
INTRODUCTION
Breast cancer remains a leading cause of death in women throughout the world. Triple negative breast cancer (TNBC) accounts for only 15-20% of all breast cancer, but is disproportionally associated with the majority of breast cancer related deaths [1]. Chemotherapy is currently the mainstay of systemic medical treatment for TNBC. However, it is associated with severe off-target tissue toxicity, rapid drug-resistance, and enrichment of cancer stem cells [2, 3]. As such, development of targeted therapies for TNBC is an unmet medical need.
Over the past several decades, in vitro and in vivo preclinical research commonly uses over 27 TNBC cancer cell lines to study cancer pathogenesis, disease advancement, and drug effectiveness. However, a growing disconnection between results generated using TNBC cell lines and clinical trials has been observed. A recent example is the in vitro and in vivo results of PARP inhibitor veliparib. Veliparib is an oral inhibitor of Poly (ADP-Ribose) Polymerase (PARP) 1 and 2, which enhances the activity of DNA damaging agents in DNA repair to promote apoptosis. In vitro, veliparib is capable of suppressing the expression of Snail which promotes epithelial to mesenchymal transition, tumor metastasis and drug resistance. It also sensitizes the MDA-MB-231 TNBC cell line to chemotherapeutic drug doxorubicin, resulting in increased apoptosis [4]. In vivo, veliparib sensitizes MDA-MB-231 tumors to TMZ (temozolomide, an alkylating agent) in a SCID (severe combined immune deficiency) mouse model [5]. The effectiveness of other therapeutic combinations with veliparib has also been demonstrated in vivo xenograft mouse models using cancer cell lines [5, 6].
Clinical trials, however, failed to demonstrate the efficacy of veliparib in combination with a DNA damaging agent for the treatment of breast cancer including TNBC. The phase II clinical trial (NCT01506609) recruited 193 metastatic breast cancer patients treated with either the placebo or veliparib in a combination of carboplatin and paclitaxel. Progression-free survival in the control group (chemotherapeutic drugs alone) was 12.3 (9.3–14.5) months compared to the 14.1 (11.5–16.2) months in the combination group, showing statistically insignificant difference (p value = 0.231) [7, 8]. Overall survival in the control was 25.0 (18.1–34.8) months and the combination of veliparib and chemotherapy was 28.5 (22.4– not reported results), which was insignificant (p value = 0.148) [8]. Despite these results, veliparib in combination with paclitaxel and carboplatin followed by doxorubicin and cyclophosphamide advanced into phase III clinical trials (NCT02032277) for the treatment of TNBC [9]. 634 TNBC patients were involved in the study and treated with veliparib or placebo in combination with paclitaxel and carboplatin followed by doxorubicin and cyclophosphamide. There was no significant difference in the efficacy of treatment (53.2% veliparib + chemotherapy vs 57.5% placebo + chemotherapy, p = 0.36) [9]. This recent failure is by no means a rarity as many similar results have been reported [10–14]. This highlights the disconnection between cell lines in vitro and in vivo preclinical research and human clinical trials. The translational disparity led to the US National Cancer Institute halting the usage of 60 human cancer cell lines for drug-screening in 2016 and recommending to use patient derived xenograft (PDX) for future research. Appropriate models used in preclinical/translational studies may bridge the divide [15]. In this regard, PDXs have shone as clinically relevant models in comparison to breast cancer cell lines due to their ability to better represent the original tumor’s biology and retain the original tumor’s architecture and organization [16].
THE LIMITATIONS OF CELL LINES IN PRECLINICAL RESEARCH
Breast cancer cell lines used for conventional analysis were originally harvested and generated from patient tumor samples after in vitro culture for years or decades. The deviancies observed are thought to arise through selection of specific populations and changes over time to promote adaption to artificial culture environments. Breast cancer cell lines are capable of growing indefinitely and undergoing freezing-thawing cycles for several decades. It has been demonstrated that breast cancer cell lines possess a moderately high mutation frequency in comparison to patient tumors. Over many in vitro passages, these mutations can accumulate, possibly making the cells differ dramatically from their starting source [17–20]. Additionally, these mutations can promote certain traits which provide a survival benefit for in vitro growth in a plastic dish. This would promote clonal selection for the fittest subpopulations [21]. Continuous propagation of cells in a petri dish would also result in accumulating epigenetic alterations [22]. It has been demonstrated that human cancer cell lines possess altered methylation patterns after culture [23]. Altered DNA methylation affects gene and protein expressions, subsequently impacting signal pathways and therapeutic responses. Additional reports have shown that DNA methylation differs dramatically between cancer cell lines in comparison to patient tumors, making epigenetic studies using cell lines discordant with clinical settings [24, 25].
One example was the expression of ER/PR/HER-2 receptors in two TNBC tumors obtained from patients and cultured for 150 passages. Originally, these receptors were all absent in the primary tumors harvested from the TNBC patients [26]. Miller et al also recently showed that there were almost no overlaps in gene expression between glioblastoma samples grown in mice and cultured on a dish after 2-3 weeks, suggesting a marked modification of tumor biological features after short-term culture in petri dishes [27].
This dramatic deviance is largely associated with the disruption of the original tumor structure and microenvironment which is comprised of a heterozygous mixture of different subpopulations of tumor cells, macrophages, fibroblasts, endothelial cells, stromal cells, the extracellular matrix, etc. [28, 29]. Cancer cell lines do not represent these heterozygous components. Rather, during initial harvesting and culturing, subpopulations adapted better for in vitro petri dish environment (e.g. cancer associated fibroblast cells) are commonly selected for, overtaking the other tumor cells and resulting in a relative homozygous population overtime [30–32]. Culture methodologies which inhibit fibroblastic growth and promote epithelial proliferation, still fall victim to one dominant tumor subpopulation [31, 33]. This artificial selection makes the therapies developed highly effective on a particular cell subtype rather than the whole heterozygous tumor and its extracellular matrix and tumor microenvironment, which disconnects the bench results from the clinical trials. In vivo studies, human breast cancer cell lines are commonly mixed with matrigel and injected into mouse mammary pad to resemble the clinical settings. However, in addition to the aforementioned limitations, this sudden influx of cancerous cells bypasses the early development of a tumor in the patient and skips over the formation of the tumor microenvironment [34, 35]. This may in part, explain the divergence between the high frequency of bone metastasis for patient with breast cancer (~70% of all breast metastasis) and the very low frequency of spontaneous metastasis of breast cancer cell line implanted in the mammary fat pad [36–38]. As such, to mimic bone metastasis, breast cancer cell lines must be injected either via intracardiac, tail vein or intra-osseous, or specialized cell lines must be utilized [38–41]. Additionally, the monocultured breast cancer cell lines do not include factors commonly dysregulated in the tumor such as hypoxia, inflammation, vascularity, stromal cells, immune cell infiltration, and aberrant signalling pathways [42]. These factors work together to regulate tumor microenvironment, tumor growth and metastasis. As such, translatable research requires a breast cancer model freshly isolated from the patient without disturbing tumor structures to encompass all of these factors and retain tumor heterogeneity and microenvironment (Figure 1).
