
INTRODUCTION
Epidermal Langerhans cells (LCs) represents an exclusive subpopulation of skin-resident dendritic cells (DCs), which characteristically express the C-type lectin receptor Langerin (CD207) and its associated “tennis-racket” organelle, Birbeck granules [1]. Adult mouse LCs largely derive from embryonic fetal liver monocytes, with a small minority originate from yolk-sac macrophages [2]. They slowly self-renew in situ under steady-state whereas being supplemented by hematopoietic precursors in inflamed-state [3, 4]. Upon the capture of external or internal antigens, immature LCs leave the epidermis and migrate to nearby draining lymph nodes (LNs) where they turn into mature counterparts. Subsequently, these mature LCs induce T cell immune responses by presenting engulfed antigens to naïve T cells along with secreting cytokines for their differentiation as well as providing costimulatory signals [5]. The aberrant expression of cell-membrane costimulatory molecules on LCs has been implicated in multiple skin disorders including contact hypersensitivity, atopic dermatitis, and virus infection [6–8]. Hence, the elaboration of decisive factors during the maturation of epidermal LCs are of potential therapeutic benefit.
Transforming growth factor-β1 (TGF-β1), an important immunomodulatory and profibrotic cytokine, essentially regulates the maintenance of epidermal LCs after birth [9]. TGF-β1 constitutive null mice lacked LCs [10]. Moreover, LC-specific excision of TGF-β1 or TGF-β receptor II (TGFβRII) resulted in a profound LC loss, suggesting a direct impact of TGF-β1 on LCs through an autocrine or paracrine loop [11]. Further analysis with inducible depletion of TGF-β1 signaling in adult LCs demonstrated a crucial role of TGF-β1 in suppressing LC migration as well as preventing their spontaneous maturation [12–14]. However, the underpinning mechanism of TGF-β1-directed LC biology remained largely unknown. We have recently identified that neither Smad2, Smad3 nor Smad4 in canonical TGF-β1-Smad pathway was required for LC homeostasis [15, 16]. So far, only a limited number of TGF-β1-relevant transcription factors mediating LC development and maintenance have been reported [9].
TGFβ-inducible early gene 1 (TIEG1), also known as Krüppel-like factor 10 (Klf10), was originally discovered in normal human osteoblast cells and termed for its rapid induction by TGF-β1 without de novo protein synthesis [17]. Sequence analyses demonstrated that the evolutionarily-conserved TIEG1 gene encodes a 480 amino-acid protein comprised of a three-zinc finger motif, a Sp1-like DNA binding domain and multiple proline-rich Src homology-3 (SH3) binding domains [17, 18]. TIEG1 enhanced TGF-β1/Smad signaling by the repression of Smad7 gene and induction of Smad2 gene expression [19]. Later studies discovered that TIEG1 expression could be induced by a plethora of growth factors, cytokines, and hormones, which served as a potential marker for skeletal, heart and neoplastic diseases [17, 19–22]. However, the role of TIEG1 in TGF-β1-mediated LC homeostasis remains unexplored.
In this study, we sought to address the effect of TIEG1 deficiency on epidermal LC homeostasis, phagocytosis, maturation in vitro, and cell repopulation under inflamed condition. For the first time, we demonstrated that the ablation of TIEG1 did not hamper the survival of LCs in steady-state, cell replenishment under inflamed-state nor their capability of antigen uptake, but specifically prohibited their maturation.
RESULTS
TIEG1 is not required for epidermal LC homeostasis under the steady state
To investigate a possible regulatory role of TIEG1 in LC homeostasis, we first compared the epidermal LC ratios of TIEG1-/- C57/BL6 mice (designated as KO mice) to wild-type C57/BL6 mice (designated as WT mice), and found that the deletion of TIEG1 did not alter the density of epidermal LCs (Figure 1). Thus, our result suggested that unlike TGF-β1, TIEG1 is not required for LC homeostasis under the steady state.
