
INTRODUCTION
Pulmonary artery hypertension (PAH) is a progressive and devastating disease defined by increased pulmonary arterial pressure (>25 mmHg), vascular remodeling and narrowed pulmonary arteries [1, 2]. Without treatment, PAH leads to right ventricle dysfunction, cardiac output reduced, right ventricle failure and can lead to death [3]. Smooth muscle cell proliferation, endothelial damage and remodeling are involved in the pathogenesis of PAH [4–7]. Endothelial cell injury and apoptosis are generally considered to represent PAH initiation and they are caused by several factors which can induce pulmonary arterial smooth muscle cells (PASMCs) and fibroblast proliferation as well as vascular remodeling [8–10]. Interest in the cellular and molecular mechanisms underlying the formation and development of PAH and the key role of cell apoptosis in PAH recently has seen significant increase. Apoptosis, or programmed cell death, is an automatic and ordered cell death process that is controlled by several genes which maintains homeostasis [11]. In this process, damaged cells or unwanted cells that might be disadvantageous to the organism are removed [11]. Disturbing the balance between PASMCs proliferation and apoptosis has shown to play an important role in vascular remodeling and vessel hypertrophy [12–15]. Apoptosis-resistant endothelial cells or highly proliferative PASMCs promote pulmonary vascular lesions [5]. Previous studies have shown that therapeutic strategies for PAH were limited to vasoactive agents, including prostaglandins, endothelin receptor antagonists and a phosphodiesterase type-5 inhibitor [16]. Recently, therapies based on anti-apoptotic mechanisms have been successful in animal models of pulmonary hypertension (PH) [16]. Therefore, agents designed to induce apoptosis or inhibit PASMCs growth represent effective and promising approaches.
Connective tissue growth factor (CTGF), which is also called CCN2, is a member of the CCN family of secreted peptides [17], and is derived from human umbilical vein endothelial cells (HUVECs). It is composed of four modules: insulin-like growth factor (IGF)-binding protein, thrombospondin type I, von Willebrand type C factor and COOH-terminal domain [18]. By interacting with a variety of cell surface receptors, growth factors or extracellular matrix proteins, CTGF regulates multiple cellular functions, including cell proliferation, survival, adhesion and apoptosis as well as a wide range of other biological processes [19–21]. Increasing evidences have shown the importance of anti-apoptotic effect of CTGF in disease development. The over-expression of CTGF provides a protective effect against drug-induced apoptosis via a αvβ3/FAK/extracellular signal-regulated kinase (ERK) signaling pathway, which mediates the bcl-xl survival pathway [22]. Furthermore, CTGF might be involved in the pathogenesis of PAH. Moreover, transforming growth factor-β1 (TGF-β1) and CTGF are increased in high blood flow-induced PH, and the expression of CTGF is correlated with pulmonary vascular remodeling indicators (relative medial thickness, RMT; and relative medial area, RMA) [23]. The expression of CTGF protein and mRNA were increased in monocrotaline (MCT)-treated rats [24]. However, the detailed mechanism of CTGF in the remodeling of PASMCs in the development of PAH remains unclear.
Serotonin (5-hydroxytrytamine, 5-HT) is synthesized by tryptophan hydroxylase (TPH). TPH1 is predominantly present in the peripheral nervous system, whereas TPH2 is exclusively found in the central nervous system [25]. In idiopathic PAH (IPAH), TPH1 expression and serotonin synthesis are increased in the pulmonary vascular endothelium, and the serotonin transporter (5-HTT, SERT) is increased in PASMCs [26]. 5-HT promotes the proliferation of PASMCs and pulmonary arterial fibroblasts and leads to vasoconstriction and vascular remodeling during the development of PAH [27, 28]. Reports have demonstrated that 5-HT induces vasoconstriction via serotonin receptor-1B (SR-1B) and SR-2A [29]. Researchers have found that SR-1B agonists increase proliferation and decrease apoptosis of PASMCs by pERK1/2 and PDK [30].
Our previous study showed that 4-chloro-DL-phenylalanine (PCPA) ameliorates pulmonary artery remodeling and lung tissue inflammation and decreases the expression of TPH1, matrix metalloproteinase (MMP)/tissue inhibitor of metalloproteinase (TIMP) and pro-inflammatory factors [31]. However, the effect of PCPA on apoptosis during the pathogenesis of PAH and the roles of CTGF and ERK/p-ERK as well as their relationship remain unclear. In this study, we aim to investigate the mechanism of CTGF in PAH and determine whether PCPA can inhibit pulmonary arterial remodeling by regulating CTGF and its downstream signaling pathway.
RESULTS
Effect of PCPA on MCT-induced PAP, RVI and pulmonary arterial wall thickness
Animals were fed for 21 days. In the MCT group, pulmonary arterial pressure (PAP) markedly exceeded to other three groups. PCPA obviously inhibited the PAP which was induced by MCT (Figure 1a). The right ventricle hypertrophy index (RVI) in the MCT group was significantly increased from 0.26 ± 0.07 to 0.48 ± 0.09 (P < 0.01 compared with the control group). Compared with the MCT group, the PCPA-treated groups showed significantly reduced RVI values, with the P1 group decreasing to 0.32 ± 0.06 (P < 0.01 vs. MCT) and P2 group decreasing to 0.31 ± 0.03 (P < 0.01 vs. MCT, Figure 1b).
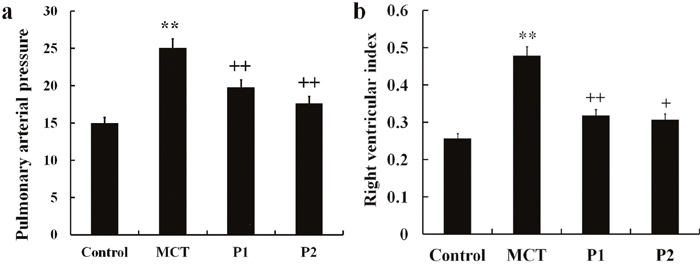
Figure 1: Effect of PCPA on MCT-induced PAP and RVI. (a) Pulmonary arterial pressure (PAP) and (b) right ventricle hypertrophy index (RVI) were compared in different groups. Data are expressed as the means ± SD. **P < 0.01 compared with the control group. + P < 0.05, ++ P < 0.01 compared with the MCT group. P, 4-Chloro-DL-phenylalanine (PCPA); MCT, monocrotaline; P1, MCT plus 50 mg/kg body mass PCPA; P2, MCT plus 100 mg/kg body mass PCPA.