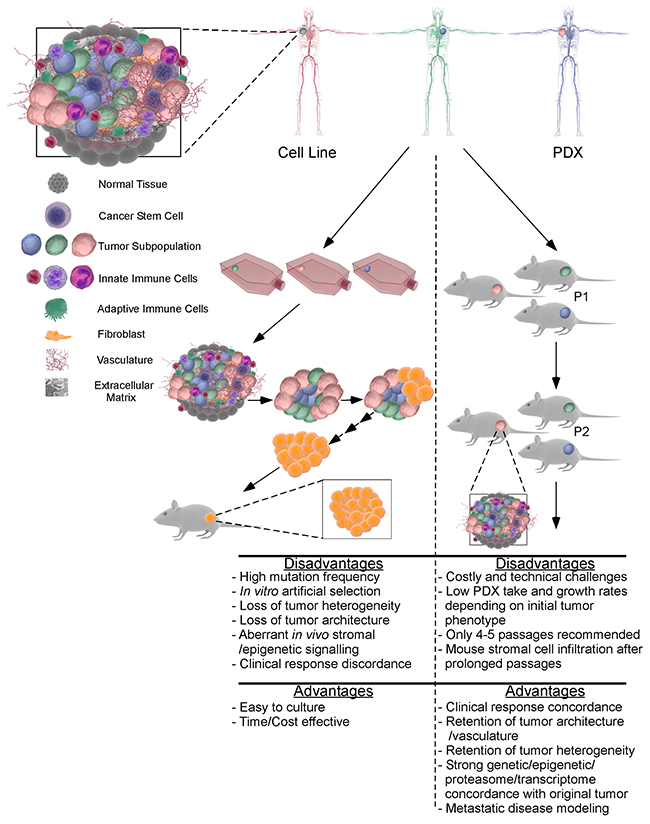
Figure 1: The main differences between PDX and cell line xenografts for preclinical research. In vitro culture of patient samples leads to a loss of tumor architecture and heterogeneity. The resultant adherent tumor cells are subject to culture selection and adaptation to artificial conditions, leading to the generation of a cell line from a subpopulation of the original patient’s tumor containing multiple cell types and subpopulations. Subsequent in vitro and in vivo experiments preformed using a subpopulation of cells may result in discrepancy between breast cell lines and clinical observations and clinical trials. In contrast, implanting breast tumor immediately after harvesting from patients into an immune deficient mouse model can preserve tumor heterogeneity, architecture and stromal and extracellular components. After in vivo expansion, the PDX tumors retain original tumor properties for up to 4-5 passages. In sharp contrast to cancer cell line xenografts, drug responses of PDX models are consistent with patients, making PDX model an invaluable tool for translational research.
THE IMPORTANCE OF USING PATIENTS TUMORS AS MODELS FOR PRECLINCIAL RESEARCH
Considerable observations obtained from patients’ tumors cannot be mimicked by using breast cancer cell lines. Acerbi et al recently demonstrated that crosstalk between the extracellular matrix and inflammation promotes invasion in 20 breast cancer patient biopsies [43]. Increased amounts of collagen were deposited within invasive breast cancer. Furthermore, the collagen was thicker, underwent a linear reorganization in the stroma of the invasive lesions, and was associated with increased mechano-signalling and increased stromal stiffness. The invasive edge of the tumors possessed the greatest stromal stiffness illustrating regional stromal heterogeneity. This stiffness at the tumor edge was caused by accumulating activated macrophages and increased TGF-β activity, suggesting a crosstalk between macrophage accumulation, stromal stiffness and tumor invasion. TNBC patient tumors possessed the greatest stromal stiffness, macrophage accumulation, and TGF-β activation at the tumor front compared to the other breast cancer subtypes. Additionally, TNBC exhibited increased YAP (Yes-associated protein) signalling that correlated with stromal stiffness, tumor aggression and invasion. YAP is a mechanically activated signaling pathway that is associated with cancer stem cells (CSCs) and poor patient prognosis [44–46]. This study highlights the multifaceted interplay between tumor cells, the extra cellular matrix and the immune system, which cannot be modeled by the cultured breast cancer cell lines and their xenografts.
Using patients’ tumor samples, Liu et al, demonstrated that there exist two pools of CSCs within the breast cancer. A mesenchymal, migratory CD44+/CD24- CSC subpopulation exists at the tumor edge, while an epithelial, proliferative ALDH+ CSC subpopulation resides within the tumor core. Moreover, interconversion (plasticity) between the fractionated two CSC subpopulations was observed, and both epithelial and mesenchymal CSCs were responsible for metastasis and tumor reconstitution at a secondary location. Controversially, in vivo xenograft analyses of breast cancer cell lines were unable to demonstrate ALDH+ or CD44+/CD24- CSC localization patterns, or demonstrate a correlation between the frequency of CD44+/CD24- CSCs and tumor metastasis as observed in patients with breast cancer [47, 48].
Recent reports demonstrated that a hybrid epithelial/mesenchymal CD44+/CD24-/ALDH+ CSC subpopulation is more tumorigenic then its pure counterpart, although its role in metastasis and secondary tumor formation remains to be elaborated [49–51]. Using patients’ metastatic breast cancer pleural effusions, Shiraishi et al demonstrated that CD44+/CD24-/ALDH+ CSCs possessed a greater hypoxic response to hypoxia inducible factor (HIF-1α) signalling [49]. This response in turn promoted an epithelial to mesenchymal transition through the inhibition of E-cadherin and stimulation of Notch-1, Jagged-1, TGF-β, Slug and Snail, which enhanced metastasis and secondary tumor formation in vivo. Interestingly, CD44+/CD24-/ALDH- CSCs in contrast, did not undergo EMT upon hypoxia. Instead, hypoxia induced HIF-1α to bind directly to the ALDH1A1 promoter, which converted CD44+/CD24- /ALDH- CSCs into CD44+/CD24- ALDH+ CSCs. The newly converted ALDH+ cells expressed angiogenic genes rather than EMT-related genes and were able to generate pulmonary metastasis [49]. In comparison to patient tumors, breast cancer cell lines differentially expressed ALDH, CD44 and/or CD24, making interpretation of experimental results difficult [52]. These studies further highlight the importance of using patients’ tumor samples over breast cancer cell lines for the studies of inter/intra tumor interactions, CSC localization and plasticity, tumor heterogeneity and metastasis in translational medicine [47, 53–55].