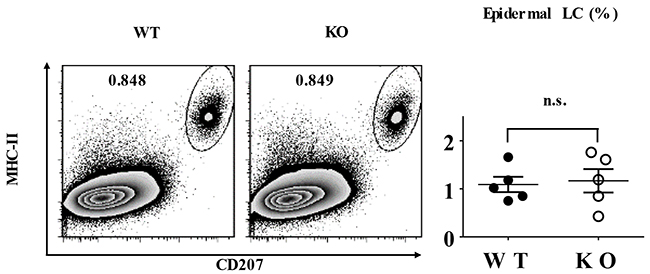
Figure 1: TIEG1 deficiency did not affect LC homeostasis. Epidermal cells were freshly-isolated from the trunk skin of TIEG1 knockout (KO) and wild-type C57BL/6 (WT) mice. Representative FACS analysis (left panel) and the ratios (right panel) of epidermal LCs from TIEG1 WT and KO mice (n=10, three independent experiments).
TIEG1 deficiency does not hamper LC phagocytosis
Typically, immature LCs capture and process external or internal antigens, migrate to the nearby draining lymph nodes where they fulfill maturation, present phagocytosed antigens to naïve T cells and subsequently induce T cell priming. Thus, the phagocytizing capacity of LCs is crucial for their immunoregulatory function. To evaluate the role of TIEG1 in LC antigen-uptake, freshly-isolated epidermal cells from TIEG1 WT and KO mice were incubated with dextran-FITC for 45 minutes at 37 °C or 4 °C (as control). The percentages of dextran-FITC+ LCs were comparable between TIEG1 WT and KO mice (Figure 2), implying that lack of TIEG1 did not affect the phagocytizing ability of LCs.
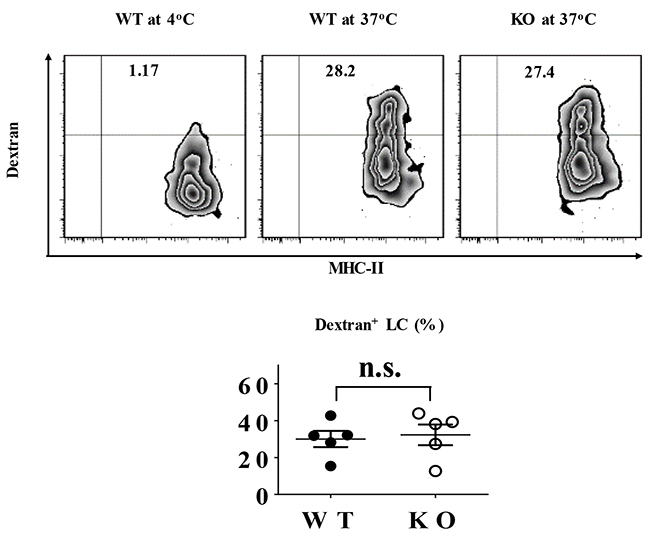
Figure 2: TIEG1 was not required for LC phagocytosis. Freshly-isolated epidermal LCs from TIEG1 WT and KO mice were incubated at 37°C or 4°C (as control) with Dextran-FITC for 45 minutes. Representative FACS analysis (upper panel) and the percentages (lower panel) of FITC+ epidermal LCs (n=10, two independent experiments).
Lack of TIEG1 prohibits LC maturation
The maturation conversion of epidermal LCs is characteristically accompanied with an upregulated expression of cell-membrane costimulatory molecules, including major histocompatibility complex-II (MHC-II), CD80 and CD86. Under steady state, the baseline expression ratios of MHC-II, CD80 or CD86 and their mean fluorescence intensity (MFI) on the freshly-isolated epidermal LCs from TIEG1 WT and KO mice were equally low (Figure 3A–3C). However, after 60 hours of in vitro culture, the expression level of MHC-II, CD80 and CD86 in TIEG1-deficient LCs were considerably lower than their normal counterparts, although the difference in CD80 expression did not reach statistical significance (Figure 3D–3F). These results indicated that in contrast to TGF-β1, TIEG1 deficiency did not disturb the immature status of epidermal LCs but impaired their capacity to acquire a mature phenotype in vitro.