The muscularization of the pulmonary arterials and lung tissues were studied via light microscopy. PCPA dramatically suppressed the remodeling of the pulmonary arteries. In the MCT group, the thickness of the aortopulmonary wall had remarkably increased (98.51 ± 5.32%, P < 0.01 vs. control 36.88 ± 6.85%). PCPA suppressed the thickness ratio in the P1 and P2 groups (52.37 ± 6.87% and 41.57 ± 9.55%, respectively, P < 0.01 vs. MCT, Figure 2A). The pulmonary arteriole wall thickness was increased from 20.48 ± 4.69% in the control group to 68.48 ± 8.76% in the MCT group (P < 0.01), and the medial wall thickness ratio was inhibited in the P1 and P2 groups (41.24 ± 7.57% and 32.42 ± 5.69%, respectively, P < 0.01 vs. MCT, Figure 2B).
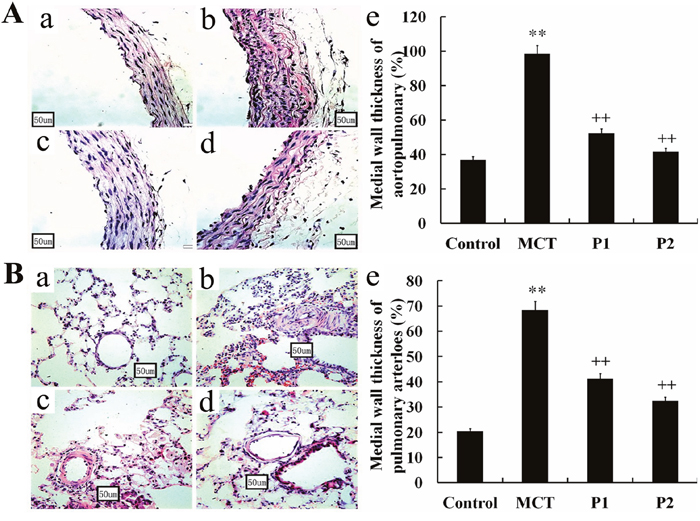
Figure 2: Effect of PCPA on the aortopulmonary and pulmonary arterioles wall thickness ratio. Aortopulmonary (A) and pulmonary arterioles (B) obtained from (a) control group, (b) monocrotaline (MCT) group, (c) MCT plus 50 mg/kg body mass PCPA group (P1), and (d) MCT plus 100 mg/kg body mass PCPA group (P2). Pulmonary vascular remodeling measured as a percentage of (e) medial wall thickness in the 4 groups. (Original magnification × 400, scale bars = 50 μm). Data are the mean ± SD (n = 3 rats). ** P < 0.01 compared with the control group. ++ P < 0.01 compared with the MCT group.
Effect of PCPA on 5-HT in plasma
To determine the role of PCPA in MCT-induced PAH, we measured 5-HT concentration in plasma in the four groups. In the control group, the 5-HT concentrations in plasma were 28.56 ± 2.5 ng/mL. In the MCT group, the 5-HT concentrations in plasma significantly increased to 58.76 ± 9.32 ng/mL (P < 0.01 vs control). At 50 mg/kg, PCPA inhibited the 5-HT concentration in plasma (36.73 ± 7.34 ng/mL, P < 0.01 vs MCT). At 100 mg/kg, PCPA markedly inhibited the plasma 5-HT concentration (30.90 ± 5.64 ng/mL, P < 0.01 vs MCT group, Figure 3).
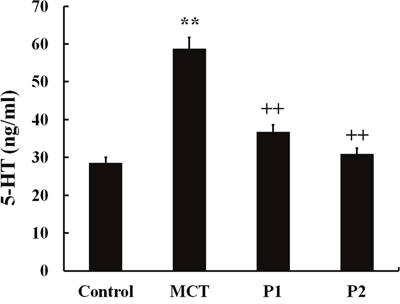
Figure 3: ELISA determination of PCPA on 5-HT concentration in plasma. Data are expressed as the means ± SD. ** P < 0.01 compared with the control group. ++ P < 0.01 compared with the MCT group. P, 4-Chloro-DL-phenylalanine (PCPA); MCT, monocrotaline; P1, MCT plus 50 mg/kg body mass PCPA; P2, MCT plus 100 mg/kg body mass PCPA.
Effect of PCPA on CTGF expression in aortopulmonary and pulmonary arterioles
Immunohistochemistry and Western blotting analyses were used to evaluate the CTGF expression in the aortopulmonary and pulmonary arterioles. The immunohistochemistry analysis showed strong positive CTGF expression in the MCT group compared with control group. The low dosage of PCPA caused a mild reduction of CTGF expression in the aortopulmonary and pulmonary arterioles, whereas the high dosage of PCPA seemingly suppressed the MCT-induced CTGF overexpression (Figure 4).
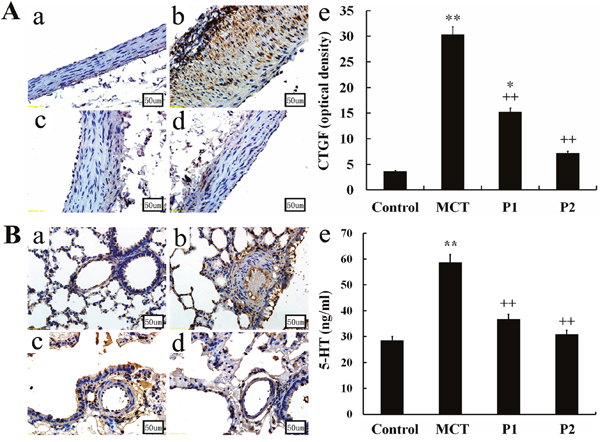
Figure 4: Immunohistochemical determination of CTGF expression in the aortopulmonary and pulmonary arterioles. Aortopulmonary (A) and pulmonary arterioles (B) obtained from (a) control group, (b) monocrotaline (MCT) group, (c) MCT plus 50 mg/kg body mass 4-Chloro-DL-phenylalanine (PCPA) group (P1), and (d) MCT plus 100 mg/kg body mass PCPA group (P2). (Original magnification × 400, scale bars = 50 μm). (e) Average optical density of CTGF in rat aortopulmonary. Data are the mean ± SD (n = 3 rats). * P < 0.05, **P < 0.01 compared with the control group. ++P < 0.01 compared with the MCT group.
The Western blotting analysis showed that the levels of CTGF protein were strongly up-regulated in the MCT group (1.04 ± 0.08 vs. 0.46 ± 0.11, respectively, P < 0.01). At the 50 mg/kg dose, PCPA inhibited the MCT-induced expression of CTGF (0.89 ± 0.13, P > 0.05 vs. MCT). At a higher dose (100 mg/kg), PCPA markedly suppressed the MCT-induced CTGF expression (0.64 ± 0.13, P < 0.01 vs. MCT, Figure 5).