PATIENT-DERIVED XENOGRAFT MODELS
While fresh patients’ tumors are a great model for cancer research, their availability, quantity and quality are limiting factors for widespread usage [56]. Patient-derived xenograft (PDX) models become an excellent alternative and are readily available for researchers. PDX models are generated through the transplantation of patients’ tumor tissues into an immunocompromised mouse [57]. The implanted tumors are expanded and serially passaged in mice. PDX procedures exclude tissue dissociation and in vitro culture, which prevents cell adaptation to artificial culture system, clonal selection, and homogeneity (Figure 1) [58].
Another advantage of the PDX model over cell lines is the preservation of the original tumor architecture and organization such as vasculature and stromal components [16]. This is thought to represent the original tumor’s biology and retain the interactions between the tumor and its microenvironment [16, 59]. PDX models also retain intra/inter-tumor heterogeneity, gene expression, single nucleotide polymorphisms, copy number variants and chromosomal architecture of the original tumors [16, 58–63].
The ability of the PDX models to simulate the original patients’ tumors may explain the strong correlation between PDX models and actual patient responses [64–67]. Zhang et al demonstrated this through implanting a series of human breast tumor tissues into the mammary fat pad of immunodeficient mice [68]. The tumor growth was correlated with tumor grade and the absence of estrogen (ER)/progesterone (PR) expression. After successful engraftment and growth, it was found that all PDXs retained the primary tumors’ histologic phenotypes. PDXs were also evaluated at the transcriptome, proteasome, and genome levels across multiple generations, and all closely resembling the original tumors [68]. Moreover, in a close resemblance to actual breast cancer progression, 48% of PDX tumors exhibited pulmonary metastasis after implantation into mammary fat pad. More importantly, clinical relevance was compared by assessing PDX response to the same treatment regime that had been used in the same patients giving rise to the PDX. Of 13 PDX tumors, 12 (92%) showed the same response as did patients to the chemotherapeutic drugs such as doxorubicin, paclitaxel or dasatinib amongst others, illustrating a high correlation between patients and PDX models [68].
In another report, Marangoni et al implanted 200 breast adenocarcinoma samples into the fat pad of athymic mice and stably generated 22 PDXs. They demonstrated that high breast grade tumors were superior to lower grade counterparts for engraftment and growth. Again, the original patient tumor histology, genomic rearrangements, chromosomal amplifications, and gene expression profiles were preserved in PDXs. Spontaneous metastasis was observed in 10/22 PDXs (45%), which also exhibited similar histology to the original tumors. Similar responses to chemotherapy (e.g. docetaxel/5-flurouracil/trastuzumab) between patients and their PDXs were also demonstrated in five out of seven cases [69, 70].
PDX models also retain the epigenetic patterns of the original patient tumor. Guilhamon et al demonstrated that in osteosarcoma and colon cancer, methylation profiles of PDXs were well preserved compared to the primary patient tumor with only 2.7% of CpG sites undergoing a major methylation shift in PDXs [71]. The second passage of PDXs showed only 0.07% of alternations in CpG methylation sites in comparison to the first passage [71]. Tomar et al also demonstrated that only 0.66-1.17% of CpGs were significantly altered after 3 passages compared to the original patient tumor in high-grade serious ovarian cancer PDXs [72]. While chemotherapy did not alter the DNA methylation pattern, treatment with decitabine (a demethylation agent) significantly demethylated 10.6% CpG sites and inhibited in vivo PDX tumor growth. Together, these studies suggest the epigenetic stability of PDX models and their suitability for epigenetic studies in comparison to cancer cells lines [72].
Short-term ex vivo cultured PDXs have also been used for pre-clinical high-throughput drug screening. Bruma et al showed that all PDX tumor tissues they tested could be successfully cultured ex vivo for a short period (n=27). These short-term ex vivo cultured PDX tissues retained tissue architecture, molecular and genetic features of in vivo PDXs. Of 40 ex vivo cultured PDX tissues used for drug screening, 33 (82.5%) were verified by in vivo PDX models, suggesting that ex vivo cultured PDX tissues can be used for high-throughput preclinical drug screening [73].
The predictive power of PDX models has led to the development of co-clinical trials, where patients and mice implanted with PDX tumors developed from the patient will be treated simultaneously or retrospectively. This allows for validation of the PDX results generated, and determination of factors affecting drug response/efficacy/resistance [74]. These personalized approaches are currently being investigated for various cancer types in multiple ongoing clinical trials [75]. One particular ongoing co-clinical trial for the treatment of TNBC is to study the effects of neoadjuvant docetaxel in combination with carboplatin in patients with stage 2-3 TNBC who have not achieved a pathologic complete response due to chemotherapeutic resistance (NCT02124902) [76]. The PDX models in this study will be developed simultaneously to determine chemotherapeutic response between patients, PDX take rates and to identify signatures of chemotherapy resistance and response [76]. Table 1 summarizes current active clinical trials using both PDX models and patients, investigating mechanisms underlying tumor progression, metastasis, and drug response and resistance.
Table 1: List of ongoing clinical trials using PDX models
|
Rank |
NCT number |
Title |
Recruitment |
Conditions |
|---|---|---|---|---|
|
1 |
NCT03164863 |
Onco4D(TM) Biodynamic Chemotherapy Selection for Breast Cancer Patients |
Recruiting |
Breast Cancer |
|
2 |
NCT02752893 |
Estrogen Receptor-Positive Breast Cancer Patient-Derived Xenografts |
Recruiting |
Breast Cancer |
|
3 |
NCT02732860 |
Personalized Patient Derived Xenograft (pPDX) Modeling to Test Drug Response in Matching Host |
Enrolling by invitation |
Colorectal Neoplasms|Colorectal Cancer|Breast Cancer|Breast Neoplasms |
|
4 |
NCT02455882 |
Tissue Procurement Protocol for Patients Undergoing Treatment for Early-Stage Breast Cancer |
Recruiting |
Breast Cancer |
|
5 |
NCT02315196 |
Pegylated Liposomal Doxorubicin Hydrochloride and Carboplatin Followed by Surgery and Paclitaxel in Treating Patients With Triple Negative Stage II-III Breast Cancer |
Recruiting |
Estrogen Receptor-negative Breast |
|
6 |
NCT02247037 |
Patient-derived Xenograft (PDX) Modeling of Treatment Response for Triple Negative Breast Cancer |
Recruiting |
Triple Negative Breast Cancer |
|
7 |
NCT02124902 |
Neoadjuvant Treatment of Triple Negative Breast Cancer Patients With Docetaxel and Carboplatin to Assess Anti-tumor Activity |
Recruiting |
Triple Negative Breast Neoplasms |
Despite these advantages, PDX models are not perfect (Advantages/Disadvantages being summarized in Figure 1). The growth rate of PDX models are very slow compared to cell culture and xenografts generated using cancer cell lines. PDX implanted will take around 4-8 months for the development of a preclinical research specimen [74, 77]. Low engraftment rate persists as a critical challenge for PDX models. It was reported that TNBC possessed 53.8% of engraftment compared to 15.6% for hormone receptor positive breast cancer [78]. However, the established PDX samples exhibit over 90% engraftment rate despite low success for the primary PDX. The considerable established PDX samples that have been well characterized are currently available from research institutes or companies. The growth rate in each PDX mouse can also be highly variable depending on the quality and location of the tissues prepared from the same tumor.