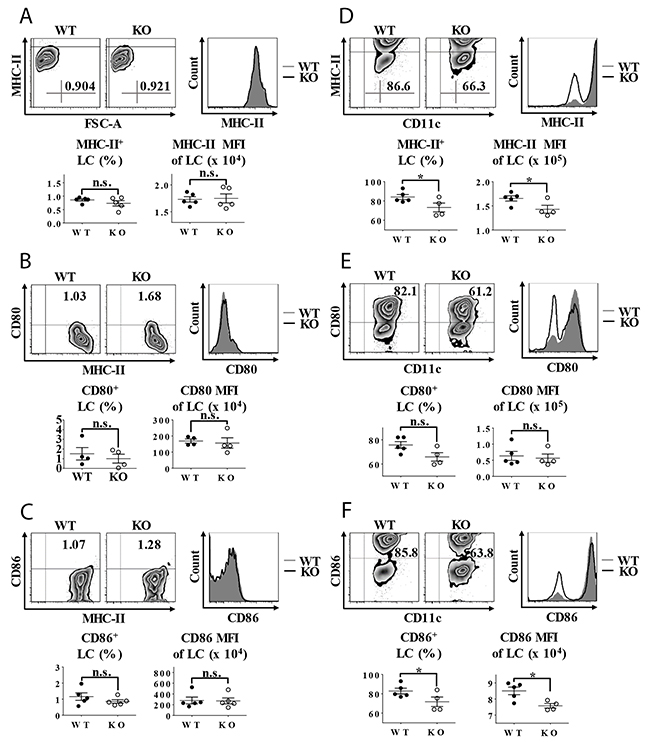
Figure 3: Impaired LC maturation in TIEG1 knockout mice. (A) Epidermal suspensions freshly isolated from the trunk skin were stained with anti-MHC-II, CD45.2, CD80, and CD86 antibodies and analyzed by flow cytometry. Representative FACS analysis (upper-left panel), histogram (upper-right panel) and the ratios (lower panel) of MHC-II+ epidermal LCs from TIEG1 WT and KO mice (n=10, three independent experiments); (B) Epidermal cells were stained as described in (3a). Representative FACS analysis, histogram and the ratios of CD80+ epidermal LCs from TIEG1 WT and KO mice (n=8, two independent experiments); (C) Epidermal cells were stained as described in (3a). Representative FACS analysis, histogram and the ratios of CD86+ epidermal LCs from TIEG1 WT and KO mice (n=10, three independent experiments); (D) Epidermal suspensions from the trunk skin of TIEG1 WT and KO mice were cultured in RPMI for 60 hours, and then stained as described in (3a). Representative FACS analysis, histogram and the ratios of MHC-II+ epidermal LCs from TIEG1 WT and KO mice (n= 9, three independent experiments); (E) Epidermal cells were treated as in (3d). Representative FACS analysis, histogram and the ratios of CD80+ epidermal LCs from TIEG1 WT and KO mice (n=9, three independent experiments); (F) Epidermal cells were treated as in (3d). Representative FACS analysis, histogram and the ratios of CD86+ epidermal LCs from TIEG1 WT and KO mice (n=9, three independent experiments).
TIEG1 deficiency enhanced contact hypersensitivity
To assess the role of TIEG1 in contact hypersensitivity (CHS), we sensitized TIEG1 WT and KO mice by topically applying 0.5% dinitrofluorobenzene (DNFB) on abdominal skin, and then challenged with 0.2% DNFB five days later. The percentages of increased ear thickness caused by the challenge in TIEG1 KO mice were significantly higher than in TIEG1 WT mice at different time points (24 hours, 48 hours, 72 hours, 96 hours) (Figure 4). This result indicated that TIEG1 deficiency enhanced CHS response.
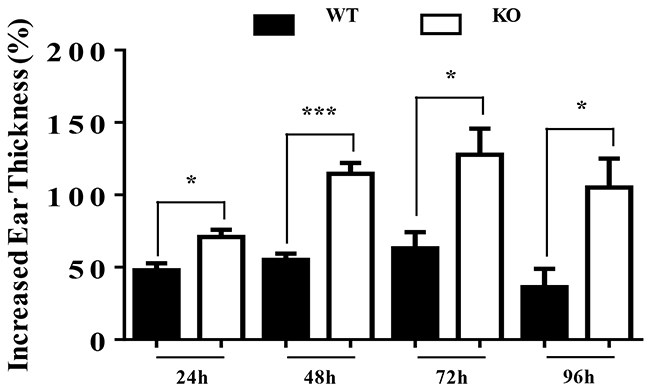
Figure 4: TIEG1 deficiency enhanced contact hypersensitivity response. Mice were sensitized with DNFB (0.5%) in the abdominal area and then challenged with DNFB (0.2%) 5 days later to induce delayed type hypersensitivity in the ears. Ear thicknesses were measured at different time points after challenge (24 hours, 48 hours, 72 hours, 96 hours) by comparing challenged and unchallenged ears using a thickness gauge in a blinded manner. Ten mice were analyzed.
