A review of triple-negative breast cancer. Cancer Control. 2010; 17:173–6.
12. De Laurentiis M, Cianniello D, Caputo R, Stanzione B, Arpino G, Cinieri S, Lorusso V, De Placido S. Treatment of triple negative breast cancer (TNBC): current options and future perspectives. Cancer Treat Rev. 2010; 36:S80–6.
13. Carey L, Winer E, Viale G, Cameron D, Gianni L. Triple-negative breast cancer: disease entity or title of convenience? Nat Rev Clin Oncol. 2010; 7:683–92.
14. Perez-Neut M, Rao VR, Gentile S. hERG1/Kv11.1 activation stimulates transcription of p21waf/cip in breast cancer cells via a calcineurin-dependent mechanism. Oncotarget. 2015; 7:58893–58902. https://doi.org/10.18632/oncotarget.3797.
15. Perez-Neut M, Shum A, Cuevas BD, Miller R, Gentile S. Stimulation of hERG1 channel activity promotes a calcium-dependent degradation of cyclin E2, but not cyclin E1, in breast cancer cells. Oncotarget. 2015; 6:1631–1639. https://doi.org/10.18632/oncotarget.2829.
16. Burton DG, Krizhanovsky V. Physiological and pathological consequences of cellular senescence. Cell Mol Life Sci. 2014; 71:4373–86.
17. Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013; 75:685–705.
18. Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961; 25:585–621.
19. Munoz-Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014; 15:482–96.
20. Prieur A, Peeper DS. Cellular senescence in vivo: a barrier to tumorigenesis. Curr Opin Cell Biol. 2008; 20:150–5.
21. Ewald JA, Desotelle JA, Wilding G, Jarrard DF. Therapy-induced senescence in cancer. J Natl Cancer Inst. 2010; 102:1536–46.
22. te Poele RH, Okorokov AL, Jardine L, Cummings J, Joel SP. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res. 2002; 62:1876–83.
23. Chang BD, Broude EV, Dokmanovic M, Zhu H, Ruth A, Xuan Y, Kandel ES, Lausch E, Christov K, Roninson IB. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res. 1999; 59:3761–7.
24. Raj S, Franco VI, Lipshultz SE. Anthracycline-induced cardiotoxicity: a review of pathophysiology, diagnosis, and treatment. Curr Treat Options Cardiovasc Med. 2014; 16:315.
25. Wojtacki J, Lewicka-Nowak E, Lesniewski-Kmak K. Anthracycline-induced cardiotoxicity: clinical course, risk factors, pathogenesis, detection and prevention--review of the literature. Med Sci Monit. 2000; 6:411–20.
26. Lefrak EA, Pitha J, Rosenheim S, Gottlieb JA. A clinicopathologic analysis of adriamycin cardiotoxicity. Cancer. 1973; 32:302–14.
27. Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. N Engl J Med. 1998; 339:900–5.
28. Octavia Y, Tocchetti CG, Gabrielson KL, Janssens S, Crijns HJ, Moens AL. Doxorubicin-induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. J Mol Cell Cardiol. 2012; 52:1213–25.
29. Agostinho P, Duarte CB, Carvalho AP, Oliveira CR. Oxidative stress affects the selective ion permeability of voltage-sensitive Ca2+ channels in cultured retinal cells. Neurosci Res. 1997; 27:323–34.
30. Patel R, Sesti F. Oxidation of ion channels in the aging nervous system. Brain Res. 2016; 1639:174–85.
31. Su Z, Limberis J, Martin RL, Xu R, Kolbe K, Heinemann SH, Hoshi T, Cox BF, Gintant GA. Functional consequences of methionine oxidation of hERG potassium channels. Biochem Pharmacol. 2007; 74:702–11.
32. Ciorba MA, Heinemann SH, Weissbach H, Brot N, Hoshi T. Modulation of potassium channel function by methionine oxidation and reduction. Proc Natl Acad Sci U S A. 1997; 94:9932–7.
33. Chen J, Avdonin V, Ciorba MA, Heinemann SH, Hoshi T. Acceleration of P/C-type inactivation in voltage-gated K(+) channels by methionine oxidation. Biophys J. 2000; 78:174–87.