Additionally, passaging the PDX samples in mice requires more resources, time and expertise in comparison to cell lines. Long-term passaging of PDX samples also affects PDX characteristics. Pearson et al demonstrated that PDXs of human head and neck squamous cell carcinoma increased their growth rate and displayed histopathological features of a higher tumor grade after prolonged in vivo passages [79]. To avoid deviations, it is recommended to use low passages (less than 5 passages). McAuliffe et al showed that high passages of breast cancer PDX exhibited some aberrations in P13K/mTOR signalling, an abrupt loss of human DNA in the PDX tumor and an increase in murine DNA. This was followed by the spontaneous generation of murine mammary adenocarcinoma [78]. Additionally, after 3-5 passages, the tumor stroma has been found to be replaced by the host mouse stroma which could influence stromal signalling, tumor rigidity, macrophage infiltration, autocrine and paracrine signalling, possibly deviating PDX from the original patients’ tumors [74].
Another limitation for current PDX models and for cancer cell line-xenografts is the requirement for the tumor to be implanted into an immunodeficient mouse for tumor engraftment and growth. Due to the lack of an immune system, the PDX model is not practical for immunological research. New PDX models have been proposed to address these issues by humanizing the immune deficient mice (e.g. JAX NSG). The human immune system will be generated through early transplantation of human hematopoietic stem cells into immunodeficient mice, followed by PDX implantation. This model will allow for assessment of immuno-tumor interactions in PDX [80]. This advancement can finally allow for the studies of human chimeric antigen receptor T cell, anti-PDL/PDL-1 and CTLA-4 in a PDX model.
PDX MODELS AND NANOMEDICINE
Different from normal vascular system, the presence of endothelial gaps and transcellular holes in tumors increases blood vessel leakiness [81]. It is also found that tumor vasculature lacks vascular hierarchy, and possesses architectural abnormalities (heterogeneous, disorganized, branched/overlapped, and/or loosely connected) that resist blood flow and promote the extravascular erythrocyte accumulation (blood lakes) [81–84]. This in turn promotes improper nutrient translocation to the tumor and insufficient metabolite clearance, resulting in ischemia, hypoxia, acidic tumor environment, and necrosis. Increased HIF-1 in the tumor further enhances abnormal angiogenesis and tumor growth [82, 85].
PDX models have been demonstrated to be capable of representing human tumor angiogenesis [62]. The tumor vasculature comprised of human endothelial cells has been shown to mirror the donor patients’ tumor angiogenesis up to 35 days after implantation [56, 86]. In contrast, cancer cell line xenografts exhibit different vasculature from patients’ tumors, leading to contradictory effectiveness in angiogenic therapy [87–89]. Since angiogenesis is not only regulated by human tumor cells but also by human stromal cells and extracellular matrix, this might be a possible cause for the discrepancy as cancer cell line xenografts lack the components of human stromal cells and human extracellular matrix [90–92].
Abnormal tumor vasculature plays a key role in nanoparticle-based therapy. Nanotechnology applications in cancer have revolutionized the landscape of cancer drug development by their uniquely appealing features, such as improved blood circulation, higher tumor accumulation and reduced toxicities leading to a higher therapeutic index. Upon systemic administration, therapeutic nanoparticles have been shown to accumulate in tumors as a result of a multitude of biological processes involving mainly leaky tumor vasculature, poor lymphatic drainage and other minor events as well as enhanced permeability, and retention properties of the nanoparticle itself [93–98]. As such, considerable nanomedicine based therapies are undergoing clinical trials today [99].
One of challenges for nanoparticle-based therapy is to determine treatment efficacy using a model system that resembles patients’ tumor. PDX as a model meets this requirement. It has been demonstrated that PDX TNBC models are highly vascularized in comparison to cell line xenografts, resembling original patients’ tumors [100]. Although this field is advancing rapidly, specificity of nanoparticle-drug accumulation within TNBC PDX tumor as opposed to surrounding tissues and other organs due to enhanced permeability and retention effect has yet to be investigated. Using PDX model to determine the therapeutic efficacy of nanomedicine will provide novel, translatable and tangible approaches for the clinical treatment of TNBC patients.
To conclude, a hierarchy of patient tumors, in vivo PDX and short-term ex vivo cultured PDX tissues have been depicted with their respective overlapping or distinctive features (Figure 2). Since the development of PDX TNBC models is crucial for experimentation, we have included the procedures for the expansion/generation of PDX in NOD-SCID mice (Supplementary Materials and Supplementary Figure 1) [101].
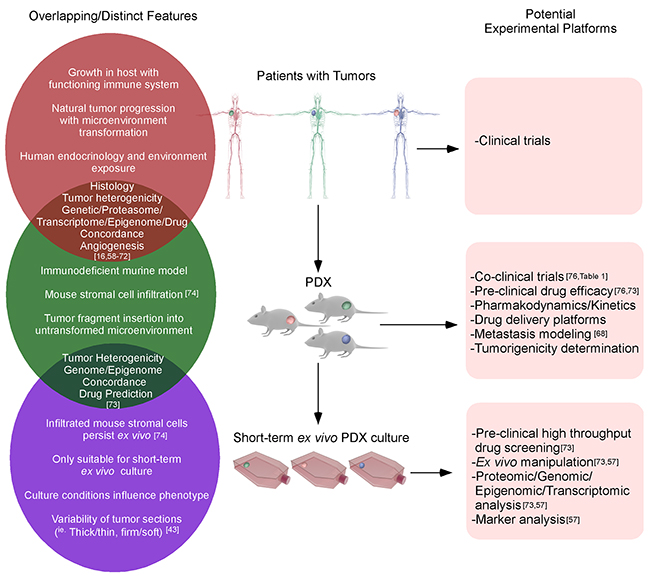
Figure 2: The similarities and differences between clinical, PDX and ex vivo cultured PDX tissues. The growth of original tumors in patients will be influenced by the tumor microenvironment, immune system, etc. When the primary tumor tissues harvested from patients are inserted into immunodeficient mice, the majority of tumor properties could be retained. After in vivo passages, murine stromal cells could gradually infiltrate the tumor, although this will not significantly alter the tumor phenotypes until later passages. When PDX tissues are excised and cultured ex vivo for a short-term, genetic/epigenetic/drug predictions are still highly correlated with the original patients’ tumors.