TIEG1 is not involved in LC replenishment
To explore the role of TIEG1 in LC repopulation, we provoked skin inflammation in TIEG1 WT and KO mice by 15 minutes of ultraviolet (UV) irradiation. As expected, UV treatment caused a transient loss of LCs which were replenished by short-term LCs (MHC-II+ langerinlow) (Figure 5) and long-term LCs (MHC-II+ langerin+) successively (data not shown) [4]. In detail, the epidermal LC ratios one week after UV irradiation were comparable between TIEG1 WT and KO mice (Figure 5A), and the percentages of short-term and long-term LCs were also not altered by TIEG1 deficiency (Figure 5B). Hence, these results implied that TIEG1 was not required for the repopulation of LCs under inflamed state.
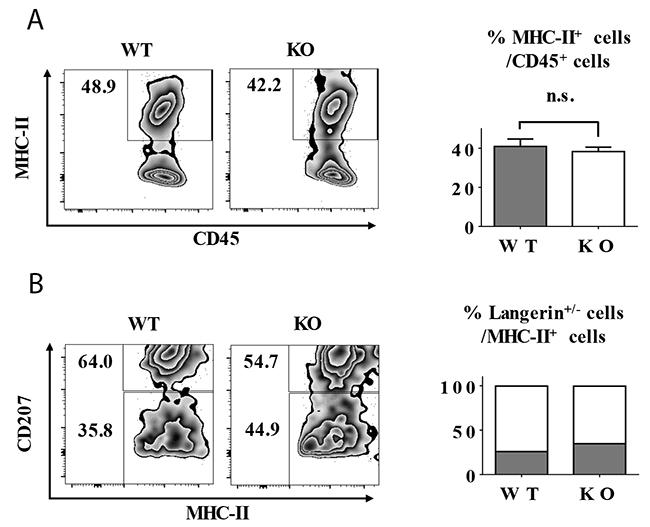
Figure 5: TIEG1 deficiency did not impair LC repopulation in inflamed state. The mice were treated with 15 minutes of UV exposure. (A) Representative FACS analysis (left panel) and the ratios (right panel) of epidermal LCs (MHC-II+) one week after UV treatment. (B) Representative FACS analysis (left panel) and the frequencies (right panel) of short-term LCs (MHC-II+ langerinlow) and long-term (MHC-II+ langerin+) cells from each genotype. The data represent the result of 12 mice.
DISCUSSION
As one of the TGF-β1 down-stream transcription factors, TIEG1 enhanced TGF-β1 signaling by inhibiting Smad7 gene and promoting Smad2 gene expression [19]. Besides, TIEG1 could also directly increased TGFβRII gene expression through histone H3 acetylation [23]. Hence, TIEG1 might be a major positive regulator in TGF-β1 signaling. Previous studies suggested that TGF-β1 is a prerequisite for LC homeostasis and maintenance in skin, and TGF-β1 inhibits LC maturation and migration, but does not affect LC replenishment under inflamed-state [10–14]. Our results demonstrated that unlike TGF-β1, TIEG1 was not essential for LC homeostasis but is required for the full maturation of LCs. The discrepancy in LC phenotype between TGF-β1- and TIEG1-deficient mice suggested that TIEG1 may not be a key molecule involved in TGF-β1-directed LC homeostasis, while TIEG1-related signaling pathways modulate LC maturation. It has been reported that TIEG-1 overexpression leads to the activation of p38 MAPK, which could induces costimulatory molecule expression on the LCs and enhanced their T-cell stimulatory capacity [24–26]. Whether TIEG1 mediates the maturation process of LCs through p38 MAPK or other factors awaits future investigation.