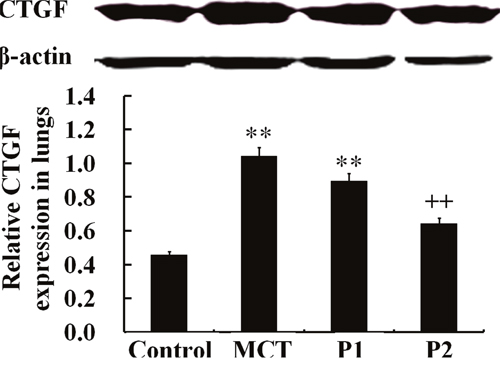
Figure 5: Western blot analysis of CTGF expression in lung tissues. The control group, the monocrotaline (MCT)-induced pulmonary arterial hypertension group, P1 group with 4-Chloro-DL-phenylalanine (PCPA) treated 50 mg/kg/day, and P2 group with PCPA treated 100 mg/kg/day. Data are expressed as mean ± SD (n = 5 rats). ** P < 0.01 compared with the control group. ++ P < 0.01 compared with the MCT group.
Effect of PCPA on SR-1B in rat lungs
The Western blotting analysis using anti-SR-1B antibodies showed that the levels of SR-1B protein significantly increased in the MCT-induced lungs compared with the control group (from 0.45 ± 0.09 to 1.77 ± 0.11, P < 0.01). Treatment with PCPA (50 mg/kg) caused a small improvement in SR-1B protein expression (0.96 ± 0.08, P < 0.01 vs. MCT). At 100 mg/kg, PCPA significantly attenuated the MCT-induced expression of SR-1B in lung tissues (0.68 ± 0.07, P < 0.01 vs. MCT, Figure 6).
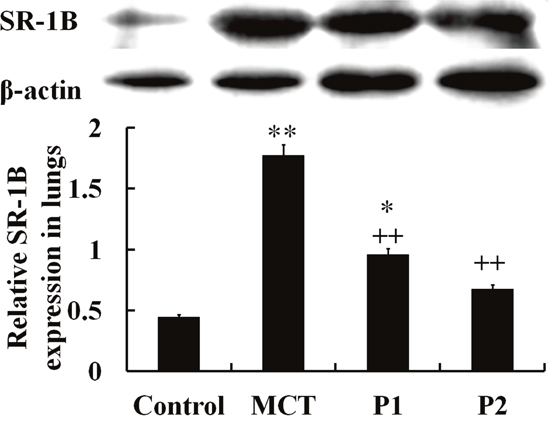
Figure 6: Western blot analysis of SR-1B expression in lung tissues. The control group, the monocrotaline (MCT)-induced pulmonary arterial hypertension group, P1 group with 4-Chloro-DL-phenylalanine (PCPA) treated 50 mg/kg/day, and P2 group with PCPA treated 100 mg/kg/day. Data are expressed as mean ± SD (n = 5 rats). * P < 0.05, **P < 0.01 compared with the control group. ++P < 0.01 compared with the MCT group.
Effect of PCPA on ERK and p-ERK expression in rat lungs
The Western blotting analysis of ERK phosphorylation demonstrated that MCT-induced ERK phosphorylation was significantly up-regulated compared with that of the control group from 0.58 ± 0.05 to 1.53 ± 0.13 (P < 0.01). PCPA inhibited the induction of ERK phosphorylation by MCT. The levels of p-ERK/ERK decreased to 1.18 ± 0.05 (P < 0.05 vs. MCT) in the P1 group and 0.82 ± 0.07 (P < 0.01 vs. MCT) in the P2 group. Together, these results show that PCPA inhibited the MCT-induced activation of ERK in rat lungs (Figure 7).
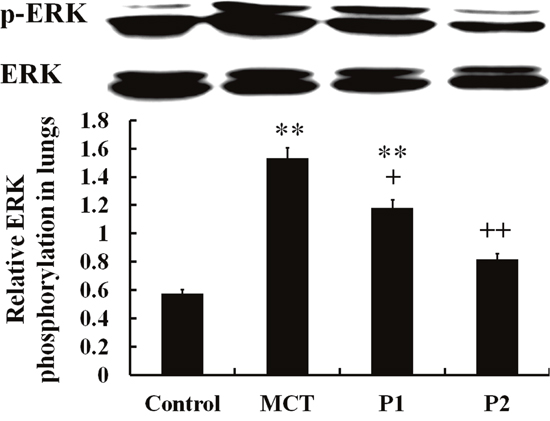
Figure 7: Comparison of ERK phosphorylation in rat lung tissues. The control group, the monocrotaline (MCT)-induced pulmonary arterial hypertension group, P1 group with 4-Chloro-DL-phenylalanine (PCPA) treated 50 mg/kg/day, and P2 group with PCPA treated 100 mg/kg/day. Data are expressed as mean ± SD (n = 5 rats). ** P < 0.01 compared with the control group. + P < 0.05, ++P < 0.01 compared with the MCT group.
Effect of PCPA on apoptosis in rat lungs
As shown in Figure 8, in MCT group, the percentage of TUNEL-positive cells were slightly decreased (vs control group). PCPA treatment evidently enhanced the percentage of TUNEL-positive cells, especially with high dosage.
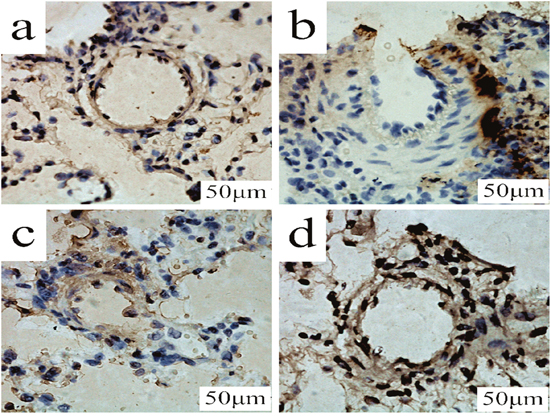
Figure 8: TUNEL determination of PCPA on apoptosis in lung tissues. Lung tissues obtained from (a) control group, (b) monocrotaline (MCT) group, (c) MCT plus 50 mg/kg body mass 4-Chloro-DL-phenylalanine (PCPA) group (P1), and (d) MCT plus 100 mg/kg body mass PCPA group (P2). (Original magnification × 400, scale bars = 50 μm).