Author contributions
AS and LW conceived and designed the study. AS drafted the manuscript. LW and AS edited the manuscript. All authors approved the final version of the manuscript.
ACKNOWLEDGMENTS
We thank Dr. Luk Cox and Dr. Idoya Lahortiga from Somersault 18:24 to allow the use of their Library of Science and Medical Illustrations (http://www.somersault1824.com/resources/) for the creation of the Figure 1 and 2. We also thank the Huntsman Cancer Institute in Salt Lake City, UT for the use of the Preclinical Research Resource (PRR) to provide the PDX samples and Dr. Christiano Souza and Sarah McGarry for their help in developing our PDX protocol.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This work is supported by operating grants from Canadian Breast Cancer Foundation-Ontario Region and the Canadian Institutes of Health Research MOP-111224 to LW.
REFERENCES
1. Anders CK, Carey LA. Biology, metastatic patterns, and treatment of patients with triple-negative breast cancer. Clin Breast Cancer. 2009; 9:S73–S81.
2. Gómez-Miragaya J, Palafox M, Paré L, Yoldi G, Ferrer I, Vila S, Galván P, Pellegrini P, Pérez-Montoyo H, Igea A. Resistance to taxanes in triple-negative breast cancer associates with the dynamics of a CD49f+ tumor-initiating population. Stem Cell Reports. 2017; 8:1392–407.
3. Jia D, Tan Y, Liu H, Ooi S, Li L, Wright K, Bennett S, Addison CL, Wang L. Cardamonin reduces chemotherapy-enriched breast cancer stem-like cells in vitro and in vivo. Oncotarget. 2016; 7:771-785. https://doi.org/10.18632/oncotarget.5819.
4. Mariano G, Ricciardi MR, Trisciuoglio D, Zampieri M, Ciccarone F, Guastafierro T, Calabrese R, Valentini E, Tafuri A, Del Bufalo D. PARP inhibitor ABT-888 affects response of MDA-MB-231 cells to doxorubicin treatment, targeting Snail expression. Oncotarget. 2015; 6:15008-15021. https://doi.org/10.18632/oncotarget.3634.
5. Palma JP, Wang YC, Rodriguez LE, Montgomery D, Ellis PA, Bukofzer G, Niquette A, Liu X, Shi Y, Lasko L. ABT-888 confers broad in vivo activity in combination with temozolomide in diverse tumors. Clin Cancer Res. 2009; 15:7277–90.
6. Shelton JW, Waxweiler TV, Landry J, Gao H, Xu Y, Wang L, El-Rayes B, Shu HK. In vitro and in vivo enhancement of chemoradiation using the oral PARP inhibitor ABT-888 in colorectal cancer cells. Int J Radiat Oncol Biol Phys. 2013; 86:469–76.
7. Isakoff SJ, Puhalla S, Domchek SM, Friedlander M, Kaufman B, Robson M, Telli ML, Diéras V, Han HS, Garber JE. A randomized Phase II study of veliparib with temozolomide or carboplatin/paclitaxel versus placebo with carboplatin/paclitaxel in BRCA1/2 metastatic breast cancer: design and rationale. Future Oncol. 2017; 13:307–20.
8. Han H, Diéras V, Robson M, Palácová M, Marcom P, Jager A, Bondarenko I, Citrin D, Campone M, Telli M, Domchek S, Friedlander M, Kaufman B, et al. Abstract S2-05: efficacy and tolerability of veliparib (V; ABT-888) in combination with carboplatin (C) and paclitaxel (P) vs placebo (Plc)+C/P in patients (pts) with BRCA1 or BRCA2 mutations and metastatic breast cancer: a randomized, phase 2 study. Cancer Res. 2017; 77:S2-05.
9. Geyer CE, O’Shaughnessy J, Untch M, Sikov W, Rugo HS, McKee MD, Huober JB, Golshan M, Giranda VL, Minckwitz GV, Maag D, Sullivan DM, Wolmark N, et al. Phase 3 study evaluating efficacy and safety of veliparib (V) plus carboplatin (Cb) or Cb in combination with standard neoadjuvant chemotherapy (NAC) in patients (pts) with early stage triple-negative breast cancer (TNBC). J Clin Oncol. 2017; 35:520.
10. Wakelee HA, Dahlberg SE, Keller SM, Tester WJ, Gandara DR, Graziano SL, Adjei A, Leighl N, Aisner SC, Rothman JM. Randomized phase III trial of adjuvant chemotherapy with or without bevacizumab in resected non-small cell lung cancer (NSCLC): results of E1505. J Thorac Oncol. 2015.
11. Sinha G. (2014). Downfall of iniparib: a PARP inhibitor that doesn’t inhibit PARP after all. Oxford University Press US.
12. Flinn IW, Miller CB, Ardeshna KM, Tetreault S, Assouline SE, Zinzani PL, Mayer J, Merli M, Lunin SD, Pettitt AR. Dynamo: a phase 2 study demonstrating the clinical activity of duvelisib in patients with relapsed refractory indolent non-hodgkin lymphoma. Blood. 2016; 128:1218.
13. Soria JC, Wu YL, Nakagawa K, Kim SW, Yang JJ, Ahn MJ, Wang J, Yang JC, Lu Y, Atagi S. Gefitinib plus chemotherapy versus placebo plus chemotherapy in EGFR-mutation-positive non-small-cell lung cancer after progression on first-line gefitinib (IMPRESS): a phase 3 randomised trial. Lancet Oncol. 2015; 16:990–8.
14. Taplin ME, Antonarakis ES, Ferrante KJ, Horgan K, Blumenstein BA, Saad F, Luo J, De Bono JS. Clinical factors associated with AR-V7 detection in ARMOR3-SV, a randomized trial of galeterone (Gal) vs enzalutamide (Enz) in men with AR-V7+ metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol. 2017; 35:5005.
15. Ledford H. US cancer institute overhauls cell lines: veteran cells to be replaced by human tumours grown in mice. Nature. 2016; 530:391–2.
16. Williams SA, Anderson WC, Santaguida MT, Dylla SJ. Patient-derived xenografts, the cancer stem cell paradigm, and cancer pathobiology in the 21st century. Lab Invest. 2013; 93:970.