34. Tang XD, Daggett H, Hanner M, Garcia ML, McManus OB, Brot N, Weissbach H, Heinemann SH, Hoshi T. Oxidative regulation of large conductance calcium-activated potassium channels. J Gen Physiol. 2001; 117:253–74.
35. Berube J, Caouette D, Daleau P. Hydrogen peroxide modifies the kinetics of HERG channel expressed in a mammalian cell line. J Pharmacol Exp Ther. 2001; 297:96–102.
36. Cotella D, Hernandez-Enriquez B, Wu X, Li R, Pan Z, Leveille J, Link CD, Oddo S, Sesti F. Toxic role of K+ channel oxidation in mammalian brain. J Neurosci. 2012; 32:4133–44.
37. Yu W, Parakramaweera R, Teng S, Gowda M, Sharad Y, Thakker-Varia S, Alder J, Sesti F. Oxidation of KCNB1 Potassium Channels Causes Neurotoxicity and Cognitive Impairment in a Mouse Model of Traumatic Brain Injury. J Neurosci. 2016; 36:11084–11096.
38. Gorlach A, Bertram K, Hudecova S, Krizanova O. Calcium and ROS: A mutual interplay. Redox Biol. 2015; 6:260–71.
39. Szasz AM, Lanczky A, Nagy A, Forster S, Hark K, Green JE, Boussioutas A, Busuttil R, Szabo A, Gyorffy B. Cross-validation of survival associated biomarkers in gastric cancer using transcriptomic data of 1,065 patients. Oncotarget. 2016; 7:49322–49333. https://doi.org/10.18632/oncotarget.10337.
40. Gluck S, Ross JS, Royce M, McKenna EF Jr, Perou CM, Avisar E, Wu L. TP53 genomics predict higher clinical and pathologic tumor response in operable early-stage breast cancer treated with docetaxel-capecitabine +/- trastuzumab. Breast Cancer Res Treat. 2012: 132:781–91.
41. Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, Chen H, Omeroglu G, Meterissian S, Omeroglu A, Hallett M, Park M. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med. 2008; 14:518–27.
42. Zhao H, Langerod A, Ji Y, Nowels KW, Nesland JM, Tibshirani R, Bukholm IK, Karesen R, Botstein D, Borresen-Dale AL, Jeffrey SS. Different gene expression patterns in invasive lobular and ductal carcinomas of the breast. Mol Biol Cell. 2004; 15:2523–36.
43. Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001; 98:10869–74.
44. Richardson AL, Wang ZC, De Nicolo A, Lu X, Brown M, Miron A, Liao X, Iglehart JD, Livingston DM, Ganesan S. X chromosomal abnormalities in basal-like human breast cancer. Cancer Cell. 2006; 9:121–32.
45. Gonzalez C. Drosophila melanogaster: a model and a tool to investigate malignancy and identify new therapeutics. Nat Rev Cancer. 2013: 13:172–83.
46. Tipping M, Perrimon N. Drosophila as a model for context-dependent tumorigenesis. J Cell Physiol. 2014; 229:27–33.
47. Rudrapatna VA, Cagan RL, Das TK. Drosophila cancer models. Dev Dyn. 2012; 241:107–18.
48. Dahal GR, Pradhan SJ, Bates EA. Inwardly rectifying potassium channels regulate Dpp release in the Drosophila wing disc. Development. 2017; 144:2771–2783.
49. Warmke JW, Ganetzky B. A family of potassium channel genes related to eag in Drosophila and mammals. Proc Natl Acad Sci U S A. 1994; 91:3438–42.
50. Titus SA, Warmke JW, Ganetzky B. The Drosophila erg K+ channel polypeptide is encoded by the seizure locus. J Neurosci. 1997; 17:875–81.
51. Lee J, Wu CF. Orchestration of stepwise synaptic growth by K+ and Ca2+ channels in Drosophila. J Neurosci. 2010; 30:15821–33.
52. Willecke M, Toggweiler J, Basler K. Loss of PI3K blocks cell-cycle progression in a Drosophila tumor model. Oncogene. 2011; 30:4067–74.
53. Vidal M, Cagan RL. Drosophila models for cancer research. Curr Opin Genet Dev. 2006; 16:10–6.
54. Narita M, Krizhanovsky V, Nunez S, Chicas A, Hearn SA, Myers MP, Lowe SW. A novel role for high-mobility group a proteins in cellular senescence and heterochromatin formation. Cell. 2006; 126:503–14.
