
Introduction
Recent reports have demonstrated synergistic effects of HDAC inhibitors with cancer immunotherapy. Whereas the direct cytotoxic effects of HDAC inhibitors on cancer cells are well known, the effects of HDAC inhibitors on the immune system are less well understood. Here, we review the effects of HDAC inhibitors on immune cell function in relation to anti-tumor immunity. Furthermore, we discuss the mechanisms of HDAC inhibitors in combination with immunotherapy in the treatment of patients with cancer and provide future directions for research.
Only a few gene mutations may lead to the change of a healthy normal cell into a malignant cancer cell. The genetic instability of a developing tumor leads to further accumulation of genomic mutations/alterations but also to changes in the epigenetic code, the epigenome[1]. The epigenome regulates the heritable patterns of gene expression without changing the sequence of the genome[2]. In cancer, alterations to the epigenome may lead to gene expression profiles that support tumorigenesis and thereby play an important role in cancer initiation and progression[3-6]. Epigenetic changes can occur at multiple levels including direct modifications of the DNA itself as well as modifications of the DNA associated histone proteins[7]. Histone proteins can be chemically modified by acetylation, methylation, phosphorylation, and ubiquitination[8]. Hyperacetylation of histone proteins results in increased transcriptional activity, whilst histone hypoacetylation is associated with repression of gene transcription[9]. Hypoacetylation of histones was shown to occur specifically in a variety of human cancer cell lines as well as in primary lymphoma and colon carcinoma tissue samples[10].
Immunotherapy is a promising novel cancer therapy for multiple cancer types, including melanoma and neuroblastoma[11, 12]. Immunotherapy can lead to clinical benefit and active anti-tumor immune responses have been observed in selected patients[13]. However, in most patients, the clinical response is still limited or even absent following immunotherapy[14]. Accumulating evidence indicates that tumors evade immune responses by down regulation of MHC molecules and tumor antigens or active suppression of anti-tumor immune responses at the site of the tumor by creating an immune suppressive tumor microenvironment (TME)[15-18]. Therefore, immunotherapy should best be combined with other therapies in so-called immunocombination therapy to overcome these tumor induced immune escape mechanisms[19].
One potential type of therapies that could be combined with immunotherapy are therapies that target the epigenetic code. Epigenetic alterations are dynamic and generally reversible and for this reason epigenetic manipulation has emerged as an attractive novel treatment for cancer[20]. Small molecule inhibitors were identified that target the enzymes responsible for the deacetylation of histones, the histone deacetylases (HDACs). These so-called HDAC inhibitors, are now regarded as a group of anti-cancer drugs with high clinical potential[9, 21, 22]. Inhibition of HDACs leads to genomic effects through accumulation of acetylated histone proteins, resulting in altered gene transcription[23]. Specifically in cancer cells, the altered gene transcription leads to, amongst others, the activation of and/or sensitization to intrinsic and extrinsic apoptosis pathways[24, 25]. Besides blocking the function of HDACs leading to genomic effects, HDAC inhibitors also modulate the function of many other proteins resulting in non-genomic effects. For example, p53 becomes hyper acetylated upon HDAC inhibitor treatment, resulting in tumor cell apoptosis[26, 27]. Interestingly, tumor cells appear much more sensitive to the induction of apoptosis by HDAC inhibitors than normal cells, although the responsible mechanisms are still not fully understood. Besides changes in gene transcription other possible mechanisms for the tumor specificity of HDAC inhibitors have been suggested, e.g. induction of double-strand DNA breaks[28]. Other reports link the selective sensitivity of cancer cells to HDAC inhibitors relative to normal cells to the disturbed chromatin structure in cancer cells[29].
The classical HDAC inhibitors inhibit the function of one or more of the 11 known zinc-containing HDAC enzymes. The zinc-containing HDAC enzymes can be classified into several Classes: Class I HDAC (HDAC 1,2,3,8), Class IIA (HDAC 4,5,7,9) and Class IIB (HDAC 6,10). Class III HDACs or Sirtuins, have a different catalytic mechanism and are not a target for the classical HDAC inhibitors. The most recently discovered HDAC11 is the only Class IV HDAC[9]. PanHDAC inhibitors inhibit HDACs from Class I, II and IV, while Class specific HDAC inhibitors only inhibit HDACs from either Class I or Class II. More recently, HDAC inhibitors preferentially targeting a single HDAC have been developed[30, 31]. Because each individual HDAC inhibitor has a unique chemical structure and HDAC inhibitory profile, different HDAC inhibitors can cause a large variety of biological effects in cancer cells and in normal cells[32, 33].
Although the effects of HDAC inhibitors on tumor cells have been studied extensively, the exact role of HDAC inhibitors on immune cells and in anti-tumor immunity is just emerging. HDAC inhibitors were previously reported to exhibit strong anti-inflammatory effects[34-36]. Rapamycin, however, previously viewed as a pure immune suppressant drug, was more recently also shown to mediate strong immune stimulating effects as reviewed in[37, 38]. Similarly, a recent study showed that the Class I HDAC inhibitor Entinostat markedly enhanced anti-tumor vaccination[39]. In this review we will discuss the effects of classical pan-, Class I and Class II HDAC inhibitors on tumor cells and immune cells in relation to immunotherapy. Furthermore, we will discuss the potential of combining HDAC inhibitors with immunotherapy as immunocombination therapy for cancer.
Effects of HDAC inhibitors on tumor cells; immunological consequences
HDAC inhibitors can induce tumor cell death in a specific manner through various mechanisms and with different half maximum inhibitory concentrations (IC50s) as reviewed elsewhere[9, 22, 40]. However, HDAC inhibitors also have profound effects on the remaining viable tumor cells. Here we will discuss the effects of HDAC inhibitors on the cell biology of these surviving tumor cells with a focus on the immunological consequences.
Effects of HDAC inhibitors on tumor cell recognition by T cells and NK cells
Tumor cell recognition and elimination by tumor specific T cells is, amongst others, dependent on the expression levels of both tumor associated antigens (TAAs) and MHC Class I (MHCI) molecules by the tumor cells. HDAC inhibitors can increase TAA expression by tumor cells. The Class I HDAC inhibitor Depsipeptide enhanced the expression of the tumor antigen gp100 in murine melanoma cells[41]. Combination treatment of Depsipeptide and adoptive transfer of gp100-specific cytotoxic T cells resulted in increased tumor killing by transferred tumor specific CD8 T cells. However, sometimes TAA-expression is downregulated after HDAC inhibitor treatment. The panHDAC inhibitor Valproic acid (VPA) downregulated the expression of the tumor antigen Muc1. Remarkably, it upregulated another tumor antigen NY-ESO-1 in mesothelioma cells in vitro[42].
Other reports have shown that HDAC inhibitors upregulate genes involved in the antigen presentation machinery or co-stimulatory molecule expression by tumor cells[43, 44]. The panHDAC inhibitor Trichostatin A (TSA) up-regulated MHCI surface expression in a murine cervical cell line with an impaired antigen-processing machinery[45]. TSA pre-treatment of these tumor cells resulted in more effective lysis of these tumor cells by CD8 T cells in vitro. Similarly, TSA was able to increase or induce expression of TAP-1, TAP-2, LMP-2, and Tapasin in TAP-expressing and TAP–deficient murine tumor cell lines[46]. TSA treatment of mice bearing TAP-deficient tumors delayed tumor growth due to enhanced tumor cell killing by adaptive immune effector cells. More recently, the panHDAC inhibitor Panobinostat was shown not only to enhance the expression of several TAAs, MHCI and MHCII, but also the expression of co-stimulatory molecules in several human and a murine melanoma cell lines in vitro[47]. In addition, effective therapy of B16F10 melanoma bearing mice using Panobinostat was dependent on the presence of the adaptive immune system. In another study, TSA induced MHC Class II expression in murine plasmacytoma cells through activation of the pIII-CIITA promoter, resulting in enhanced proliferation of CD4 T cells in vitro[48]. In summary, HDAC inhibitors can modulate TAA expression and many components of the tumor antigen processing and MHC presentation pathway in surviving tumor cells, overall resulting in enhanced tumor cell recognition and killing by tumor specific T cells (Table 1).
NK cells are innate immune cells that exert important anti-tumor effector functions in cancer immunotherapy[49]. The outcome of an interaction between a tumor cell and a NK cell is balanced by the expression of activating and inhibitory ligands by the tumor cell. Several stress-induced activating ligands, like MHC class I-related chain A (MICA) and B molecules (MICB), and UL16-binding proteins (ULBPs), expressed by tumor cells are recognized by the activating NKG2D receptor on NK cells[50]. HDAC inhibitors were shown to increase the expression of activating ligands for this NK cell receptor by tumor cells. The Class I HDAC inhibitor VPA induced the expression of MICA, MICB and ULBPs in human hepatocellular carcinoma cells resulting in enhanced recognition and killing by NK cells in vitro[51]. Importantly, in non-malignant primary human hepatocytes, VPA treatment did not induce the expression of these NKG2D ligands. Although the mechanisms for the tumor specific induction of NKG2D ligands remained unclear, this study confirms the selective effect of this panHDAC inhibitor on the expression of NK cell activating ligands by malignant cells. Lopez-Soto et al. showed that TSA increased the expression of ULBPs in epithelial tumor cells by releasing HDAC3 mediated repression on ULBP promotors[52]. Similar effects have been reported for other panHDAC inhibitors when added to osteosarcoma, leukemia and Ewing sarcoma cell lines in vitro and to primary myeloid leukemia cells ex vivo[53-56]. These studies collectively show that panHDAC inhibitors can increase the expression of activating NKG2D ligands resulting in enhanced tumor cell recognition and elimination by NK cells.