17. Jiang G, Zhang S, Yazdanparast A, Li M, Pawar AV, Liu Y, Inavolu SM, Cheng L. Comprehensive comparison of molecular portraits between cell lines and tumors in breast cancer. BMC Genomics. 2016; 17:525.
18. Osborne CK, Hobbs K, Trent JM. Biological differences among MCF-7 human breast cancer cell lines from different laboratories. Breast Cancer Res Treat. 1987; 9:111–21.
19. Wenger SL, Senft JR, Sargent LM, Bamezai R, Bairwa N, Grant SG. Comparison of established cell lines at different passages by karyotype and comparative genomic hybridization. Biosci Rep. 2004; 24:631–9.
20. Briske-Anderson MJ, Finley JW, Newman SM. The influence of culture time and passage number on the morphological and physiological development of Caco-2 cells. Proc Soc Exp Biol Med. 1997; 214:248–57.
21. Pastrana E. Stem cells: the survival of the fittest. Nat Methods. 2012; 9:16.
22. Nestor CE, Ottaviano R, Reinhardt D, Cruickshanks HA, Mjoseng HK, McPherson RC, Lentini A, Thomson JP, Dunican DS, Pennings S. Rapid reprogramming of epigenetic and transcriptional profiles in mammalian culture systems. Genome Biol. 2015; 16:11.
23. Varley KE, Gertz J, Bowling KM, Parker SL, Reddy TE, Pauli-Behn F, Cross MK, Williams BA, Stamatoyannopoulos JA, Crawford GE. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 2013; 23:555–67.
24. Houshdaran S, Hawley S, Palmer C, Campan M, Olsen MN, Ventura AP, Knudsen BS, Drescher CW, Urban ND, Brown PO. DNA methylation profiles of ovarian epithelial carcinoma tumors and cell lines. PLoS One. 2010; 5:e9359.
25. Poirier JT, Gardner EE, Connis N, Moreira AL, De Stanchina E, Hann CL, Rudin CM. DNA methylation in small cell lung cancer defines distinct disease subtypes and correlates with high expression of EZH2. Oncogene. 2015; 34:5869–78.
26. Kamalidehghan B, Houshmand M, Kamalidehghan F, Jafarzadeh N, Azari S, Akmal SN, Rosli R. Establishment and characterization of two human breast carcinoma cell lines by spontaneous immortalization: discordance between estrogen, progesterone and HER2/neu receptors of breast carcinoma tissues with derived cell lines. Cancer Cell Int. 2012; 12:43.
27. Miller TE, Liau BB, Wallace LC, Morton AR, Xie Q, Dixit D, Factor DC, Kim LJ, Morrow JJ, Wu Q, Mack SC, Hubert CG, Gillespie SM, et al. Transcription elongation factors represent in vivo cancer dependencies in glioblastoma. Nature. 2017; 547:355–9.
28. Balkwill FR, Capasso M, Hagemann T. (2012). The tumor microenvironment at a glance. The Company of Biologists Ltd.
29. Binnemars-Postma K, Storm G, Prakash J. Nanomedicine strategies to target tumor-associated macrophages. Int J Mol Sci. 2017; 18:979.
30. Speirs V, Green A, Walton D, Kerin M, Fox J, Carleton P, Desai S, Atkin S. Short-term primary culture of epithelial cells derived from human breast tumours. Br J Cancer. 1998; 78:1421.
31. Wang CS, Goulet F, Tremblay N, Germain L, Auger F, Têtu B. Selective culture of epithelial cells from primary breast carcinomas using irradiated 3T3 cells as feeder layer. Pathol Res Pract. 2001; 197:175–81.
32. Nishikata T, Ishikawa M, Matsuyama T, Takamatsu K, Fukuhara T, Konishi Y. Primary culture of breast cancer: a model system for epithelial-mesenchymal transition and cancer stem cells. Anticancer Res. 2013; 33:2867–73.
33. Janik K, Popeda M, Peciak J, Rosiak K, Smolarz M, Treda C, Rieske P, Stoczynska-Fidelus E, Ksiazkiewicz M. Efficient and simple approach to in vitro culture of primary epithelial cancer cells. Biosci Rep. 2016; 36:e00423.
34. Mullen P, Ritchie A, Langdon SP, Miller WR. Effect of matrigel on the tumorigenicity of human breast and ovarian carcinoma cell lines. Int J Cancer. 1996; 67:816–20.
35. Vargo-Gogola T, Rosen JM. Modelling breast cancer: one size does not fit all. Nat Rev Cancer. 2007; 7:659.
36. Simmons J, Hildreth B 3rd, Supsavhad W, Elshafae S, Hassan B, Dirksen W, Toribio RE, Rosol TJ. Animal models of bone metastasis. Vet Pathol. 2015; 52:827–41.
37. Coleman R, Rubens R. The clinical course of bone metastases from breast cancer. Br J Cancer. 1987; 55:61.
38. Lelekakis M, Moseley JM, Martin TJ, Hards D, Williams E, Ho P, Lowen D, Javni J, Miller FR, Slavin J. A novel orthotopic model of breast cancer metastasis to bone. Clin Exp Metastasis. 1999; 17:163–70.
39. Thibaudeau L, Quent VM, Holzapfel BM, Taubenberger AV, Straub M, Hutmacher DW. Mimicking breast cancer-induced bone metastasis in vivo: current transplantation models and advanced humanized strategies. Cancer Metastasis Rev. 2014; 33:721–35.
40. Fantozzi A, Christofori G. Mouse models of breast cancer metastasis. Breast Cancer Res. 2006; 8:212.
41. Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordón-Cardo C, Guise TA, Massagué J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003; 3:537–49.
42. Prasetyanti PR, Medema JP. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol Cancer. 2017; 16:41.
43. Acerbi I, Cassereau L, Dean I, Shi Q, Au A, Park C, Chen Y, Liphardt J, Hwang E, Weaver V. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr Biol (Camb). 2015; 7:1120–34.
44. Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C, Inui M, Montagner M, Parenti Anna R, Poletti A, Daidone Maria G, Dupont S, Basso G, et al. The hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell. 2011; 147:759–72.
45. Xu Y, Stamenkovic I, Yu Q. CD44 attenuates activation of the hippo signaling pathway and is a prime therapeutic target for glioblastoma. Cancer Res. 2010; 70:2455–64.
46. Kim T, Yang SJ, Hwang D, Song J, Kim M, Kim SK, Kang K, Ahn J, Lee D, Kim MY. A basal-like breast cancer-specific role for SRF-IL6 in YAP-induced cancer stemness. Nat Commun. 2015; 6:10186.
47. Liu Y, Nenutil R, Appleyard M, Murray K, Boylan M, Thompson A, Coates P. Lack of correlation of stem cell markers in breast cancer stem cells. Br J Cancer. 2014; 110:2063.