The Western blotting analysis showed that the levels of bcl-2 and bcl-xl in the MCT group were noticeably up-regulated compared with that of the control group. In the MCT group, bcl-2 expression increased from 0.72 ± 0.39 to 1.99 ± 0.24 (P < 0.01 vs. control) and bcl-xl increased from 0.53 ± 0.09 to 2.95 ± 0.05 (P < 0.01 vs. control). PCPA inhibited bcl-2 and bcl-xl expression. In the P1 group, bcl-2 and bcl-xl protein decreased to 1.77 ± 0.06 and 2.16 ± 0.07 (P < 0.05 vs. MCT, respectively). In the P1 group, bcl-2 and bcl-xl protein were reduced to 1.39 ± 0.21 (P < 0.05 vs. MCT) and 0.89 ± 0.30 (P < 0.01 vs. MCT), respectively. However, the levels of capase-3 significantly decreased in the MCT group from 2.09 ± 0.41 in the control group to 0.84 ± 0.16 (P < 0.01). PCPA up regulated the expression of capase-3, especially at the higher dose. In the P1 and P2 groups, capase-3 expression increased to 1.37 ± 0.13 (P < 0.05 vs. MCT) and 1.94 ± 0.06 (P < 0.01 vs. MCT), respectively (Figure 9).
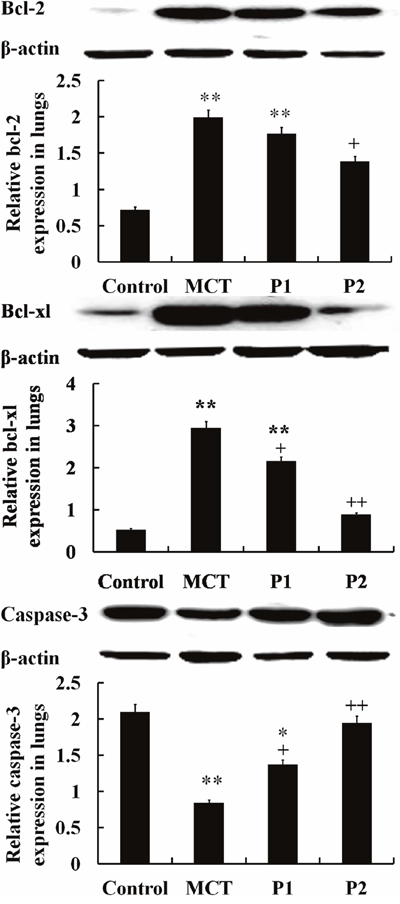
Figure 9: Western blot analysis of bcl-2, bcl-xl and caspase-3 protein expression in lungs. The control group, the monocrotaline (MCT)-induced pulmonary arterial hypertension group and the 50 and 100 mg/kg 4-Chloro-DL-phenylalanine (PCPA) treated groups (P1 and P2, respectively). Data are expressed as mean ± SD (n = 5 rats). **P < 0.01 compared with the control group. +P < 0.05, ++P < 0.01 compared with the MCT group.
DISCUSSION
In this study, we showed that PCPA can ameliorate MCT-induced PAH which might associate with down-regulated SR-1B, CTGF and its downstream signaling pathway. Our results indicated that the expression of 5-HT, SR-1B, CTGF, p-ERK/ERK, bcl-2 and bcl-xl is significantly increased and the expression of caspase-3 is decreased in the MCT group. PCPA inhibits pulmonary artery remodeling, attenuated the expression of 5-HT, SR-1B, CTGF, p-ERK/ERK, bcl-2 and bcl-xl and increasing the expression of caspase-3. These results suggest PCPA contributes to preventing the pathogenesis of PAH by suppressing remodeling and inducing apoptosis, and these effects are likely associated with the CTGF and ERK signaling pathway.
Studies have found that disturbing the balance between proliferation and apoptosis of PASMCs is important for vascular remodeling [12–15]. MCT is a toxic alkaloid that can induce PAH via endothelial damage and PASMCs proliferation and hypertrophy. Previous results from our laboratory showed that an i.p. injection of MCT induces vascular remodeling, increases PASMC proliferation and decreases apoptosis by up-regulating anti-apoptotic proteins [31, 32]. PCPA, as an inhibitor of 5-HT synthesis, has amelioration effect on remodeling in the PAH [31, 33]. But whether PCPA can regulate the apoptosis and its associated signaling pathway is unknown. Therefore, we aim to study the involvement of CTGF and p-ERK/ERK signaling in the pathogenesis of PAH for further investigation.
Serotonin plays an important role in PAH. Results from our laboratories showed that the rate-limiting enzyme of serotonin synthesis, TPH1 and SERT expression is increased in PAH [31, 33]. Serotonin mediates PAH by inducing pulmonary vascular remodeling and vasoconstriction [27]. 5-HT leads to vascular remodeling by stimulating PASMCs proliferation and reducing apoptosis through SR or SERT [30]. The 5-HT signaling pathway is an important mechanism of human PASMCs proliferation in which SR-1B mediates MEK/ERK activation and increases the expression of many transcription factors, which then induce calcium-dependent PASMC proliferation [34]. Moreover, agents targeting 5-HT receptors or SERT may alter apoptotic pathways and ameliorate the development of vascular remodeling [34]. In addition, researchers have used SB2116641, an antagonist of SR-1B, to study cellular functions and found that it could induce apoptosis by decreasing caspase-3, -8 and -9 and poly ADP-ribose polymerase (PARP) [35]. Furthermore, 5-HT promotes vasoconstriction via SR-1B in human pulmonary arteries [36], and knockdown of the SR-1B gene and treatment with neutralizing antibodies promote effective results in a hypoxia-induced PAH animal model [36]. The data from other studies are consistent with those of our study in which the expression of TPH-1 and SR-1B were up-regulated in MCT-induced PAH and TPH-1, SR-1B and MCT-induced pulmonary arterial remodeling were decreased by PCPA. However, the regulatory mechanisms underlying these effects are not clear.
CTGF regulates a variety of cellular functions, including adhesion, proliferation, differentiation, extracellular matrix (ECM) remodeling as well as apoptosis by interacting with multiple integrins [19–21]. Increasing evidences have shown the important role of CTGF in apoptosis during disease development. The up-regulation of CTGF reduced chemotherapy-induced apoptosis in breast cancer by increasing the expression of bcl-xl and cellular inhibitor of apoptosis proteins (cIAP1) [22]. Silencing CTGF expression suppresses the growth and differentiation of rhabdomyosarcoma cells by inducing apoptosis [37]. In human chondrosarcoma, micro-environmental stress, such as hypoxia, induce the expression of CTGF via interactions between the 3’-untranslated region and a cellular protein [38]. Conversely, CTGF protects tumor cell growth by suppressing hypoxia-induced apoptosis [39]. Knockdown of CTGF in fibroblast-like synoviocytes of rheumatoid arthritis induce apoptosis and inhibits survivin mRNA expression [40]. These results indicate that CTGF is an important survival (or apoptosis) factor and could be a therapeutic target in various diseases. The important role of CTGF in the pathogenesis of PAH can be observed from many aspects. For example, Zhu et al. [23] performed a dynamic evaluation and found that the expression of TGF-β1 and CTGF was correlated with the severity of PAH and indicated that CTGF might be involved with vascular remodeling in high blood flow-induced PH. Another report indicated that the expression of CTGF protein and mRNA was increased in MCT-treated rats compared with the control groups, and this effect could be reversed by simvastatin [24]. These findings are consistent with our results. The expression of CTGF and apoptosis-associated factors was remarkably elevated in the MCT group. PCPA inhibited the expression of CTGF, bcl-2, and bcl-xl and up-regulated the expression of caspase-3 protein. Therefore, we infer that CTGF may participate in pulmonary arteries remodeling and apoptosis in MCT-induced PAH and PCPA may inhibit pulmonary artery remodeling and induce apoptosis through suppressing the TPH-1, SR-1B, which influence CTGF and the downstream signaling pathways. However, the specific mechanisms underlying these changes require further investigation.