HDAC inhibitors, however, do not always increase tumor cell recognition by immune cells. Fiegler et al. showed that the panHDAC inhibitor Vorinostat and other panHDAC inhibitors down regulated the B7 family member B7-H6, a ligand for the activating NKp30 receptor on NK cells, in multiple human cancer cell lines, both at the mRNA and protein level[57]. The decreased surface expression of B7-H6 resulted in decreased degranulation of primary NK cells in an NKp30 dependent manner. Thus, HDAC inhibitors can also mediate the downregulation of activating ligands for NK cells by tumor cells. Furthermore, HDAC inhibitor induced increased MHCI expression, leading to increased tumor cell recognition by T cells, at the same time will negatively affect NK cell recognition. Besides lymphocytic NK and T cells, macrophages and other myeloid cells are also able to recognize and kill tumor cells directly[58]. However, the effects of HDAC inhibitors on tumor cell recognition by myeloid cells have not been reported to date. We conclude that, in general, HDAC inhibitors lead to enhanced recognition and elimination of tumor cells by effector lymphocytes, but this effect may vary between tumor types and HDAC inhibitors used.
HDAC inhibitors and immunogenic cell death
Selected chemotherapeutics can induce so-called “immunogenic cell death”, a process in which dying tumor cells can stimulate cellular uptake, activation and cross-presentation by antigen presenting cells (APCs), thereby inducing antitumor T cell responses[59]. Christiansen et al. showed that MC38 colon carcinoma cells, treated with the panHDAC inhibitor Vorinostat, were efficiently taken up by dendritic cells (DCs) in vitro[60]. In addition, in other studies, Vorinostat stimulated the release of important mediators of immunogenic cell death, like HMGB1 and ATP, as well as the expression of cell surface Calreticulin, an important ‘eat-me’ signal, by dying tumor cells[61, 62]. However, the precise role of immunogenic cell death upon Vorinostat therapy in vivo was not investigated in these studies. AK7 pancreatic carcinoma cells, pretreated with Vorinostat or other cytotoxic drugs, have also been used together with the adjuvant BCG as a vaccine. Only the vaccine consisting of the Vorinostat treated tumor cells was able to inhibit tumor growth upon tumor challenge and resulted in increased CD8 T cell infiltration in this experimental setting[63]. These studies imply that Vorinostat, like a subset of chemotherapeutic compounds, induces a form of cell death with immunogenic properties. It remains to be determined whether other panHDAC or Class specific HDAC inhibitors also induce immunogenic cell death. Furthermore, it will be interesting to compare the potency of Vorinostat and other HDAC inhibitors to induce immunogenic cell death with that of chemotherapeutic agents previously demonstrated to induce immunogenic cell death[64].
Effect of HDAC inhibitors on immune cells
So far, we discussed the effects of HDAC inhibitors on tumor cell biology and the immunological consequences. In the next paragraphs we will review the effects of HDAC inhibitors on immune cell viability and function and address the mechanisms and critical factors of successful combinations of HDAC inhibitors and immunotherapy in vivo. Immune cells can have both pro- and anti-tumor effects, depending on cell lineage and environmental cues. For example, in a recent study by West et al, successful treatment of MC38 colon carcinoma tumors using the panHDAC inhibitors Vorinostat and Panobinostat was fully dependent on the presence of an intact immune system[61].
Effect of HDAC inhibitors on antigen presenting cells and cytokine production
In order to generate effective and long-lasting immune responses, activation of both the innate and adaptive arms of the immune system is required. APCs, like monocytes, macrophages and DCs are the first innate immune cells to sense danger signals coming from foreign or damaged self[65]. Upon encounter of these signals, APCs become activated and start to recruit and activate other immune cells and thereby initiate antigen specific immunity[66]. DCs are professional APCs and are therefore especially capable of inducing adaptive immunity. Besides signal 1 coming from specific recognition of antigens presented in MHCI and MHCII molecules, co-stimulatory and cytokine signals (signals 2 and 3, respectively) are required to initiate and direct adaptive immune responses[67].
HDAC inhibitors were repeatedly shown to down regulate both co-stimulatory and cytokine signals coming from APCs. The panHDAC inhibitors Vorinostat and TSA profoundly down-regulated genes involved in co-stimulation and the production of cytokines in murine bone marrow derived macrophages (BMDM) and dendritic cells (BMDC)[68, 69]. Upon treatment with TSA, surface expression of CD40, CD80, CD86 and CCR7 as well as the production of pro-inflammatory cytokines IL-6, IL-12 and TNF-α was down regulated in BMDC[69]. In contrast, TSA and the panHDAC inhibitor Panobinostat increased IL-12 production in peritoneal elicitated macrophages, which was associated with a downregulation of the anti-inflammatory cytokine IL-10[70]. One explanation for these different findings could be that the timing of HDAC inhibitor exposure differed between these studies: in the first studies the APCs were exposed to the HDAC inhibitor one hour before the immune stimulus, whereas in the latter study the HDAC inhibitor and the immune stimulus were administered together. These observations suggest that the pre-activation of these APCs leads to differential effects upon HDAC inhibitor treatment. Also, in contrast to the panHDAC inhibitor TSA, the Class I specific HDAC inhibitor Entinostat did not down regulate IL-12 mRNA in BMDM, suggesting Entinostat may prevent the reduced production of IL-12 in these APCs[71]. In addition, HDAC6 was required for the production of IL-10 in murine APCs, suggesting panHDAC and Class II HDAC inhibitors may downregulate IL-10 production by APCs[72].
Support for a role of individual HDACs in the initiation of inflammatory cytokine production by APCs comes from studies using HDAC3-/- murine BMDM. These cells were shown to be incapable of a pro-inflammatory gene response when stimulated with LPS[73]. The lack of expression of pro-inflammatory genes could be largely explained by a lack of IFN-β expression in these macrophages. This was most probably due to the constitutive over expression of Cox-1 in the se macrophages, which interfered with pro-inflammatory signaling cascades. In addition, other studies have reported on panHDAC inhibitor mediated downregulation of systemic inflammation by reducing pro-inflammatory cytokine production in mice[34, 35, 74-77]. Similar suppressive effects of HDAC inhibitors on pro-inflammatory cytokine production were also reported for human macrophages and DCs[76, 78-81]. In human monocyte derived DCs, the reduced cytokine production upon HDAC inhibitor exposure was suggested to be mediated through decreased NF-kB and type I interferon signaling as suggested by the reduced nuclear translocation of NF-kB RelB, IRF-3 and IRF-8[82]. Similarly, upon exposure to Panobinostat, RelB was down-regulated in a dose-dependent manner in human monocyte derived DCs[83]. Cytokine production by immune cells was also reduced upon administration of the panHDAC inhibitor Givinostat to healthy human subjects[84]. Givinostat treatment reduced the pro-inflammatory cytokine production by endotoxin stimulated PBMC, four hours after Givinostat administration ex vivo. Twelve hours after Givinostat administration, however, cytokine production by the PBMC had already returned to baseline levels, suggesting a transient reduction in cytokine production. This observation was in agreement with the half-life of Givinostat of around 6 hours as determined in the same study. This transient reduction of pro-inflammatory cytokine production following Givinostat treatment, could imply that the timing of HDAC inhibitor treatment may be crucial when combining HDAC inhibitors with immunotherapy (Figure 1). In this respect, short pulses of Givinostat might circumvent prolonged immune suppression in immunocombination therapy regimens. Overall, these data suggest that HDAC inhibitors suppress macrophage and DC functions in terms of reduced expression of co-stimulatory molecules and reduced production of pro-inflammatory cytokines.
The effect of HDAC inhibitors on the (cross-)presentation of antigens by APCs has not been studied extensively. TSA reduced the capacity of murine BMDC to induce T cell proliferation in mixed-lymphocyte-reactions, but the contribution of antigen (cross-)presentation was not investigated[85].
Besides down-regulating pro-inflammatory cytokines, HDAC inhibitors can also reduce the production of pro-tumorigenic soluble factors or cytokines. Vorinostat reduced the production of the tumorigenic factors Nitric Oxide (NO) and M-CSF & MMP-9 by murine peritoneal macrophages and primary mammary tumor cells, respectively[86]. Administration of Vorinostat delayed tumor onset in a spontaneous mammary tumor model, which was associated with reduced numbers of tumor infiltrating macrophages. Similarly, several panHDAC inhibitors reduced the production of the tumorigenic cytokine macrophage migration inhibitory factor (MIF) in several human cancer cell lines as well as in mouse blood[87]. These studies suggest that the production of tumorigenic cytokines or other soluble factors by myeloid cells and tumor cells are also reduced upon panHDAC inhibitor treatment. The net outcome of HDAC inhibitor treatment on anti-tumor immune responses will likely be a delicate balance in the production of pro- and anti-inflammatory cytokines and other soluble factors in the blood, lymphoid organs and, more importantly, locally in the tumor.