48. Sheridan C, Kishimoto H, Fuchs RK, Mehrotra S, Bhat-Nakshatri P, Turner CH, Goulet R, Badve S, Nakshatri H. CD44+/CD24-breast cancer cells exhibit enhanced invasive properties: an early step necessary for metastasis. Breast Cancer Res. 2006; 8:R59.
49. Shiraishi A, Tachi K, Essid N, Tsuboi I, Nagano M, Kato T, Yamashita T, Bando H, Hara H, Ohneda O. Hypoxia promotes the phenotypic change of aldehyde dehydrogenase activity of breast cancer stem cells. Cancer Sci. 2017; 108:362–72.
50. Sulaiman A, Yao Z, Wang L. Re-evaluating the role of epithelial-mesenchymal-transition in cancer progression. J Biomed Res. 2017.
51. Shao J, Fan W, Ma B, Wu Y. Breast cancer stem cells expressing different stem cell markers exhibit distinct biological characteristics. Mol Med Rep. 2016; 14:4991–8.
52. Ricardo S, Vieira AF, Gerhard R, Leitão D, Pinto R, Cameselle-Teijeiro JF, Milanezi F, Schmitt F, Paredes J. Breast cancer stem cell markers CD44, CD24 and ALDH1: expression distribution within intrinsic molecular subtype. J Clin Pathol. 2011; 64:937–46.
53. Horimoto Y, Arakawa A, Sasahara N, Tanabe M, Sai S, Himuro T, Saito M. Combination of cancer stem cell markers CD44 and CD24 is superior to ALDH1 as a prognostic indicator in breast cancer patients with distant metastases. PLoS One. 2016; 11:e0165253.
54. Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007; 1:555–67.
55. Velasco-Velázquez MA, Popov VM, Lisanti MP, Pestell RG. The role of breast cancer stem cells in metastasis and therapeutic implications. Am J Pathol. 2011; 179:2–11.
56. Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM, Arcaroli JJ, Messersmith WA, Eckhardt SG. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol. 2012; 9:338–50.
57. Du L, Li YJ, Fakih M, Wiatrek RL, Duldulao M, Chen Z, Chu P, Garcia-Aguilar J, Chen Y. Role of SUMO activating enzyme in cancer stem cell maintenance and self-renewal. Nat Commun. 2016; 7.
58. Daniel VC, Marchionni L, Hierman JS, Rhodes JT, Devereux WL, Rudin CM, Yung R, Parmigiani G, Dorsch M, Peacock CD. A primary xenograft model of small-cell lung cancer reveals irreversible changes in gene expression imposed by culture in vitro. Cancer Res. 2009; 69:3364–73.
59. Choi SY, Lin D, Gout PW, Collins CC, Xu Y, Wang Y. Lessons from patient-derived xenografts for better in vitro modeling of human cancer. Adv Drug Deliv Rev. 2014; 79:222–37.
60. Jin K, Teng L, Shen Y, He K, Xu Z, Li G. Patient-derived human tumour tissue xenografts in immunodeficient mice: a systematic review. Clin Transl Oncol. 2010; 12:473–80.
61. McEvoy J, Ulyanov A, Brennan R, Wu G, Pounds S, Zhang J, Dyer MA. Analysis of MDM2 and MDM4 single nucleotide polymorphisms, mRNA splicing and protein expression in retinoblastoma. PLoS One. 2012; 7:e42739.
62. DeRose YS, Wang G, Lin YC, Bernard PS, Buys SS, Ebbert MT, Factor R, Matsen C, Milash BA, Nelson E. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat Med. 2011; 17:1514–20.
63. Bertotti A, Migliardi G, Galimi F, Sassi F, Torti D, Isella C, Corà D, Di Nicolantonio F, Buscarino M, Petti C. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011; 1:508–23.
64. Owonikoko TK, Zhang G, Kim HS, Stinson RM, Bechara R, Zhang C, Chen Z, Saba NF, Pakkala S, Pillai R. Patient-derived xenografts faithfully replicated clinical outcome in a phase II co-clinical trial of arsenic trioxide in relapsed small cell lung cancer. J Transl Med. 2016; 14:111.
65. Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, Zhang C, Schnell C, Yang G, Zhang Y. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med. 2015; 21:1318.
66. Berger D, Fiebig H, Winterhalter B, Wallbrecher E, Henss H. Preclinical phase II study of ifosfamide in human tumour xenografts in vivo. Cancer Chemother Pharmacol. 1990; 26:S7–11.
67. Julien S, Merino-Trigo A, Lacroix L, Pocard M, Goéré D, Mariani P, Landron S, Bigot L, Nemati F, Dartigues P. Characterization of a large panel of patient-derived tumor xenografts representing the clinical heterogeneity of human colorectal cancer. Clin Cancer Res. 2012.
68. Zhang X, Claerhout S, Prat A, Dobrolecki LE, Petrovic I, Lai Q, Landis MD, Wiechmann L, Schiff R, Giuliano M. A renewable tissue resource of phenotypically stable, biologically and ethnically diverse, patient-derived human breast cancer xenograft models. Cancer Res. 2013; 73:4885–97.
69. Whittle JR, Lewis MT, Lindeman GJ, Visvader JE. Patient-derived xenograft models of breast cancer and their predictive power. Breast Cancer Res. 2015; 17:17.
70. Marangoni E, Vincent-Salomon A, Auger N, Degeorges A, Assayag F, de Cremoux P, De Plater L, Guyader C, De Pinieux G, Judde JG. A new model of patient tumor-derived breast cancer xenografts for preclinical assays. Clin Cancer Res. 2007; 13:3989–98.
71. Guilhamon P, Butcher LM, Presneau N, Wilson GA, Feber A, Paul DS, Schütte M, Haybaeck J, Keilholz U, Hoffman J. Assessment of patient-derived tumour xenografts (PDXs) as a discovery tool for cancer epigenomics. Genome Med. 2014; 6:116.
72. Tomar T, de Jong S, Alkema NG, Hoekman RL, Meersma GJ, Klip HG, Zee AG, Wisman GB. Genome-wide methylation profiling of ovarian cancer patient-derived xenografts treated with the demethylating agent decitabine identifies novel epigenetically regulated genes and pathways. Genome Med. 2016; 8:107.
73. Bruna A, Rueda OM, Greenwood W, Batra AS, Callari M, Batra RN, Pogrebniak K, Sandoval J, Cassidy JW, Tufegdzic-Vidakovic A. A biobank of breast cancer explants with preserved intra-tumor heterogeneity to screen anticancer compounds. Cell. 2016; 167:260-74.e22.