ERKs are members of the MAPK family and widely expressed protein kinases that are involved in multiple functions, including growth and differentiation [41]. Previous studies showed that the role of ERK1/2 as an important protein kinase intracellular signaling molecules was critical in the resistance to apoptosis in various types of cells [42]. Additionally, an ERK-activating kinase inhibitor was used to block the ERK activation and reverse its protective effect against DNA strand damage and apoptosis in hyperoxic lung injury, thus indicating that this pathway is ERK dependent [43]. Furthermore, studies have found the Ras-MAPK pathway promotes cell survival and inhibits apoptosis by increasing the transcription of pro-survival genes and suppressing various apoptotic regulatory molecules, including Bad, Bim, Mcl-1, caspase-9 and bcl-2, via phosphorylation [41, 44]. In dexamethasone-induced thymocyte cells apoptosis, ERK1/2 is suppressed, which causes calcium ion pool to dysfunction and prevents the phosphorylation of bcl-2, thereby accelerating the apoptotic process [45, 46]. Moreover, ERK/MAPK had been demonstrated to be involved in MCT-induced PAH and aspirin treatment attenuates PAH by suppressing ERK1/2 signaling pathway in rats [47]. Our present study showed that in MCT-induced PAH the expression of p-ERK/ERK and apoptosis-resistant associated proteins such as bcl-2, bcl-xl were up-regulated, but pro-apoptosis protein caspase-3 was inhibited. PCPA inhibited the p-ERK/ERK signal pathway and induced the apoptosis, which indicated that the ERK signaling pathway may regulate the process of apoptotic in the MCT-induced PAH and therefore ameliorate the pulmonary arteries remodeling.
Additionally, previous research had shown that the COOH-terminal domain of CTGF binds to integrin αvβ3 which activates FAK and ERK1/2 and increases the expression of bcl-xl in MCF7 cells [22]. Αvβ3 integrin is necessary for CTGF-induced ERK activation [22]. Evidence had also shown that anti-apoptotic effect of CCR7 is related to the downstream bcl-2, bax and caspase-3; however, treatment with PD98059 indirectly inhibited ERK activation via the MEK pathway and reverses the CCR7 effect [48]. During cisplatin treatment of human osteosarcoma, CTGF could protect against apoptosis by enhancing the expression of bcl-xl and survivin [49]. Further investigations had shown that CTGF exerted anti-apoptotic effects via the FAK, MEK, and ERK signaling pathways [49]. A recent study showed that the up-regulation of CTGF significantly reduced its apoptotic effect in 5-FU-treated CRC cells [50]. Factors upstream of CTGF can enhance the phosphorylation of ERK signaling pathways as well as the expression of bcl-xl and survivin [50]. These results indicated that CTGF can regulate the apoptosis by activating the phosphorylation of ERK, and based on our experimental results, we conclude that PCPA may suppress the expression of CTGF via the p-ERK/ERK signal pathway and then promote apoptosis and inhibit pulmonary artery remodeling. Thus, by targeting defects in anti-apoptosis signaling pathways, PCPA might exert apoptotic effects in PAH rats.
Accumulating experimental studies and computational models have shown that non-coding RNAs (ncRNAs) are involved in the life cycle of cells through different mechanism and play an important role in various biological processes [51–56]. NcRNAs can be divided into long ncRNAs (lncRNA) and small ncRNA (including siRNA, miRNA and piRNA, et al) [52]. Considering the different functions of ncRNAs, it is no hard to find that the dysregulations and mutations of ncRNAs are tightly associated to the progression of remodeling in PAH [57–59]. Although the roles of ncRNAs in PAH has been some progress, but the specific mechanisms need to be further studied.
In conclusion, the increased proliferation and decreased apoptosis of pulmonary arteries plays an important role in PASMC accumulation and pulmonary blood vessel remodeling. Considerable evidence support the effect of 5-HT on pulmonary vasculature in the pathobiology of PAH. PCPA acts as an inhibitor of TPH1 to reduce the levels of TPH and synthesis of 5-HT. Our results indicated that decreased levels of TPH1 suppresses pulmonary arterial remodeling and induces apoptosis which is most likely mediated by SR-1B, and this may involve CTGF and p-ERK/ERK signaling. Extensive research is necessary in order to investigate the specific mechanism underlying these effects, their relationship, and uncovering further potential therapeutic targets to treat PAH.
MATERIALS AND METHODS
Animal models
Forty Sprague-Dawley (SD) rats weighing 180 ± 10 g were obtained from the Animal Resource Center in China Medical University (Certificate No. Liaoning 034). All rats were randomly divided into four groups: the control group, in which rats received a vehicle (0.9% physiological saline); the MCT group (Sigma-Aldrich, St Louis, MO, USA) group, in which rats received MCT (60 mg/kg); the P1 group, in which rats received MCT (60 mg/kg) once and PCPA (50 mg/kg) daily; and the P2 group, in which rats received MCT (60 mg/kg) once and PCPA (100 mg/kg) daily. All rats in the MCT, P1 and P2 groups received intraperitoneal (i.p.) injections of MCT dissolved in vehicle (1:4 mixture of dehydrated ethanol-normal saline]. Rats in the PCPA-treated groups were administered PCPA (i.p.) once daily for 21 days. The control group rats received the same volume of vehicle as the other groups during the same period. All rats were provided food and water ad libitum and placed under a natural day/night cycle at 18-22°C and 50-70% humidity. All measurements taken throughout the entire experiment were performed in a blinded fashion. All animal experiments were conducted in strict accordance with international ethical guidelines and the National Institute of Health Guide concerning the Care and Use of Laboratory Animals. The protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of China Medical University. The approval reference number is SYXK (Liao) 2013-0007. All research was accepted by the local authority and conducted according to the guidelines of China Medical University.