Effect of HDAC inhibitors on effector lymphocytes
CD4 T cells are essential in the induction of adaptive anti-tumor immunity by maintaining and skewing immune responses[88-90]. HDAC inhibitors were reported to have inhibitory effects on CD4 T cell viability and function. The panHDAC inhibitor TSA inhibited PMA/Ionomycin induced NF-kB nuclear translocation in murine CD4 T cells after 8 hours and impaired their viability after 20 hours in vitro[91]. TSA also potently decreased the antigen specific proliferation of murine CD4 T cells, which was associated with upregulation of the cyclin-dependent kinase inhibitor 1 p21Cip1[92, 93]. In human CD4 T cells, Vorinostat had limited to no effect on viability and the expression of genes involved in proliferation, differentiation, and apoptosis after a 24 hour exposure period[94]. These opposite observations may be species related or due to intrinsic differences between the panHDAC inhibitors used in these studies. Indeed, the panHDAC inhibitor TSA has a higher inhibitory potency for most of the individual HDACs compared to Vorinostat, which could explain the more pronounced effects of TSA in the murine CD4 T cells[95-97]. In patients treated with the Class I HDAC inhibitor Romidepsin, the percentages of CD4 and CD8 T cells in patient’s blood were decreased by around 50%[98]. In a study by Schmudde et al, Vorinostat affected the proliferation and function of naïve human PBMC and T cells, but not of IL-2 pre-activated PBMC or previously primed T cells[99]. These studies suggest that pan- and Class I HDAC inhibitors reduce the viability and function of naive CD4 T cells, thus hampering the induction of anti-tumor T cell responses. The inhibitory effect of HDAC inhibitors, however, seems less severe or absent following activation of these CD4+ lymphocytes. Therefore, the timing of HDAC inhibitor administration in combination with immunotherapy could critically determine the outcome of immunocombination therapy, combining HDAC inhibitors with immunotherapy (Figure 1).
For effective anti-tumor immunity the function of cytotoxic CD8 T cells is of crucial importance[13]. Several studies suggest HDAC inhibitors enhance the function of this CD8+ T cell subset. Treatment of mice with the panHDAC inhibitor Panobinostat resulted in increased serum levels of IFN-γ and TNF-α and an accelerated graft-versus-host disease, which was associated with higher numbers of CD8 T cells in the affected organs[100]. Strikingly, in murine CD8 T cells, the panHDAC inhibitor TSA mimicked the effect of the pro-inflammatory cytokines IL-12 and IFN-α at the level of gene expression[101]. TSA up-regulated genes involved in CD8 T cell activation and memory similar to those induced by these pro-inflammatory cytokines. This finding suggests that part of the immune inhibitory effect of HDAC inhibitors on pro-inflammatory cytokine production by APC may be compensated by the direct induction of a set of genes in these effector lymphocytes. Murine memory CD8 T cells were capable of producing IFN-γ only when CD4 T cell help was present during priming[102]. Interestingly, only in these CD8 T cell memory cells, the IFN-γ locus was hyper acetylated, allowing for the rapid production of IFN-γ. More recently, the function of exhausted CD8 T cells in chronic viral infection was shown to be restored upon treatment with panHDAC inhibitors[103]. The effect of Class II HDAC inhibitors on CD4 and CD8 T cells have not been reported to date. These studies collectively suggest that panHDAC inhibitors have the ability to stimulate CD8 T cell activation and function.
Although HDAC inhibitor treated tumor cells are more efficiently recognized by effector NK cells, not much is known regarding the effects of HDAC inhibitors on NK cell function itself. Ogbomo et al. first showed that the Class I HDAC inhibitor VPA and the panHDAC inhibitor Vorinostat inhibited the proliferation and cytotoxic capacity of human NK cells, treated with IL-2 and the HDAC inhibitor simultaneously[104]. The NK cells down regulated the expression of activating receptors on the cell surface upon Vorinostat exposure. In another study, upon exposure to similar concentrations of Vorinostat, NK cells were still able to degranulate upon co-culture with tumor cells[99]. The NK cells from the latter study, however, had received the activating cytokine three days prior to the addition of the HDAC inhibitor and were thus pre-activated. These studies imply that IL-2 pre-activated NK cells retain their functionality whereas NK cells receiving IL-2 simultaneous with the HDAC inhibitor show reduced functionality. In a more recent study, the Class I HDAC inhibitor Entinostat increased the expression of the activating receptor NKG2D in IL-2 and IL-21 activated as well as in freshly isolated human NK cells. Entinostat also increased the expression of NKG2D ligands on human tumor cell lines, but not on normal cells, in this study[105]. Furthermore, Entinostat treated NK cells showed increased cytotoxicity upon co-culture with tumor cells and the combination of Entinostat treatment and adoptive NK cell transfer resulted in synergistic inhibition of tumor growth in vivo. Thus, the Class I HDAC inhibitor Entinostat increased activating ligand expression by tumor cells as well as NK cell cytotoxicity. The effects of Class II HDAC inhibitors on NK cells have not been reported to date. These studies suggest that panHDAC inhibitors have distinct effects on NK cells, at least partially depending on their activation status, whereas Class I HDAC inhibition seems to enhance NK cell function.
In conclusion, HDAC inhibitors can either inhibit or promote effector lymphocyte function depending on the cell type, activation status and the type of HDAC inhibitor used.
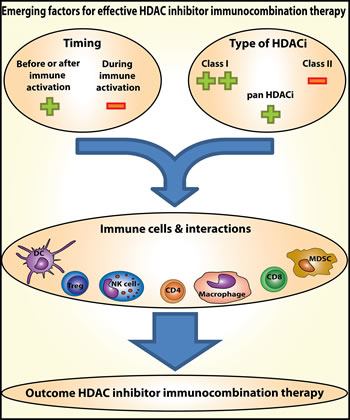
Figure 1: Emerging factors for effective HDAC inhibitor immunocombination therapy. Timing of HDAC inhibitor administration and the type of HDAC inhibitor used, determine the effect on the various types of immune cells and their interactions. Therefore, these are emerging factors determining the outcome of combinations of HDAC inhibitors with immunotherapy in the treatment of cancer.
Effect of HDAC inhibitors on regulatory immune cells
After induction of effective immunity through activation of both innate and adaptive arms of the immune system, feedback loops are in place to control the ongoing inflammation. Specialized immune cells like CD4+ regulatory T cells (Treg) and specific subsets of regulatory myeloid cells can actively dampen immune responses. Tumors actively recruit Treg, myeloid derived suppressor cells (MDSC) and tumor associated macrophages (TAM)[106]. These immune cells all contribute to an immune suppressive tumor microenvironment (TME), abrogating anti-tumor immune responses.
Several panHDAC inhibitors were shown to promote the expansion and function of CD4+ Treg in multiple mouse studies, often after prolonged daily administrations of panHDAC inhibitors[107-111]. HDAC 6 and HDAC9, both Class II HDAC enzymes, were shown to be expressed by murine CD4+FoxP3+ Treg and acted as inhibitors of the suppressive function of these regulatory immune cells[107, 112, 113]. HDAC9-/- mice showed increased numbers of Treg with increased suppressive capacity[112]. In line with these observations, exposure to Class II HDAC inhibitors was repeatedly shown to directly enhance the suppressive function of murine Treg[113-115]. These studies demonstrate that Class II HDAC enzymes are important regulators of Treg function and that Class II HDAC inhibition results in increased Treg functionality. The effect of Class I HDAC inhibition on Treg proliferation and function is less clear. The Class I HDAC inhibitor Entinostat enhanced Treg numbers and FoxP3 expression by Treg[116, 117]. In another study, however, the numbers of Treg were equal and FoxP3 expression by the Treg was down-regulated following Entinostat treatment, leading to improved anti-tumor vaccination in vivo[118]. In addition, Bridle et al. showed both reduced Treg numbers as well as reduced FoxP3 expression in Treg upon Entinostat treatment[39]. These studies collectively indicate that pan- and Class II HDAC inhibitors enhance Treg numbers and function, whereas Class I HDAC inhibitors show more complex effects on Treg, including decreased numbers and function, that need to be studied in more detail. In this respect, Class I HDAC or panHDAC inhibitors might be more suited to combine with immunotherapy than Class II HDAC inhibitors (Figure 1).
MDSC are immature myeloid cells that can actively suppress T cell responses and contribute to an immune suppressive TME[106, 119]. Murine bone marrow precursor cells cultured for 7 days in the presence of GM-CSF & TSA or GM-CSF alone, revealed striking differences in myeloid cell differentiation. The presence of the panHDAC inhibitor TSA throughout this culture period resulted in the accumulation of a pool of undifferentiated myeloid cells. These cells were CD11b(+)Ly6C(+)F4/80(int)CD115(+) and showed immune suppressive properties in vitro, thus mimicking MDSC[120]. Treatment of naïve mice with GM-CSF and TSA resulted in a similar accumulation of CD11b(+)Gr1(+) cells in the spleens of these mice, showing immune suppressive activity ex vivo. Youn et al. showed that the Class I HDAC inhibitor VPA could differentiate tumor induced MDSC into macrophages and DC following in vitro culture[121]. These MDSC were isolated from the BM of tumor bearing mice and cultured with GM-CSF and tumor conditioned medium in the presence or absence of VPA. Thus, panHDAC inhibition affects myeloid cell differentiation from precursors towards MDSC, whereas Class I inhibition directs MDSC to more differentiated macrophages and DC. In the latter study, the Rb1 gene was shown to regulate the differentiation from MDSC to macrophages and DC. Rb1 expression in turn was regulated by HDAC2, showing a role for HDAC2 in MDSC differentiation. The effects of Class II specific HDAC inhibitors on MDSC have not been reported.