74. Hidalgo M, Amant F, Biankin AV, Budinská E, Byrne AT, Caldas C, Clarke RB, de Jong S, Jonkers J, Mælandsmo GM. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov. 2014; 4:998–1013.
75. Byrne AT, Alferez DG, Amant F, Annibali D, Arribas J, Biankin AV, Bruna A, Budinska E, Caldas C, Chang DK, Clarke RB, Clevers H, Coukos G, et al. Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nat Rev Cancer. 2017; 17:254–68.
76. Ademuyiwa FO, Li S, Skinner T, Hoshower J, Luo J, Ma CX, Weilbaecher KN, Naughton M, Hernandez-Aya LF, Mardis ER. A co-clinical phase II trial of carboplatin and docetaxel as neoadjuvant treatment for triple negative breast cancer with genomic discovery analysis. J Clin Oncol. 2016.
77. Paez-Ribes M, Man S, Xu P, Kerbel RS. Development of patient derived xenograft models of overt spontaneous breast cancer metastasis: a cautionary note. PLoS One. 2016; 11:e0158034.
78. McAuliffe PF, Evans KW, Akcakanat A, Chen K, Zheng X, Zhao H, Eterovic AK, Sangai T, Holder AM, Sharma C. Ability to generate patient-derived breast cancer xenografts is enhanced in chemoresistant disease and predicts poor patient outcomes. PLoS One. 2015; 10:e0136851.
79. Pearson AT, Finkel KA, Warner KA, Nör F, Tice D, Martins MD, Jackson TL, Nör JE. Patient-derived xenograft (PDX) tumors increase growth rate with time. Oncotarget. 2016; 7:7993-8005. https://doi.org/10.18632/oncotarget.6919.
80. Simpson-Abelson MR, Sonnenberg GF, Takita H, Yokota SJ, Conway TF, Kelleher RJ, Shultz LD, Barcos M, Bankert RB. Long-term engraftment and expansion of tumor-derived memory T cells following the implantation of non-disrupted pieces of human lung tumor into NOD-scid IL2Rγnull mice. J Immunol. 2008; 180:7009–18.
81. Hashizume H, Baluk P, Morikawa S, McLean JW, Thurston G, Roberge S, Jain RK, McDonald DM. Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol. 2000; 156:1363–80.
82. Hlushchuk R, Barré S, Djonov V. Morphological aspects of tumor angiogenesis. Methods Mol Biol. 2016; 1464:13–24.
83. Less JR, Skalak TC, Sevick EM, Jain RK. Microvascular architecture in a mammary carcinoma: branching patterns and vessel dimensions. Cancer Res. 1991; 51:265–73.
84. Baluk P, Hashizume H, McDonald DM. Cellular abnormalities of blood vessels as targets in cancer. Curr Opin Genet Dev. 2005; 15:102–11.
85. Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996; 16:4604–13.
86. Rofstad EK, Huang R, Galappathi K, Andersen L, Wegner CS, Hauge A, Gaustad JV, Simonsen TG. Functional intratumoral lymphatics in patient-derived xenograft models of squamous cell carcinoma of the uterine cervix: implications for lymph node metastasis. Oncotarget. 2016; 7:56986–97. https://doi.org/10.18632/oncotarget.10931.
87. Lieu CH, Tan AC, Leong S, Diamond JR, Eckhardt SG. From bench to bedside: lessons learned in translating preclinical studies in cancer drug development. J Natl Cancer Inst. 2013; 105:1441–56.
88. Hecht JR, Trarbach T, Hainsworth JD, Major P, Jäger E, Wolff RA, Lloyd-Salvant K, Bodoky G, Pendergrass K, Berg W. Randomized, placebo-controlled, phase III study of first-line oxaliplatin-based chemotherapy plus PTK787/ZK 222584, an oral vascular endothelial growth factor receptor inhibitor, in patients with metastatic colorectal adenocarcinoma. J Clin Oncol. 2011; 29:1997–2003.
89. Drevs J, Hofmann I, Hugenschmidt H, Wittig C, Madjar H, Müller M, Wood J, Martiny-Baron G, Unger C, Marmé D. Effects of PTK787/ZK 222584, a specific inhibitor of vascular endothelial growth factor receptor tyrosine kinases, on primary tumor, metastasis, vessel density, and blood flow in a murine renal cell carcinoma model. Cancer Res. 2000; 60:4819–24.
90. Montecinos VP, Godoy A, Hinklin J, Vethanayagam RR, Smith GJ. Primary xenografts of human prostate tissue as a model to study angiogenesis induced by reactive stroma. PLoS One. 2012; 7:e29623.
91. Garber K. Angiogenesis inhibitors suffer new setback. Nat Biotechnol. 2002; 20:1067–8.
92. Jain RK, Duda DG, Clark JW, Loeffler JS. Lessons from phase III clinical trials on anti-VEGF therapy for cancer. Nat Clin Pract Oncol. 2006; 3:24–40.
93. Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer. 2017; 17:20–37.
94. Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014; 66:2–25.
95. Min KH, Lee HJ, Kim K, Kwon IC, Jeong SY, Lee SC. The tumor accumulation and therapeutic efficacy of doxorubicin carried in calcium phosphate-reinforced polymer nanoparticles. Biomaterials. 2012; 33:5788–97.
96. Guo S, Wang Y, Miao L, Xu Z, Lin CM, Zhang Y, Huang L. Lipid-coated cisplatin nanoparticles induce neighboring effect and exhibit enhanced anticancer efficacy. ACS Nano. 2013; 7:9896–904.
97. Miao L, Huang L. (2015). Exploring the tumor microenvironment with nanoparticles. Nanotechnology-Based Precision Tools for the Detection and Treatment of Cancer: Springer, pp. 193–226.
98. McDonald DM, Thurston G, Baluk P. Endothelial gaps as sites for plasma leakage in inflammation. Microcirculation. 1999; 6:7–22.
99. Bobo D, Robinson KJ, Islam J, Thurecht KJ, Corrie SR. Nanoparticle-based medicines: a review of FDA-approved materials and clinical trials to date. Pharm Res. 2016; 33:2373–87.
100. Pham E, Yin M, Peters CG, Lee CR, Brown D, Xu P, Man S, Jayaraman L, Rohde E, Chow A. Preclinical efficacy of bevacizumab with CRLX101, an investigational nanoparticle–drug conjugate, in treatment of metastatic triple-negative breast cancer. Cancer Res. 2016; 76:4493–503.
101. DeRose YS, Gligorich KM, Wang G, Georgelas A, Bowman P, Courdy SJ, Welm AL, Welm BE. Patient - derived models of human breast cancer: protocols for in vitro and in vivo applications in tumor biology and translational medicine. Curr Protoc Pharmacol. 2013.