Detection of pulmonary arterial morphometry and evaluation of right ventricular hypertrophy
On day 22, the rats were anaesthetized via the administration of 3% pentobarbital sodium (40 mg/kg), and then the PAP and systemic arterial pressure were measured using methods detailed in our previous study [31]. Then, the rats were euthanized with pentobarbital sodium (60 mg/kg, absolute lethal dose, i.p.), and heart and lung tissues were excised. The hearts were separated into the left ventricle plus septum (LV + S) and the right ventricle (RV). The RVI was calculated using the formula RVI = RV/(LV + S) to evaluate heart remodeling.
We used 4% paraformaldehyde and sterile saline to exsanguinate the rats. The pulmonary arteries and lungs tissues were cut and placed in 4% paraform. Paraffin-embedded pulmonary arteries and lung tissues were prepared in 5-μm sections and then stained with hematoxylin-eosin (H&E) for further research. For each rat, 15 pulmonary arteries were observed using a MetaMorph (Universal Imaging Corporation, PA, USA)-DP10/BX51 system (Olympus, Tokyo, Japan). We used the following formula to calculate the medial thickness for measuring pulmonary arterial remodeling: pulmonary arterial wall thickness (%) = (external diameter – internal diameter)/external diameter × 100% [31].
Immunohistochemical staining
Paraffin-embedded aortopulmonary and lung tissues sections were dyed using ultrasensitive S-P and diaminobenzidine (DAB) staining kits (Maxin-Bio, Fuzhou, China). The primary rabbit polyclonal antibody against CTGF (BA0752, Boster biotechnology) was diluted to 1:200. The negative control was incubated in 0.01 M phosphate-buffered saline (PBS) instead of the primary antibody. A BX51 microscope (Olympus) was used to analyze the digital pictures. Fifteen pulmonary arteries (external diameter 50~100 μm) from at least 3 rats were analyzed in each group, and at least 3 aortopulmonary sections were studied among the 4 groups. The levels of CTGF were assessed as the average optical density. Immunohistochemical staining followed a basic indirect protocol using a citrate antigen-retrieval method [31].
ELISA analysis
We collected blood samples from different groups of rats in the chilled tubes containing 2 mg/ml EDTA. Then the tubes were centrifuged at 2400 g for 10 min (4°C). The plasma was collected frozen and stored at −80°C for ELISA. The concentrations of 5-HT in plasma were measured using a rat 5-HT ELISA kit (CSB-E08364r, Cusabio, Wuhan, China) according to the manufacturer’s instructions. The limit of detection of the assay for 5-HT protein was 0.02 ng/ml.
TUNEL
Apoptotic were determined by the terminal deoxyribonucleotidyl transferase-mediated dUTP–digoxigenin nick end-labelling (TUNEL) method of an in situ cell death detection kit (KGA702, Keygenbio, Nanjing, China) in terms of manufacturer’s instruction. The number of TUNEL-positive cells in 12 fields for each section of small-pulmonary arteries was quantitatively estimated as a percentage of total smooth muscle cells (×400) by researcher blinded to the treatment group.
Total protein preparation
Total protein was prepared as previously described [31]. The lung samples were homogenized to extract the total protein content using a polytron homogenizer (Kinematica, Lucerne, Switzerland). Then, we centrifuged the homogenate at 15000 g for 30 min at 4°C. The supernatant was collected and stored at −80°C until it was used in the experiments. The total protein concentration was determined by the BCA method (Beyotime Biotechnology, Inc).
Western blotting analysis
Equal amounts of protein were separated by a reducing SDS-PAGE and electrotransferred onto a polyvinylidene difluoride (PVDF) membrane [31, 32]. After incubation in blocking buffer (5% nonfat dry milk, TBS and 0.05% Tween-20) at room temperature for 2 h, the membranes were incubated with primary antibodies overnight at 4°C. The antibodies included rabbit polyclonal anti-CTGF antibody (1:400, BA0752, Boster Biotechnology), rabbit polyclonal anti-SR-1B antibody (1:500, BS2765, Bioworld Technology, Inc.), rabbit polyclonal anti-ERK1/2 antibody (1:500, sc-292838, Santa Cruz Biotechnology), mouse monoclonal anti-p-ERK1/2 (1:500, sc-81492, Santa Cruz Biotechnology), rabbit polyclonal anti-bcl-2 antibody (1:500, BS1511, Bioworld Technology, Inc.), rabbit polyclonal anti-bcl-xl antibody (1:500, BS1032, Bioworld Technology, Inc.), rabbit polyclonal anti-caspase-3 antibody (1:400, BS61583, Bioworld Technology, Inc.) and mouse polyclonal anti-β-actin antibody (1:2000, sc-47778, Santa Cruz Biotechnology]. The immunoreactive bands were visualized using the corresponding horseradish peroxidase-conjugated secondary antibodies and super ECL plus (Thermo Fisher Scientific, Waltham, MA, USA). The relative protein expression was quantified by densitometry using Quantity One (Bio-Rad Laboratories, California, USA).
Statistical analysis
Data are shown as the mean with the standard error. All statistical analyses were performed using SPSS, version 16.0 (SPSS, Inc, Chicago, IL, USA). Statistical comparisons were conducted using a one-way ANOVA and Fisher’s least significant difference (LSD) or Dunnett’s T3 tests. Values of P < 0.05 were considered statistically significant.
ACKNOWLEDGMENTS AND FUNDING
This research was supported by the science research projects of Education Department of Liaoning Province (No. LK201640), the Doctor Startup Foundation from Liaoning Province (No. 20170520217), Innovation Team Project (No. LT201511), and National Natural Science Foundation of China (No. 81273511 and 61772531).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Barst RJ, McGoon M, Torbicki A, Sitbon O, Krowka MJ, Olschewski H, Gaine S. Diagnosis and differential assessment of pulmonary arterial hypertension. J Am Coll Cardiol. 2004; 43: 40S–7S.
2. Leopold JA, Maron BA. Molecular mechanisms of pulmonary vascular remodeling in pulmonary arterial hypertension. Int J Mol Sci. 2016; 17:E761.
3. Lai YC, Potoka KC, Champion HC, Mora AL, Gladwin MT. Pulmonary arterial hypertension: the clinical syndrome. Circ Res. 2014; 115: 115-30.
4. Stenmark KR, Meyrick B, Galie N, Mooi WJ, McMurtry IF. Animal models of pulmonary arterial hypertension: the hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol. 2009; 297: L1013–32.
5. Sakao S, Tatsumi K, Voelkel NF. Endothelial cells and pulmonary arterial hypertension: apoptosis, proliferation, interaction and transdifferentiation. Respir Res. 2009; 10: 95.
6. Sumpio BE, Riley JT, Dardik A. Cells in focus: endothelial cell. Int J Biochem Cell Biol. 2002; 34: 1508–12.
7. Zhang S, Dong H, Rubin LJ, Yuan JX. Upregulation of Na+/Ca2+ exchanger contributes to the enhanced Ca2+entry in pulmonary artery smooth muscle cells from patients with idiopathic pulmonary arterial hypertension. Am J Physiol Cell Physiol. 2007; 292: C2297–305.