TAM expressing low levels of MHCII also accumulate in tumors and are associated with tumor progression[122]. Similarly to MDSCs, TAM have been reported to be sensitive to the Class I HDAC inhibitor VPA and the panHDAC inhibitor TSA, resulting in restoration of MHC class II expression, reversal of immune suppression and delayed tumor growth[123, 124]. The effects of Class II specific HDAC inhibitors on TAM have not been reported.
Overall, the available data suggest that the effect of HDAC inhibitors on regulatory immune cells differ between the immune cell type studied, the differentiation status and the HDAC inhibitor used. The precise effects of HDAC inhibitors on myeloid cells in cancer, like MDSC and TAM, deserve further exploration.
Table 1 : Overview of the observed effects of HDAC inhibitors on tumor cells and immune cells
HDAC inhibitor (class) |
Observations |
References |
|
Tumor cells |
|||
Depsipeptide (Class I) Valproic Acid (panHDAC) Trichostatin A (panHDAC) LAQ824 (panHDAC) Panobinostat (panHDAC) |
TAA MHCI MHCII Co-stimulatory molecules Recognition by T cells |
||
Valproic Acid (panHDAC) Trichostatin A (panHDAC) |
Expression NKG2D ligands/ Recognition by NK cells |
||
Vorinostat (panHDAC) |
Immunogenic cell death |
||
Effector lymphocytes |
|||
CD4 T cells |
Trichostatin A (panHDAC) N-butyrate(panHDAC) Scriptaid (panHDAC) Vorinostat (panHDAC) Romidepsin (Class I) |
Viability Proliferation Pro-inflammatory cytokines |
|
Vorinostat (panHDAC) |
Viability= |
[94] |
|
Activated CD4 T cells |
Vorinostat (panHDAC) |
Viability= Cytotoxicity= |
[99] |
CD8 T cells |
Panobinostat (panHDAC) Trichostatin A (panHDAC) |
Pro-inflammatory cytokines Cytotoxicity Memory function |
|
NK cells |
Valproic Acid (panHDAC) |
Proliferation Cytotoxicity |
[104] |
Entinostat (Class I) |
Cytotoxicity |
[105] |
|
Activated NK cells |
Vorinostat (panHDAC) |
Cytotoxicity = |
[99] |
APC / Cytokine production |
|||
Macrophages /DC |
Trichostatin A (panHDAC) Vorinostat (panHDAC) LAQ824 (panHDAC) Panobinostat (panHDAC) Valproic Acid (panHDAC) Entinostat (Class I) |
Co-stimulatory molecules Pro-inflammatory cytokines APC function |
|
Cytokines / inflammation |
Vorinostat (panHDAC) Givinostat (panHDAC) |
Pro-inflammatory cytokine production Inflammation |
|
Macrophages Tumor cells |
Vorinostat (panHDAC) Trichostatin A (panHDAC) Valproic Acid (panHDAC) |
Tumorigenic sol. factors / cytokines |
|
Regulatory Immune Cells |
|||
Treg |
Trichostatin A (panHDAC) Vorinostat (panHDAC) Valproic Acid (panHDAC) Tubacin (Class II) Entinostat (Class I) |
Cell numbers FoxP3 expression Immune suppressive capacity |
|
Entinostat (Class I) |
Numbers =/ FoxP3 expression Immune suppressive capacity |
||
Bone marrow cells |
Trichostatin A (panHDAC) Vorinostat (panHDAC) |
Differentiation MDSC Macrophages/DC |
[120] |
MDSC |
Valproic Acid (panHDAC) |
Differentiation MDSC Macrophages/DC |
[121] |
TAM |
Trichostatin A (panHDAC) Valproic Acid (panHDAC) |
MHCII expression |
|
HDAC inhibitors in immunocombination therapy in vivo
HDAC inhibitors can impact the immune cascade by influencing different cell types in various life-cycle stages including activation, differentiation, and proliferation. The complex interactions of immune cells in this cascade makes the influence of HDAC inhibitors on the overall outcome of an immune response difficult to predict. In the tumor setting, the growing tumor influences the various immune cells locally and systemically, which further increases the complexity. Thus, the effect of HDAC inhibitor treatment combined with immunotherapy should be investigated by the use of autologous and immunocompetent preclinical models.
As can be concluded from previous sections, HDAC inhibitors can have immune suppressive as well as immune stimulating effects in vitro. There are not many studies reporting on detrimental effects of HDAC inhibitors in combination with cancer immunotherapy in vivo. HDAC inhibitors, however, have been used to limit cytokine production and immune damage in autoimmune diseases, for example in rheumatoid arthritis[78, 125]. The few studies that have reported synergistic effects of treatments combining HDAC inhibitors and immunotherapy in the treatment of cancer, will be discussed here.
The panHDAC inhibitors Vorinostat or Panobinostat showed a synergistic effect in combination with the immune cell stimulating antibodies anti-CD40 and anti-CD137 in immunocompetent models of mammary, renal and colon carcinoma[60]. This synergistic inhibition of tumor growth was highly dependent on CD8 T cells. Vo et al. demonstrated that the panHDAC inhibitor LAQ824 potentiated both adoptive transfers of tumor specific T cells as well as a prime/boost vaccination scheme in mice bearing B16 melanoma tumors[126]. The authors showed that the adoptively transferred T cells were more abundant in the tumor when LAQ824 was co-administered. Similar to what has been reported in vitro[99], naïve T cells were more sensitive to LAQ824 mediated cell death in vivo, suggesting a survival advantage of the transferred CD8 T cells. In addition, upon LAQ824 treatment, the tumor cells expressed higher levels of the tumor antigen gp100 and MHCI presenting molecules, implying enhanced recognition by the tumor specific T cells. The adoptively transferred T cells in LAQ824 treated mice produced higher levels of IFN-γ upon re-stimulation ex vivo, indicating also direct enhancement of CD8 T cell function by this HDAC inhibitor. In addition to the panHDAC inhibitor LAQ824, also the Class I inhibitor Entinostat showed synergistic anti-tumor effects when combined with IL-2 in mice bearing established RENCA tumors[127]. This synergistic effect was also dependent on the presence of CD8 T cells. More recently, Bridle et al showed that administration of Entinostat enhanced tumor specific T cell function only when Entinostat was given at the time of the booster vaccination, but not at the prime vaccination[39]. Surprisingly, the tumor specific CD8 T cell expansion was not enhanced directly by Entinostat in this study. Instead, Entinostat enhanced functionality of the tumor specific CD8 T cells by creating a prolonged state of lymphopenia following vaccination. The selective elimination of unwanted precursor lymphocytes from the BM resulted in tumor specific CD8 cytotoxic lymphocytes exhibiting enhanced functionality. This study elegantly shows that the timing of HDAC inhibitor treatment is essential for the combination with immunotherapy in order to boost anti-tumor immune responses. Taken together, these studies indicate that carefully designed regimens of HDAC inhibitor treatment and immunotherapy have the potential to be synergistic in the treatment of cancer. The underlying mechanisms of these successful immunocombination therapies are, however, complex and diverse and may include direct tumor cell killing, reprogramming of the tumor microenvironment as well as different effects on innate and adaptive immune cells like depletion of bystander lymphocytes and activation of effector lymphocytes.
Conclusions and future directions
Although HDAC inhibitors have negative/detrimental effects on immune cell viability and function, increasing evidence also supports a rationale to combine HDAC inhibitors with immunotherapy to obtain synergistic anti-tumor effects. HDAC inhibitors induce tumor cell specific apoptosis already resulting in tumor debulking. Selective elimination of tumor cells also reduces the tumor induced immune suppression and makes the tumor more accessible for immune cells. In addition, HDAC inhibitors can increase tumor cell recognition by NK and T cells. More recently, it was shown that HDAC inhibitors can have both stimulatory as well as detrimental effects on immune cell viability and function, depending on cell type and activation status. Timing is therefore emerging as a crucial factor in obtaining synergistic effects with immunotherapy (Figure 1). For example, based on present data, the administration of HDAC inhibitors should take place following immune activation/CD8 T cell priming, as activated lymphocytes seem less affected and CD8 T cells become more activated by HDAC inhibitors. Besides timing, the Class of the HDAC inhibitor is very important. Inhibition or downregulation of Class II HDACs enhanced Treg numbers and function, whereas the Class I HDAC inhibitor Entinostat enhanced NK cell and CD8 T cell functions. Finally, due to the vast complexity of molecular and cellular events, rational combination therapies of immunotherapy with HDAC inhibitors should be designed and tested using autologous and immunocompetent preclinical models, to elucidate the complex underlying mechanisms. Understanding the mechanisms of such synergistic combinations will be instrumental to efficiently translate these findings into effective immunocombination therapies for cancer patients.
Acknowledgements
The authors have no relevant affiliations or financial involvements with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the present manuscript. This includes employment, consultancies, honorees, stock ownership or options, expert testimony, grants, or patents received or pending, or royalties. No writing assistance was utilized in the production of this manuscript. This work was supported by the foundations ‘Vrienden KOC Nijmegen’, ‘Villa Joep’, STOPhersentumoren foundation and the RUNMC RUCO institute from The Netherlands.