55. Boo LM, Lin HH, Chung V, Zhou B, Louie SG, O'Reilly MA, Yen Y, Ann DK. High mobility group A2 potentiates genotoxic stress in part through the modulation of basal and DNA damage-dependent phosphatidylinositol 3-kinase-related protein kinase activation. Cancer Res. 2005; 65:6622–30.
56. Sharma A, Singh K, Almasan A. Histone H2AX phosphorylation: a marker for DNA damage. Methods Mol Biol. 2012; 920:613–26.
57. Tanaka T, Halicka D, Traganos F, Darzynkiewicz Z. Cytometric analysis of DNA damage: phosphorylation of histone H2AX as a marker of DNA double-strand breaks (DSBs). Methods Mol Biol. 2009; 523:161–8.
58. Wei H, Yu X. Functions of PARylation in DNA Damage Repair Pathways. Genomics Proteomics Bioinformatics. 2016; 14:131–139.
59. Evans MD, Dizdaroglu M, Cooke MS. Oxidative DNA damage and disease: induction, repair and significance. Mutat Res. 2004; 567:1–61.
60. Imlay JA, Linn S. DNA damage and oxygen radical toxicity. Science. 1988; 240:1302–9.
61. Yan Y, Wei CL, Zhang WR, Cheng HP, Liu J. Cross-talk between calcium and reactive oxygen species signaling. Acta Pharmacol Sin. 2006; 27:821–6.
62. Vassiliadis E, Barascuk N, Didangelos A, Karsdal MA. Novel cardiac-specific biomarkers and the cardiovascular continuum. Biomark Insights. 2012; 7:45–57.
63. Dubyak GR. Ion homeostasis, channels, and transporters: an update on cellular mechanisms. Adv Physiol Educ. 2004; 28:143–54.
64. Litan A, Langhans SA. Cancer as a channelopathy: ion channels and pumps in tumor development and progression. Front Cell Neurosci. 2015; 9:86.
65. Peruzzo R, Biasutto L, Szabo I, Leanza L. Impact of intracellular ion channels on cancer development and progression. Eur Biophys J. 2016; 45:685–707.
66. Yang M, Brackenbury WJ. Membrane potential and cancer progression. Front Physiol. 2013; 4:185.
67. Okada Y, Doida Y, Roy G, Tsuchiya W, Inouye K, Inouye A. Oscillations of membrane potential in L cells. I. Basic characteristics. J Membr Biol. 1977; 35:319–35.
68. Okada Y, Roy G, Tsuchiya W, Doida Y, Inouye A. Oscillations of membrane potential in L cells. II. Effect of monovalent ion concentrations and conductance changes associated with oscillations. J Membr Biol. 1977; 35:337–50.
69. Okada Y, Tsuchiya W, Inouye A. Oscillations of membrane potential in L cells. IV. Role of intracellular Ca2+ in hyperpolarizing excitability. J Membr Biol. 1979; 47:357–76.
70. Pardo LA. Voltage-gated potassium channels in cell proliferation. Physiology (Bethesda). 2004; 19:285–92.
71. D'Amico M, Gasparoli L, Arcangeli A. Potassium channels: novel emerging biomarkers and targets for therapy in cancer. Recent Pat Anticancer Drug Discov. 2013; 8:53–65.
72. Lastraioli E, Perrone G, Sette A, Fiore A, Crociani O, Manoli S, D'Amico M, Masselli M, Iorio J, Callea M, Borzomati D, Nappo G, Bartolozzi F, et al. hERG1 channels drive tumour malignancy and may serve as prognostic factor in pancreatic ductal adenocarcinoma. Br J Cancer. 2015; 112:1076–87.
73. Lobikin M, Chernet B, Lobo D, Levin M. Resting potential, oncogene-induced tumorigenesis, and metastasis: the bioelectric basis of cancer in vivo. Phys Biol. 2012; 9:065002.
74. Lansu K, Gentile S. Potassium channel activation inhibits proliferation of breast cancer cells by activating a senescence program. Cell Death Dis. 2013; 4:e652.