8. Winn RK, Harlan JM. The role of cell apoptosis in inflammatory and immune diseases. J Thromb Haemost. 2005; 3: 1815-24.
9. Sage E, Mercier O, Van den Eyden F, de Perrot M, Barlier-Mur AM, Dartevelle P, Eddahibi S, Herve P, Fadel E. Endothelial cell apoptosis in chronically obstructed and reperfused pulmonary artery. Respir Res. 2008; 9: 19.
10. Budhiraja R, Tuder RM, Hassoun PM. Endothelial dysfunction in pulmonary hypertension. Circulation. 2004; 109: 159-65.
11. Wyllie AH, Kerr JF, Currie AR. Cell death: the significance of apoptosis. Int Rev Cytol. 1980; 68: 251–306.
12. Gurbanov E, Shiliang X. The key role of apoptosis in the pathogenesis and treatment of pulmonary hypertension. Eur J Cardiothorac Surg. 2006; 30: 499-507.
13. Archer S, Rich S. Primary pulmonary hypertension: a vascular biology and translational research “work in progress”. Circulation. 2000; 102: 2781-91.
14. Zhang S, Fantozzi I, Tigno DD, Yi ES, Platoshyn O, Thistlethwaite PA, Kriett JM, Yung G, Rubin LJ, Yuan JX. Bone morphogenetic proteins induce apoptosis in human pulmonary vascular smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2003; 285: L740-54.
15. McMurtry MS, Bonnet S, Wu X, Dyck JR, Haromy A, Hashimoto K, Michelakis ED. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ Res. 2004; 95: 830-40.
16. Morrell NW, Archer SL, Defelice A, Evans S, Fiszman M, Martin T, Saulnier M, Rabinovitch M, Schermuly R, Stewart D, Truebel H, Walker G, Stenmark KR. Anticipated classes of new medications and molecular targets for pulmonary arterial hypertension. Pulm Circ. 2013; 3: 226-44.
17. Bradham DM, Igarashi A, Potter RL, Grotendorst GR. Connective tissue growth factor: a cystein-rich mitogen secreted by human vascular endothelial cells is related to the SRC-induced immediate early gene product CEF-10. J Cell Biol. 1991; 114: 1285–94.
18. Kono M, Nakamura Y, Suda T, Kato M, Kaida Y, Hashimoto D, Inui N, Hamada E, Miyazaki O, Kurashita S, Fukamachi I, Endo K, Ng PS, et al. Plasma CCN2 (connective tissue growth factor; CTGF) is a potential biomarker in idiopathic pulmonary fibrosis (IPF). Clin Chim Acta. 2011; 412: 2211-5.
19. Jun JI, Lau LF. Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat Rev Drug Discov. 2011; 10: 945-63.
20. Rachfal AW, Brigstock DR. Structural and functional properties of CCN proteins. Vitam Horm. 2005; 70: 69-103.
21. Chen CC, Lau LF. Functions and mechanisms of action of CCN matricellular proteins. Int J Biochem Cell Biol. 2009; 41:771–83.
22. Wang MY, Chen PS, Prakash E, Hsu HC, Huang HY, Lin MT, Chang KJ, Kuo ML. Connective tissue growth factor confers drug resistance in breast cancer through concomitant up-regulation of Bcl-xL and cIAP1. Cancer Res. 2009; 69: 3482–91.
23. Zhu R, He L, Xu J, Zhang Y, Hu Y. [Changes of TGF-β1 and CTGF in rats with increased blood flow-induced pulmonary artery hypertension]. [Article in Chinese]. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2012; 37:1013–20.
24. Liu B, Wang XM, Zhou TF, Hua YM, Liu HM, Wei L, Qiao LN, Wang XQ, Zhao SS, Shi K. [Expression of connective tissue growth factor and its down-regulation by simvastatin administration in pulmonary hypertensive rats]. [Article in Chinese]. Zhonghua Er Ke Za Zhi. 2008; 46:359–65.
25. Izikki M, Hanoun N, Marcos E, Savale L, Barlier-Mur AM, Saurini F, Eddahibi S, Hamon M, Adnot S. Tryptophan hydroxylase 1 knockout and tryptophan hydroxylase 2 polymorphism: effects on hypoxic pulmonary hypertension in mice. Am J Physiol Lung Cell Mol Physiol. 2007; 293: L1045-52.
26. Eddahibi S, Guignabert C, Barlier-Mur AM, Dewachter L, Fadel E, Dartevelle P, Humbert M, Simonneau G, Hanoun N, Saurini F, Hamon M, Adnot S. Cross talk between endothelial and smooth muscle cells in pulmonary hypertension: critical role for serotonin-induced smooth muscle hyperplasia. Circulation. 2006; 113: 1857-64.
27. MacLean MR, Herve P, Eddahibi S, Adnot S. 5-hydroxytryptamine and the pulmonary circulation: receptors, transporters and relevance to pulmonary arterial hypertension. Br J Pharmacol. 2000; 131: 161–8.
28. Welsh DJ, Harnett M, MacLean M, Peacock AJ. Proliferation and signaling in fibroblasts: role of 5-hydroxytryptamine 2A receptor and transporter. Am J Respir Crit Care Med. 2004; 170: 252–9.
29. Dempsie Y, MacLean MR. Pulmonary hypertension: therapeutic targets within the serotonin system. Br J Pharmacol. 2008; 155: 455-62.
30. Liu Y, Tian HY, Yan XL, Fan FL, Wang WP, Han JL, Zhang JB, Ma Q, Meng Y, Wei F. Serotonin inhibits apoptosis of pulmonary artery smooth muscle cell by pERK1/2 and PDK through 5-HT1B receptors and 5-HT transporters. Cardiovasc Pathol. 2013; 22:451-7.
31. Bai Y, Wang HM, Liu M, Wang Y, Lian GC, Zhang XH, Kang J, Wang HL. 4-Chloro-DL-phenylalanine protects against monocrotaline-induced pulmonary vascular remodeling and lung inflammation. Int J Mol Med. 2014; 33: 373-82.
32. Zhai FG, Zhang XH, Wang HL. Fluoxetine protects against monocrotalien-induced pulmonary arterial hypertension: potential roles of induction of apoptosis and upregulation of Kv1.5 channels in rat. Clin Exp Pharmacol Physiol. 2009; 36: 850-6.
33. Bai Y, Li ZX, Wang HL, Lian GC, Wang Y. The protective effects of PCPA against monocrotaline-induced pulmonary arterial hypertension are mediated through the downregulation of NFAT-1 and NF-κB. Int J Mol Med. 2017; 40: 155-63.