Abbreviations
| APC |
: Antigen presenting cells |
| BM |
: Bone marrow |
| BMDM |
: Bone marrow derived macrophage |
| BMDC |
: Bone marrow derived dendritic cell |
| CIITA |
: Class II trans-activator |
| DcR3 |
: Decoy Receptor 3 |
| DC |
: Dendritic cells |
| FasL |
: Fas ligand |
| HAT |
: Histone acetylases |
| HDAC |
: Histone deacetylases |
| LPS |
: Lipopolysaccharide |
| mAb |
: monoclonal antibody |
| MDSC |
: Myeloid derived suppressor cells |
| MICA |
: MHC class I-related chain A |
| MICB |
: MHC class I-related chain B |
| MIF NK cell |
: Migration inhibitory factor : Natural Killer cell |
| NO |
: Nitric oxide |
| PBMC |
: Peripheral blood mononuclear cells |
| TAA |
: Tumor associated antigen |
| TAM |
: Tumor associated macrophage |
| TME |
: Tumor microenvironment |
| TRAIL |
:Tumor necrosis factor-related apoptosis-inducing ligand |
| Treg |
: T regulatory cells |
| TSA |
: Trichostatin A |
| ULBP |
: UL16-binding proteins |
| VPA |
: Valproic Acid |
References
1. You JS and Jones PA. Cancer genetics and epigenetics: two sides of the same coin? Cancer Cell. 2012; 22(1):9-20.
2. Iglesias-Bartolome R, Callejas-Valera JL and Gutkind JS. Control of the epithelial stem cell epigenome: the shaping of epithelial stem cell identity. Curr Opin Cell Biol. 2013; 25(2):162-169.
3. Xu Z, Bolick SC, DeRoo LA, Weinberg CR, Sandler DP and Taylor JA. Epigenome-wide association study of breast cancer using prospectively collected sister study samples. J Natl Cancer Inst. 2013; 105(10):694-700.
4. Cicek MS, Koestler DC, Fridley BL, Kalli KR, Armasu SM, Larson MC, Wang C, Winham SJ, Vierkant RA, Rider DN, Block MS, Klotzle B, Konecny G, Winterhoff BJ, Hamidi H, Shridhar V, et al. Epigenome-wide ovarian cancer analysis identifies a methylation profile differentiating clear-cell histology with epigenetic silencing of the HERG K+ channel. Hum Mol Genet. 2013; 22(15):3038-3047.
5. Akhtar-Zaidi B, Cowper-Sal-lari R, Corradin O, Saiakhova A, Bartels CF, Balasubramanian D, Myeroff L, Lutterbaugh J, Jarrar A, Kalady MF, Willis J, Moore JH, Tesar PJ, Laframboise T, Markowitz S, Lupien M, et al. Epigenomic enhancer profiling defines a signature of colon cancer. Science. 2012; 336(6082):736-739.
6. Lin PC, Giannopoulou EG, Park K, Mosquera JM, Sboner A, Tewari AK, Garraway LA, Beltran H, Rubin MA and Elemento O. Epigenomic alterations in localized and advanced prostate cancer. Neoplasia. 2013; 15(4):373-383.
7. Baylin SB and Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011; 11(10):726-734.
8. Campbell MJ and Turner BM. Altered histone modifications in cancer. Adv Exp Med Biol. 2013; 754:81-107.
9. Witt O, Deubzer HE, Milde T and Oehme I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009; 277(1):8-21.
10. Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, Iyer NG, Perez-Rosado A, Calvo E, Lopez JA, Cano A, Calasanz MJ, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005; 37(4):391-400.
11. Jacobs JF, Punt CJ, Lesterhuis WJ, Sutmuller RP, Brouwer HM, Scharenborg NM, Klasen IS, Hilbrands LB, Figdor CG, de Vries IJ and Adema GJ. Dendritic cell vaccination in combination with anti-CD25 monoclonal antibody treatment: a phase I/II study in metastatic melanoma patients. Clin Cancer Res. 2010; 16(20):5067-5078.
12. Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, Smith M, Anderson B, Villablanca JG, Matthay KK, Shimada H, Grupp SA, Seeger R, Reynolds CP, Buxton A, Reisfeld RA, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. 2010; 363(14):1324-1334.
13. de Vries IJ, Bernsen MR, Lesterhuis WJ, Scharenborg NM, Strijk SP, Gerritsen MJ, Ruiter DJ, Figdor CG, Punt CJ and Adema GJ. Immunomonitoring tumor-specific T cells in delayed-type hypersensitivity skin biopsies after dendritic cell vaccination correlates with clinical outcome. J Clin Oncol. 2005; 23(24):5779-5787.
14. Aarntzen EH, Bol K, Schreibelt G, Jacobs JF, Lesterhuis WJ, Van Rossum MM, Adema GJ, Figdor CG, Punt CJ and De Vries IJ. Skin-test infiltrating lymphocytes early predict clinical outcome of dendritic cell-based vaccination in metastatic melanoma. Cancer Res. 2012; 72(23):6102-6110.
15. Hanahan D and Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144(5):646-674.
16. Goodyear O, Agathanggelou A, Novitzky-Basso I, Siddique S, McSkeane T, Ryan G, Vyas P, Cavenagh J, Stankovic T, Moss P and Craddock C. Induction of a CD8+ T-cell response to the MAGE cancer testis antigen by combined treatment with azacitidine and sodium valproate in patients with acute myeloid leukemia and myelodysplasia. Blood. 2010; 116(11):1908-1918.
17. Ramachandran IR, Martner A, Pisklakova A, Condamine T, Chase T, Vogl T, Roth J, Gabrilovich D and Nefedova Y. Myeloid-derived suppressor cells regulate growth of multiple myeloma by inhibiting T cells in bone marrow. J Immunol. 2013; 190(7):3815-3823.
18. Norde WJ, Maas F, Hobo W, Korman A, Quigley M, Kester MG, Hebeda K, Falkenburg JH, Schaap N, de Witte TM, van der Voort R and Dolstra H. PD-1/PD-L1 interactions contribute to functional T-cell impairment in patients who relapse with cancer after allogeneic stem cell transplantation. Cancer Res. 2011; 71(15):5111-5122.
19. Kroesen M, Lindau D, Hoogerbrugge P and Adema GJ. Immunocombination therapy for high-risk neuroblastoma. Immunotherapy. 2012; 4(2):163-174.
20. Lawlor ER and Thiele CJ. Epigenetic changes in pediatric solid tumors: promising new targets. Clin Cancer Res. 2012; 18(10):2768-2779.
21. Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R and Pavletich NP. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 1999; 401(6749):188-193.
22. Khan O and La Thangue NB. HDAC inhibitors in cancer biology: emerging mechanisms and clinical applications. Immunol Cell Biol. 2012; 90(1):85-94.
23. Hahn CK, Ross KN, Warrington IM, Mazitschek R, Kanegai CM, Wright RD, Kung AL, Golub TR and Stegmaier K. Expression-based screening identifies the combination of histone deacetylase inhibitors and retinoids for neuroblastoma differentiation. Proc Natl Acad Sci U S A. 2008; 105(28):9751-9756.
24. Bolden JE, Shi W, Jankowski K, Kan CY, Cluse L, Martin BP, MacKenzie KL, Smyth GK and Johnstone RW. HDAC inhibitors induce tumor-cell-selective pro-apoptotic transcriptional responses. Cell Death Dis. 2013; 4:e519.
25. Wood TE, Dalili S, Simpson CD, Sukhai MA, Hurren R, Anyiwe K, Mao X, Suarez Saiz F, Gronda M, Eberhard Y, MacLean N, Ketela T, Reed JC, Moffat J, Minden MD, Batey RA, et al. Selective inhibition of histone deacetylases sensitizes malignant cells to death receptor ligands. Mol Cancer Ther. 2010; 9(1):246-256.
26. Condorelli F, Gnemmi I, Vallario A, Genazzani AA and Canonico PL. Inhibitors of histone deacetylase (HDAC) restore the p53 pathway in neuroblastoma cells. Br J Pharmacol. 2008; 153(4):657-668.
27. Terui T, Murakami K, Takimoto R, Takahashi M, Takada K, Murakami T, Minami S, Matsunaga T, Takayama T, Kato J and Niitsu Y. Induction of PIG3 and NOXA through acetylation of p53 at 320 and 373 lysine residues as a mechanism for apoptotic cell death by histone deacetylase inhibitors. Cancer Res. 2003; 63(24):8948-8954.
28. Lee JH, Choy ML, Ngo L, Foster SS and Marks PA. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc Natl Acad Sci U S A. 2010; 107(33):14639-14644.
29. Nalabothula N and Carrier F. Cancer cells’ epigenetic composition and predisposition to histone deacetylase inhibitor sensitization. Epigenomics. 2011; 3(2):145-155.
30. Krennhrubec K, Marshall BL, Hedglin M, Verdin E and Ulrich SM. Design and evaluation of ‘Linkerless’ hydroxamic acids as selective HDAC8 inhibitors. Bioorg Med Chem Lett. 2007; 17(10):2874-2878.