75. Perez-Neut M, Haar L, Rao V, Santha S, Lansu K, Rana B, Jones WK, Gentile S. Activation of hERG3 channel stimulates autophagy and promotes cellular senescence in melanoma. Oncotarget. 2016; 7:21991–22004. https://doi.org/10.18632/oncotarget.7831.
76. Accardi A. CELL SIGNALING. Lipids link ion channels and cancer. Science. 2015; 349:789–90.
77. Asher V, Sowter H, Shaw R, Bali A, Khan R. Eag and HERG potassium channels as novel therapeutic targets in cancer. World J Surg Oncol. 2010; 8:113.
78. Chen SZ, Jiang M, Zhen YS. HERG K+ channel expression-related chemosensitivity in cancer cells and its modulation by erythromycin. Cancer Chemother Pharmacol. 2005; 56:212–20.
79. Cone CD Jr. Electroosmotic interactions accompanying mitosis initation in sarcoma cells in vitro. Trans N Y Acad Sci. 1969; 31:404–27.
80. Cone CD Jr, Tongier M Jr. Control of somatic cell mitosis by simulated changes in the transmembrane potential level. Oncology. 1971; 25:168–82.
81. Stillwell EF, Cone CM, Cone CD Jr. Stimulation of DNA synthesis in CNS neurones by sustained depolarisation. Nat New Biol. 1973; 246:110–1.
82. Djamgoz MB, Coombes RC, Schwab A. Ion transport and cancer: from initiation to metastasis. Philos Trans R Soc Lond B Biol Sci. 2014; 369:20130092.
83. Huang X, He Y, Dubuc AM, Hashizume R, Zhang W, Reimand J, Yang H, Wang TA, Stehbens SJ, Younger S, Barshow S, Zhu S, Cooper MK, et al. EAG2 potassium channel with evolutionarily conserved function as a brain tumor target. Nat Neurosci. 2015; 18:1236–46.
84. Blackiston DJ, McLaughlin KA, Levin M. Bioelectric controls of cell proliferation: ion channels, membrane voltage and the cell cycle. Cell Cycle. 2009; 8:3527–36.
85. Curigliano G, Criscitiello C. Successes and limitations of targeted cancer therapy in breast cancer. Prog Tumor Res. 2014; 41:15–35.
86. Holmstrom KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol. 2014; 15:411–21.
87. Lepple-Wienhues A, Berweck S, Bohmig M, Leo CP, Meyling B, Garbe C, Wiederholt M. K+ channels and the intracellular calcium signal in human melanoma cell proliferation. J Membr Biol. 1996; 151:149–57.
88. Wang Z. Roles of K+ channels in regulating tumour cell proliferation and apoptosis. Pflugers Arch. 2004; 448:274–86.
89. Berul CI, Perry JC. Contribution of long-QT syndrome genes to sudden infant death syndrome: is it time to consider newborn electrocardiographic screening? Circulation. 2007; 115:294–6.
90. Salvi V, Karnad DR, Panicker GK, Kothari S. Update on the evaluation of a new drug for effects on cardiac repolarization in humans: issues in early drug development. Br J Pharmacol. 2010; 159:34–48.
91. van Noord C, Eijgelsheim M, Stricker BH. Drug- and non-drug-associated QT interval prolongation. Br J Clin Pharmacol. 2010; 70:16–23.
92. Ganapathi SB, Kester M, Elmslie KS. State-dependent block of HERG potassium channels by R-roscovitine: implications for cancer therapy. Am J Physiol Cell Physiol. 2009; 296:C701–10.
93. Wang Y, Zhang Y, Yang L, Cai B, Li J, Zhou Y, Yin L, Yang BF, Lu YJ. Arsenic trioxide induces the apoptosis of human breast cancer MCF-7 cells through activation of caspase-3 and inhibition of HERG channels. Exp Ther Med. 2011; 2:81–486.
94. Bentzen BH, Bahrke S, Wu K, Larsen AP, Odening KE, Franke G, Storm vans Gravesande K, Biermann J, Peng X, Koren G, Zehender M, Bode C, Grunnet M, Brunner M. Pharmacological activation of Kv11.1 in transgenic long QT-1 rabbits. J Cardiovasc Pharmacol. 2011; 57:223–30.