34. Thomas M, Ciuclan L, Hussey MJ, Press NJ. Targeting the serotonin pathway for the treatment of pulmonary arterial hypertension. Pharmacol Ther. 2013; 138: 409-17.
35. Gurbuz N, Asoglu MR, Ashour AA, Salama S, Kilic GS, Ozpolat B. A selective serotonin 5-HT1B receptor inhibition suppresses cells proliferation and induces apoptosis in human uterine leiomvoma cells. Eur J Obstet Gynecol Reprod Biol. 2016; 206: 114-9.
36. Keegan A, Morecroft I, Smillie D, Hicks MN, MacLean MR. Contribution of the 5-HT (1B) receptor to hypoxia-induced pulmonary hypertension: Converging evidence using 5-HT (1B)-receptor knockout mice and the 5-HT (1B/1D)-receptor antagonist GR127935. Circ Res. 2001; 89: 1231-9.
37. Croci S, Landuzzi L, Astolfi A, Nicoletti G, Rosolen A, Sartori F, Follo MY, Oliver N, De Giovanni C, Nanni P, Lollini PL. Inhibition of connective tissue growth factor (CTGF/CCN2) expression decreases the survival and myogenic differentiation of human rhabdomyosarcoma cells. Cancer Res. 2004; 64: 1730–6.
38. Kondo S, Kubota S, Mukudai Y, Moritani N, Nishida T, Matsushita H, Matsumoto S, Suqahara T, Takigawa M. Hypoxic regulation of stability of connective tissue growth factor/CCN2 mRNA by 3′-untranslated region interacting with a cellular protein in human chondrosarcoma cells. Oncogene. 2006; 25: 1099–110.
39. Bennewith KL, Huang X, Ham CM, Graves EE, Erler JT, Kambham N, Feazell J, Yang GP, Koong A, Giaccia AJ. The role of tumor cell-derived connective tissue growth factor (CTGF/CCN2) in pancreatic tumor growth. Cancer Res. 2009; 69: 775-84.
40. Ding S, Fang F, Duan H, Xiao W. Effect of CTGF siRNA on apoptosis of fibroblast-like synoviocytes of rheumatoid arthritis. Journal of China Medical University. 2016; 45: 430-3.
41. McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M, Tafuri A, Stivala F, Libra M, Basecke J, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007; 1773: 1263–84.
42. Stadheim TA, Xiao H, Eastman A. Inhibition of extracellular signal-regulated kinase (ERK) mediates cell cycle phase independent apoptosis in vinblastine-treated ML-1 cells. Cancer Res. 2001; 61: 1533-40.
43. Buckley S, Driscoll B, Barsky L, Weinberg K, Anderson K, Warburton D. ERK activation protects against DNA damage and apoptosis in hyperoxia rat AEC2. Am J Physiol. 1999; 277: L159-66.
44. Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999; 286: 1358-62.
45. Voris BP, Young DA. Glucocorticoid-induced proteins in rat thymus cells. J Biol Chem. 1981; 256: 11319-29.
46. McConkey DJ, Nicotera P, Hartzell P, Bellomo G, Wyllie AH, Orrenius S. Glucocorticoids activate a suicide process in thymocytes through an elevation of cytosolic Ca2+ concentration. Arch Biochem Biophys. 1989; 269: 365-70.
47. Gao H, Cheng Y, Zong L, Huang L, Qiao C, Li W, Gong B, Hu J, Liu H, Wang X, Zhao C. Aspirin attenuates monocrotaline-induced pulmonary arterial hypertension in rats by suppressing the ERK/MAPK pathway. Clin Exp Hypertens. 2017; 39: 34-41.
48. Xu Y, Liu L, Qiu X, Liu Z, Li H, Li Z, Luo W, Wang E. CCL21/CCR7 prevents apoptosis via the ERK pathway in human non-small cell lung cancer cells. PLoS One. 2012; 7: e33262.
49. Tsai HC, Huang CY, Su HL, Tang CH. CCN2 Enhances Resistance to Cisplatin-Mediating Cell Apoptosis in Human Osteosarcoma. PLoS One. 2014; 9: e90159.
50. Yang K, Gao K, Hu G, Wen Y, Lin C, Li X. CTGF enhances resistance to 5-FU-mediating cell apoptosis through FAK/MEK/ERK signal pathway in colorectal cancer. Onco Targets Ther. 2016; 9: 7285-95.
51. Chen X, Yan GY. Novel human lncRNA-disease association inference based on lncRNA expression profiles. Bioinformatics. 2013; 29:2617–24.
52. Chen X. Predicting lnc RNA-disease associations and constructing lncRNA functional similarity network based on the information of miRNA. Sci Rep. 2015; 5: 13186.
53. Chen X, Huang YA, Wang XS, You ZH, Chan KC. FMLNCSIM: fuzzy measure-based lncRNA functional similarity calculation model. Oncotarget. 2016; 7:45948–58. https://doi.org/10.18632/oncotarget.10008.
54. Chen X, Yan CC, Zhang X, You ZH. Long non-coding RNAs and complex diseases: from experimental results to computational models. Brief Bioinform. 2017; 18: 558-76.
55. You ZH, Huang ZA, Zhu Z, Yan GY, Li ZW, Wen Z, Chen X. PBMDA: A novel and effective path-based computational model for miRNA-disease association prediction. PLOS Comput Biol. 2017; 13:e1005455.
56. Chen X, Yan CC, Zhang X, You ZH, Deng L, Liu Y, Zhang Y, Dai Q. WBSMDA: Within and between score for miRNA-disease association prediction. Sci Rep. 2016; 6: 21106.
57. Meloche J, Lampron MC, Nadeau V, Maltais M, Potus F, Lambert C, Tremblay E, Vitry G, Breuils-Bonnet S, Boucherat O, Charbonneau E, Provencher S, Paulin R, Bonnet S. Implication of Inflammation and Epigenetic Readers in Coronary Artery Remodeling in Patients With Pulmonary Arterial Hypertension. Arterioscler Thromb Vasc Biol. 2017; 37: 1513-23.
58. Lu Z, Li S, Zhao S, Fa X. Upregulated miR-17 Regulates Hypoxia-Mediated Human Pulmonary Artery Smooth Muscle Cell Proliferation and Apoptosis by Targeting Mitofusin 2. Med Sci Monit. 2016; 22: 3301-8.
59. Bischoff FC, Werner A, John D, Boeckel JN, Melissari MT, Grote P, Glaser SF, Demolli S, Uchida S, Michalik KM, Meder B, Katus HA, Haas J, et al. Identification and Functional Characterization of Hypoxia-Induced Endoplasmic Reticulum Stress Regulating lncRNA (HypERlnc) in Pericytes. Circ Res. 2017; 121: 368-75.