31. Oehme I, Deubzer HE, Wegener D, Pickert D, Linke JP, Hero B, Kopp-Schneider A, Westermann F, Ulrich SM, von Deimling A, Fischer M and Witt O. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin Cancer Res. 2009; 15(1):91-99.
32. Lauffer BE, Mintzer R, Fong R, Mukund S, Tam C, Zilberleyb I, Flicke B, Ritscher A, Fedorowicz G, Vallero R, Ortwine DF, Gunzner J, Modrusan Z, Neumann L, Koth CM, Lupardus PJ, et al. Histone deacetylase (HDAC) inhibitor kinetic rate constants correlate with cellular histone acetylation but not transcription and cell viability. J Biol Chem. 2013; 288(37):26926-26943.
33. Chang J, Varghese DS, Gillam MC, Peyton M, Modi B, Schiltz RL, Girard L and Martinez ED. Differential response of cancer cells to HDAC inhibitors trichostatin A and depsipeptide. Br J Cancer. 2012; 106(1):116-125.
34. Leoni F, Zaliani A, Bertolini G, Porro G, Pagani P, Pozzi P, Dona G, Fossati G, Sozzani S, Azam T, Bufler P, Fantuzzi G, Goncharov I, Kim SH, Pomerantz BJ, Reznikov LL, et al. The antitumor histone deacetylase inhibitor suberoylanilide hydroxamic acid exhibits antiinflammatory properties via suppression of cytokines. Proc Natl Acad Sci U S A. 2002; 99(5):2995-3000.
35. Leoni F, Fossati G, Lewis EC, Lee JK, Porro G, Pagani P, Modena D, Moras ML, Pozzi P, Reznikov LL, Siegmund B, Fantuzzi G, Dinarello CA and Mascagni P. The histone deacetylase inhibitor ITF2357 reduces production of pro-inflammatory cytokines in vitro and systemic inflammation in vivo. Mol Med. 2005; 11(1-12):1-15.
36. Villagra A, Cheng F, Wang HW, Suarez I, Glozak M, Maurin M, Nguyen D, Wright KL, Atadja PW, Bhalla K, Pinilla-Ibarz J, Seto E and Sotomayor EM. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat Immunol. 2009; 10(1):92-100.
37. Bravo-San Pedro JM and Senovilla L. Immunostimulatory activity of lifespan-extending agents. Aging (Albany NY). 2013; 5(11):793-801.
38. Blagosklonny MV. Immunosuppressants in cancer prevention and therapy. Oncoimmunology. 2013; 2(12):e26961.
39. Bridle BW, Chen L, Lemay CG, Diallo JS, Pol J, Nguyen A, Capretta A, He R, Bramson JL, Bell JC, Lichty BD and Wan Y. HDAC inhibition suppresses primary immune responses, enhances secondary immune responses, and abrogates autoimmunity during tumor immunotherapy. Mol Ther. 2013; 21(4):887-894.
40. Dickinson M, Johnstone RW and Prince HM. Histone deacetylase inhibitors: potential targets responsible for their anti-cancer effect. Invest New Drugs. 2010; 28 Suppl 1:S3-20.
41. Murakami T, Sato A, Chun NA, Hara M, Naito Y, Kobayashi Y, Kano Y, Ohtsuki M, Furukawa Y and Kobayashi E. Transcriptional modulation using HDACi depsipeptide promotes immune cell-mediated tumor destruction of murine B16 melanoma. J Invest Dermatol. 2008; 128(6):1506-1516.
42. Roulois D, Blanquart C, Panterne C, Gueugnon F, Gregoire M and Fonteneau JF. Downregulation of MUC1 expression and its recognition by CD8(+) T cells on the surface of malignant pleural mesothelioma cells treated with HDACi. Eur J Immunol. 2012; 42(3):783-789.
43. Magner WJ, Kazim AL, Stewart C, Romano MA, Catalano G, Grande C, Keiser N, Santaniello F and Tomasi TB. Activation of MHC class I, II, and CD40 gene expression by histone deacetylase inhibitors. J Immunol. 2000; 165(12):7017-7024.
44. Khan AN, Gregorie CJ and Tomasi TB. Histone deacetylase inhibitors induce TAP, LMP, Tapasin genes and MHC class I antigen presentation by melanoma cells. Cancer Immunol Immunother. 2008; 57(5):647-654.
45. Manning J, Indrova M, Lubyova B, Pribylova H, Bieblova J, Hejnar J, Simova J, Jandlova T, Bubenik J and Reinis M. Induction of MHC class I molecule cell surface expression and epigenetic activation of antigen-processing machinery components in a murine model for human papilloma virus 16-associated tumours. Immunology. 2008; 123(2):218-227.
46. Setiadi AF, Omilusik K, David MD, Seipp RP, Hartikainen J, Gopaul R, Choi KB and Jefferies WA. Epigenetic enhancement of antigen processing and presentation promotes immune recognition of tumors. Cancer Res. 2008; 68(23):9601-9607.
47. Woods DM, Woan K, Cheng F, Wang H, Perez-Villarroel P, Lee C, Lienlaf M, Atadja P, Seto E, Weber J, Sotomayor EM and Villagra A. The antimelanoma activity of the histone deacetylase inhibitor panobinostat (LBH589) is mediated by direct tumor cytotoxicity and increased tumor immunogenicity. Melanoma Res. 2013.
48. Chou SD, Khan AN, Magner WJ and Tomasi TB. Histone acetylation regulates the cell type specific CIITA promoters, MHC class II expression and antigen presentation in tumor cells. Int Immunol. 2005; 17(11):1483-1494.
49. Tarek N, Le Luduec JB, Gallagher MM, Zheng J, Venstrom JM, Chamberlain E, Modak S, Heller G, Dupont B, Cheung NK and Hsu KC. Unlicensed NK cells target neuroblastoma following anti-GD2 antibody treatment. J Clin Invest. 2012; 122(9):3260-3270.
50. Vivier E, Tomasello E, Baratin M, Walzer T and Ugolini S. Functions of natural killer cells. Nat Immunol. 2008; 9(5):503-510.
51. Armeanu S, Bitzer M, Lauer UM, Venturelli S, Pathil A, Krusch M, Kaiser S, Jobst J, Smirnow I, Wagner A, Steinle A and Salih HR. Natural killer cell-mediated lysis of hepatoma cells via specific induction of NKG2D ligands by the histone deacetylase inhibitor sodium valproate. Cancer Res. 2005; 65(14):6321-6329.
52. Lopez-Soto A, Folgueras AR, Seto E and Gonzalez S. HDAC3 represses the expression of NKG2D ligands ULBPs in epithelial tumour cells: potential implications for the immunosurveillance of cancer. Oncogene. 2009; 28(25):2370-2382.
53. Yamanegi K, Yamane J, Kobayashi K, Kato-Kogoe N, Ohyama H, Nakasho K, Yamada N, Hata M, Nishioka T, Fukunaga S, Futani H, Okamura H and Terada N. Sodium valproate, a histone deacetylase inhibitor, augments the expression of cell-surface NKG2D ligands, MICA/B, without increasing their soluble forms to enhance susceptibility of human osteosarcoma cells to NK cell-mediated cytotoxicity. Oncol Rep. 2010; 24(6):1621-1627.
54. Kato N, Tanaka J, Sugita J, Toubai T, Miura Y, Ibata M, Syono Y, Ota S, Kondo T, Asaka M and Imamura M. Regulation of the expression of MHC class I-related chain A, B (MICA, MICB) via chromatin remodeling and its impact on the susceptibility of leukemic cells to the cytotoxicity of NKG2D-expressing cells. Leukemia. 2007; 21(10):2103-2108.
55. Berghuis D, Schilham MW, Vos HI, Santos SJ, Kloess S, Buddingh EP, Egeler RM, Hogendoorn PC and Lankester AC. Histone deacetylase inhibitors enhance expression of NKG2D ligands in Ewing sarcoma and sensitize for natural killer cell-mediated cytolysis. Clin Sarcoma Res. 2012; 2(1):8.
56. Poggi A, Catellani S, Garuti A, Pierri I, Gobbi M and Zocchi MR. Effective in vivo induction of NKG2D ligands in acute myeloid leukaemias by all-trans-retinoic acid or sodium valproate. Leukemia. 2009; 23(4):641-648.
57. Fiegler N, Textor S, Arnold A, Rolle A, Oehme I, Breuhahn K, Moldenhauer G, Witzens-Harig M and Cerwenka A. Downregulation of the activating NKp30 ligand B7-H6 by HDAC inhibitors impairs tumor cell recognition by NK cells. Blood. 2013.
58. Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, Torigian DA, O’Dwyer PJ and Vonderheide RH. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011; 331(6024):1612-1616.
59. Kroemer G, Galluzzi L, Kepp O and Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013; 31:51-72.
60. Christiansen AJ, West A, Banks KM, Haynes NM, Teng MW, Smyth MJ and Johnstone RW. Eradication of solid tumors using histone deacetylase inhibitors combined with immune-stimulating antibodies. Proc Natl Acad Sci U S A. 2011; 108(10):4141-4146.
61. West AC, Mattarollo SR, Shortt J, Cluse LA, Christiansen AJ, Smyth MJ and Johnstone RW. An intact immune system is required for the anti-cancer activities of histone deacetylase inhibitors. Cancer Res. 2013.
62. Sonnemann J, Gressmann S, Becker S, Wittig S, Schmudde M and Beck JF. The histone deacetylase inhibitor vorinostat induces calreticulin exposure in childhood brain tumour cells in vitro. Cancer Chemother Pharmacol. 2010; 66(3):611-616.
63. Guillot F, Boutin B, Blanquart C, Fonteneau JF, Robard M, Gregoire M and Pouliquen D. Vaccination with epigenetically treated mesothelioma cells induces immunisation and blocks tumour growth. Vaccine. 2011; 29(33):5534-5543.
64. Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, Schmitt E, Hamai A, Hervas-Stubbs S, Obeid M, Coutant F, Metivier D, Pichard E, Aucouturier P, Pierron G, Garrido C, et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med. 2005; 202(12):1691-1701.
65. Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994; 12:991-1045.
66. Banchereau J and Steinman RM. Dendritic cells and the control of immunity. Nature. 1998; 392(6673):245-252.
67. Medzhitov R and Janeway CA, Jr. Innate immune recognition and control of adaptive immune responses. Semin Immunol. 1998; 10(5):351-353.
68. Bode KA, Schroder K, Hume DA, Ravasi T, Heeg K, Sweet MJ and Dalpke AH. Histone deacetylase inhibitors decrease Toll-like receptor-mediated activation of proinflammatory gene expression by impairing transcription factor recruitment. Immunology. 2007; 122(4):596-606.
69. Roger T, Lugrin J, Le Roy D, Goy G, Mombelli M, Koessler T, Ding XC, Chanson AL, Reymond MK, Miconnet I, Schrenzel J, Francois P and Calandra T. Histone deacetylase inhibitors impair innate immune responses to Toll-like receptor agonists and to infection. Blood. 2011; 117(4):1205-1217.
70. Wang H, Cheng F, Woan K, Sahakian E, Merino O, Rock-Klotz J, Vicente-Suarez I, Pinilla-Ibarz J, Wright KL, Seto E, Bhalla K, Villagra A and Sotomayor EM. Histone deacetylase inhibitor LAQ824 augments inflammatory responses in macrophages through transcriptional regulation of IL-10. J Immunol. 2011; 186(7):3986-3996.
71. Halili MA, Andrews MR, Labzin LI, Schroder K, Matthias G, Cao C, Lovelace E, Reid RC, Le GT, Hume DA, Irvine KM, Matthias P, Fairlie DP and Sweet MJ. Differential effects of selective HDAC inhibitors on macrophage inflammatory responses to the Toll-like receptor 4 agonist LPS. J Leukoc Biol. 2010; 87(6):1103-1114.
72. Cheng F, Lienlaf M, Perez-Villarroel P, Wang HW, Lee C, Woan K, Woods D, Knox T, Bergman J, Pinilla-Ibarz J, Kozikowski A, Seto E, Sotomayor EM and Villagra A. Divergent roles of histone deacetylase 6 (HDAC6) and histone deacetylase 11 (HDAC11) on the transcriptional regulation of IL10 in antigen presenting cells. Mol Immunol. 2014; 60(1):44-53.
73. Chen X, Barozzi I, Termanini A, Prosperini E, Recchiuti A, Dalli J, Mietton F, Matteoli G, Hiebert S and Natoli G. Requirement for the histone deacetylase Hdac3 for the inflammatory gene expression program in macrophages. Proc Natl Acad Sci U S A. 2012; 109(42):E2865-2874.
74. Reddy P, Maeda Y, Hotary K, Liu C, Reznikov LL, Dinarello CA and Ferrara JL. Histone deacetylase inhibitor suberoylanilide hydroxamic acid reduces acute graft-versus-host disease and preserves graft-versus-leukemia effect. Proc Natl Acad Sci U S A. 2004; 101(11):3921-3926.
75. Leng C, Gries M, Ziegler J, Lokshin A, Mascagni P, Lentzsch S and Mapara MY. Reduction of graft-versus-host disease by histone deacetylase inhibitor suberonylanilide hydroxamic acid is associated with modulation of inflammatory cytokine milieu and involves inhibition of STAT1. Exp Hematol. 2006; 34(6):776-787.
76. Reddy P, Sun Y, Toubai T, Duran-Struuck R, Clouthier SG, Weisiger E, Maeda Y, Tawara I, Krijanovski O, Gatza E, Liu C, Malter C, Mascagni P, Dinarello CA and Ferrara JL. Histone deacetylase inhibition modulates indoleamine 2,3-dioxygenase-dependent DC functions and regulates experimental graft-versus-host disease in mice. J Clin Invest. 2008; 118(7):2562-2573.
77. Glauben R, Batra A, Stroh T, Erben U, Fedke I, Lehr HA, Leoni F, Mascagni P, Dinarello CA, Zeitz M and Siegmund B. Histone deacetylases: novel targets for prevention of colitis-associated cancer in mice. Gut. 2008; 57(5):613-622.
78. Grabiec AM, Krausz S, de Jager W, Burakowski T, Groot D, Sanders ME, Prakken BJ, Maslinski W, Eldering E, Tak PP and Reedquist KA. Histone deacetylase inhibitors suppress inflammatory activation of rheumatoid arthritis patient synovial macrophages and tissue. J Immunol. 2010; 184(5):2718-2728.
79. Bosisio D, Vulcano M, Del Prete A, Sironi M, Salvi V, Salogni L, Riboldi E, Leoni F, Dinarello CA, Girolomoni G and Sozzani S. Blocking TH17-polarizing cytokines by histone deacetylase inhibitors in vitro and in vivo. J Leukoc Biol. 2008; 84(6):1540-1548.
80. Salvi V, Bosisio D, Mitola S, Andreoli L, Tincani A and Sozzani S. Trichostatin A blocks type I interferon production by activated plasmacytoid dendritic cells. Immunobiology. 2010; 215(9-10):756-761.
81. Brogdon JL, Xu Y, Szabo SJ, An S, Buxton F, Cohen D and Huang Q. Histone deacetylase activities are required for innate immune cell control of Th1 but not Th2 effector cell function. Blood. 2007; 109(3):1123-1130.
82. Nencioni A, Beck J, Werth D, Grunebach F, Patrone F, Ballestrero A and Brossart P. Histone deacetylase inhibitors affect dendritic cell differentiation and immunogenicity. Clin Cancer Res. 2007; 13(13):3933-3941.
83. Song W, Tai YT, Tian Z, Hideshima T, Chauhan D, Nanjappa P, Exley MA, Anderson KC and Munshi NC. HDAC inhibition by LBH589 affects the phenotype and function of human myeloid dendritic cells. Leukemia. 2011; 25(1):161-168.
84. Furlan A, Monzani V, Reznikov LL, Leoni F, Fossati G, Modena D, Mascagni P and Dinarello CA. Pharmacokinetics, safety and inducible cytokine responses during a phase 1 trial of the oral histone deacetylase inhibitor ITF2357 (givinostat). Mol Med. 2011; 17(5-6):353-362.
85. Kim ES and Lee JK. Histone deacetylase inhibitors decrease the antigen presenting activity of murine bone marrow derived dendritic cells. Cell Immunol. 2010; 262(1):52-57.
86. Tran K, Risingsong R, Royce DB, Williams CR, Sporn MB, Pioli PA, Gediya LK, Njar VC and Liby KT. The combination of the histone deacetylase inhibitor vorinostat and synthetic triterpenoids reduces tumorigenesis in mouse models of cancer. Carcinogenesis. 2013; 34(1):199-210.
87. Lugrin J, Ding XC, Le Roy D, Chanson AL, Sweep FC, Calandra T and Roger T. Histone deacetylase inhibitors repress macrophage migration inhibitory factor (MIF) expression by targeting MIF gene transcription through a local chromatin deacetylation. Biochim Biophys Acta. 2009; 1793(11):1749-1758.
88. Steinman RM. Dendritic cells: understanding immunogenicity. Eur J Immunol. 2007; 37 Suppl 1:S53-60.
89. Aarntzen EH, De Vries IJ, Lesterhuis WJ, Schuurhuis D, Jacobs JF, Bol K, Schreibelt G, Mus R, De Wilt JH, Haanen JB, Schadendorf D, Croockewit A, Blokx WA, Van Rossum MM, Kwok WW, Adema GJ, et al. Targeting CD4(+) T-helper cells improves the induction of antitumor responses in dendritic cell-based vaccination. Cancer Res. 2013; 73(1):19-29.
90. Church SE, Jensen SM, Antony PA, Restifo NP and Fox BA. Tumor-specific CD4 T cells maintain effector and memory tumor-specific CD8 T cells. Eur J Immunol. 2013.
91. Moreira JM, Scheipers P and Sorensen P. The histone deacetylase inhibitor Trichostatin A modulates CD4+ T cell responses. BMC Cancer. 2003; 3:30.
92. Edens RE, Dagtas S and Gilbert KM. Histone deacetylase inhibitors induce antigen specific anergy in lymphocytes: a comparative study. Int Immunopharmacol. 2006; 6(11):1673-1681.
93. Dagtas AS, Edens RE and Gilbert KM. Histone deacetylase inhibitor uses p21(Cip1) to maintain anergy in CD4+ T cells. Int Immunopharmacol. 2009; 9(11):1289-1297.
94. Beliakova-Bethell N, Zhang JX, Singhania A, Lee V, Terry VH, Richman DD, Spina CA and Woelk CH. Suberoylanilide hydroxamic acid induces limited changes in the transcriptome of primary CD4(+) T cells. AIDS. 2013; 27(1):29-37.
95. Moradei O, Maroun CR, Paquin I and Vaisburg A. Histone deacetylase inhibitors: latest developments, trends and prospects. Curr Med Chem Anticancer Agents. 2005; 5(5):529-560.
96. Bradner JE, West N, Grachan ML, Greenberg EF, Haggarty SJ, Warnow T and Mazitschek R. Chemical phylogenetics of histone deacetylases. Nat Chem Biol. 2010; 6(3):238-243.
97. Moradei O, Vaisburg A and Martell RE. Histone deacetylase inhibitors in cancer therapy: new compounds and clinical update of benzamide-type agents. Curr Top Med Chem. 2008; 8(10):841-858.
98. Kelly-Sell MJ, Kim YH, Straus S, Benoit B, Harrison C, Sutherland K, Armstrong R, Weng WK, Showe LC, Wysocka M and Rook AH. The histone deacetylase inhibitor, romidepsin, suppresses cellular immune functions of cutaneous T-cell lymphoma patients. Am J Hematol. 2012; 87(4):354-360.
99. Schmudde M, Friebe E, Sonnemann J, Beck JF and Broker BM. Histone deacetylase inhibitors prevent activation of tumour-reactive NK cells and T cells but do not interfere with their cytolytic effector functions. Cancer Lett. 2010; 295(2):173-181.
100. Wang D, Iclozan C, Liu C, Xia C, Anasetti C and Yu XZ. LBH589 enhances T cell activation in vivo and accelerates graft-versus-host disease in mice. Biol Blood Marrow Transplant. 2012; 18(8):1182-1190 e1181.
101. Agarwal P, Raghavan A, Nandiwada SL, Curtsinger JM, Bohjanen PR, Mueller DL and Mescher MF. Gene regulation and chromatin remodeling by IL-12 and type I IFN in programming for CD8 T cell effector function and memory. J Immunol. 2009; 183(3):1695-1704.
102. Northrop JK, Thomas RM, Wells AD and Shen H. Epigenetic remodeling of the IL-2 and IFN-gamma loci in memory CD8 T cells is influenced by CD4 T cells. J Immunol. 2006; 177(2):1062-1069.
103. Zhang F, Zhou X, DiSpirito JR, Wang C, Wang Y and Shen H. Epigenetic Manipulation Restores Functions of Defective CD8 T Cells from Chronic Viral Infection. Mol Ther. 2014.
104. Ogbomo H, Michaelis M, Kreuter J, Doerr HW and Cinatl J, Jr. Histone deacetylase inhibitors suppress natural killer cell cytolytic activity. FEBS Lett. 2007; 581(7):1317-1322.
105. Zhu S, Denman CJ, Cobanoglu ZS, Kiany S, Lau CC, Gottschalk SM, Hughes DP, Kleinerman ES and Lee DA. The Narrow-Spectrum HDAC Inhibitor Entinostat Enhances NKG2D Expression Without NK Cell Toxicity, Leading to Enhanced Recognition of Cancer Cells. Pharm Res. 2013.
106. Lindau D, Gielen P, Kroesen M, Wesseling P and Adema GJ. The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology. 2013; 138(2):105-115.
107. Tao R, de Zoeten EF, Ozkaynak E, Chen C, Wang L, Porrett PM, Li B, Turka LA, Olson EN, Greene MI, Wells AD and Hancock WW. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med. 2007; 13(11):1299-1307.
108. Donas C, Fritz M, Manriquez V, Tejon G, Bono MR, Loyola A and Rosemblatt M. Trichostatin A promotes the generation and suppressive functions of regulatory T cells. Clin Dev Immunol. 2013; 2013:679804.
109. Johnson J, Pahuja A, Graham M, Hering B, Hancock WW and Bansal-Pakala P. Effects of histone deacetylase inhibitor SAHA on effector and FOXP3+regulatory T cells in rhesus macaques. Transplant Proc. 2008; 40(2):459-461.
110. Akimova T, Ge G, Golovina T, Mikheeva T, Wang L, Riley JL and Hancock WW. Histone/protein deacetylase inhibitors increase suppressive functions of human FOXP3+ Tregs. Clin Immunol. 2010; 136(3):348-363.
111. Saouaf SJ, Li B, Zhang G, Shen Y, Furuuchi N, Hancock WW and Greene MI. Deacetylase inhibition increases regulatory T cell function and decreases incidence and severity of collagen-induced arthritis. Exp Mol Pathol. 2009; 87(2):99-104.
112. de Zoeten EF, Wang L, Sai H, Dillmann WH and Hancock WW. Inhibition of HDAC9 increases T regulatory cell function and prevents colitis in mice. Gastroenterology. 2010; 138(2):583-594.
113. de Zoeten EF, Wang L, Butler K, Beier UH, Akimova T, Sai H, Bradner JE, Mazitschek R, Kozikowski AP, Matthias P and Hancock WW. Histone deacetylase 6 and heat shock protein 90 control the functions of Foxp3(+) T-regulatory cells. Mol Cell Biol. 2011; 31(10):2066-2078.
114. Kalin JH, Butler KV, Akimova T, Hancock WW and Kozikowski AP. Second-generation histone deacetylase 6 inhibitors enhance the immunosuppressive effects of Foxp3+ T-regulatory cells. J Med Chem. 2012; 55(2):639-651.
115. Beier UH, Wang L, Han R, Akimova T, Liu Y and Hancock WW. Histone deacetylases 6 and 9 and sirtuin-1 control Foxp3+ regulatory T cell function through shared and isoform-specific mechanisms. Sci Signal. 2012; 5(229):ra45.
116. Lucas JL, Mirshahpanah P, Haas-Stapleton E, Asadullah K, Zollner TM and Numerof RP. Induction of Foxp3+ regulatory T cells with histone deacetylase inhibitors. Cell Immunol. 2009; 257(1-2):97-104.
117. Zhang ZY and Schluesener HJ. HDAC inhibitor MS-275 attenuates the inflammatory reaction in rat experimental autoimmune prostatitis. Prostate. 2012; 72(1):90-99.
118. Shen L, Ciesielski M, Ramakrishnan S, Miles KM, Ellis L, Sotomayor P, Shrikant P, Fenstermaker R and Pili R. Class I histone deacetylase inhibitor entinostat suppresses regulatory T cells and enhances immunotherapies in renal and prostate cancer models. PLoS One. 2012; 7(1):e30815.
119. Nagaraj S, Schrum AG, Cho HI, Celis E and Gabrilovich DI. Mechanism of T cell tolerance induced by myeloid-derived suppressor cells. J Immunol. 2010; 184(6):3106-3116.
120. Rosborough BR, Castellaneta A, Natarajan S, Thomson AW and Turnquist HR. Histone deacetylase inhibition facilitates GM-CSF-mediated expansion of myeloid-derived suppressor cells in vitro and in vivo. J Leukoc Biol. 2012; 91(5):701-709.
121. Youn JI, Kumar V, Collazo M, Nefedova Y, Condamine T, Cheng P, Villagra A, Antonia S, McCaffrey JC, Fishman M, Sarnaik A, Horna P, Sotomayor E and Gabrilovich DI. Epigenetic silencing of retinoblastoma gene regulates pathologic differentiation of myeloid cells in cancer. Nat Immunol. 2013; 14(3):211-220.
122. Hagemann T, Wilson J, Burke F, Kulbe H, Li NF, Pluddemann A, Charles K, Gordon S and Balkwill FR. Ovarian cancer cells polarize macrophages toward a tumor-associated phenotype. J Immunol. 2006; 176(8):5023-5032.
123. Chang YC, Chen TC, Lee CT, Yang CY, Wang HW, Wang CC and Hsieh SL. Epigenetic control of MHC class II expression in tumor-associated macrophages by decoy receptor 3. Blood. 2008; 111(10):5054-5063.
124. Tai SK, Chang HC, Lan KL, Lee CT, Yang CY, Chen NJ, Chou TY, Tarng DC and Hsieh SL. Decoy receptor 3 enhances tumor progression via induction of tumor-associated macrophages. J Immunol. 2012; 188(5):2464-2471.
125. Grabiec AM, Korchynskyi O, Tak PP and Reedquist KA. Histone deacetylase inhibitors suppress rheumatoid arthritis fibroblast-like synoviocyte and macrophage IL-6 production by accelerating mRNA decay. Ann Rheum Dis. 2012; 71(3):424-431.
126. Vo DD, Prins RM, Begley JL, Donahue TR, Morris LF, Bruhn KW, de la Rocha P, Yang MY, Mok S, Garban HJ, Craft N, Economou JS, Marincola FM, Wang E and Ribas A. Enhanced antitumor activity induced by adoptive T-cell transfer and adjunctive use of the histone deacetylase inhibitor LAQ824. Cancer Res. 2009; 69(22):8693-8699.
127. Kato Y, Yoshimura K, Shin T, Verheul H, Hammers H, Sanni TB, Salumbides BC, Van Erp K, Schulick R and Pili R. Synergistic in vivo antitumor effect of the histone deacetylase inhibitor MS-275 in combination with interleukin 2 in a murine model of renal cell carcinoma. Clin Cancer Res. 2007; 13(15 Pt 1):4538-4546.