Epidermal LCs were initially considered as solely immunogenic that determined contact hypersensitivity (CHS) [27]. Recent in vivo studies on LC-ablative mouse models uncovered that epidermal LCs might mainly play a tolerogenic role in CHS [28]. We demonstrated here that although the maturation status of LCs was compromised in TIEG1 null mice, they developed more severe CHS response than WT mice, which further verified that LCs principally mediated immune tolerance in CHS. Moreover, our finding was same as the augmented phthalic anhydride-induced skin inflammation in TIEG1 KO mice reported by Bae et al [26]. However, the exact role of LCs in TIEG1-mediated suppression of CHS response require further examination.
LC replenishment under inflamed condition is achieved by two waves of distinct LC populations: a minor transient population of short-term LCs (MHC-II+ langerinlow) originated from circulating monocytes followed by a major persistent population of long-term LCs (MHC-II+ langerin+) of uncharacterized bone-marrow (BM) precursors [4]. Previous research demonstrated that lack of TGF-β1 exerted no impairment on the BM’s potential to generate LCs in vivo, which suggested that TGF-β1 was probably not required for the LC repopulation in inflamed state [10]. The negligible role of TIEG1 in LC replenishment we identified in this study further supported this assumption.
In summary, our results indicate that TIEG1 is not involved in TGF-β1-directed LC homeostasis, but contrarily facilitates LC maturation. Additionally, TIEG1 deficiency enhanced DNFB-induced CHS response.
MATERIALS AND METHODS
Mice
TIEG1 KO mice have been described previously [29]. C57BL/6 (WT) mice were purchased from the Jackson Laboratory. Experiments were conducted at 7 to 9 weeks of age. Mice were housed in a specific pathogen-free barrier unit. Handling of mice and experimental procedures were in accordance with the requirements of Institutional Animal Care and Use Committee.
Skin single-cell suspension preparations
The trunk skin of TIEG1 WT and KO mice was incubated in 0.5% dispase (Gibco, Life Technologies, Grand Island, NY, USA) for 1 hour at 37°C after its subcutaneous fat was scraped off. Then, epidermal sheet was peeled from the dermis, cut into tiny pieces and digested in complete culture medium containing 0.01% DNase (Sigma, St Louis, MO, USA) for 1 hour at 37°C. The epidermal single-cell suspensions were harvested after filtering through a 70 μM filter. Complete culture medium was RPMI 1640 (with 2mM L-glutamine, Gibco) supplemented with 10% heat-inactivated fetal bovine serum (Hyclone, Thermo Scientific, Pittsburgh, PA, USA), 5 x 105 M 2-mercaptoethanol, 0.15% sodium hydrogencarbonate, 1 mM sodium pyruvate, nonessential amino acids, 100U per ml penicillin and 0.01% streptomycin.
Flow cytometry and antibodies
Single-cell suspensions were pretreated with anti-FcγRII/III (clone 2.4G2) for 10 mins at 4°C, and then stained for surface and intracellular markers with the conjugated monoclonal antibodies listed as below: MHC-II (I-A/E) (M5/114.15.2), CD45.2 (104), CD11c (N418), CD80 (16-10A1), CD86 (GL1) and CD207 (929F3.01). All antibodies were purchased from eBioscience (San Diego, CA, USA) or Dendritics (Lyon, France). Cells were analyzed with FACS Aria II (BD Biosciences, San Jose, CA, USA) and FlowJo software version 7.6.1 for Microsoft (TreeStar, Sam Carlos, CA, USA).
Phagocytosis assay
Freshly-isolated epidermal cells were incubated with 0.025% Dextran-Fluorescein Isothiocyanate (FITC) (Life Technologies, Grand Island, NY, USA) for 45 mins at 37°C or 4°C. Then, cells were washed by phosphate buffered saline (PBS), and subsequently stained with anti-CD45.2 and MHC-II, the percentage of LCs that uptake antigen (CD45.2+/MHC-II+/FITC+) was determined by flow cytometry.
In vitro maturation
Freshly-isolated epidermal cells were cultured with complete culture medium at 37°C for 60 hours. The cells were then collected and stained with anti-MHC-II, CD207, CD45.2, CD80 and CD86 for flow cytometric analysis.
Hapten sensitization and elicitation of CHS
On day 0, mice were sensitized by applying 25μl of 0.5% DNFB (Sigma-Aldrich, St. Louis, MO, USA) (acetone:olive oil = 4:1) on shaved belly skin. On day 5, sensitized mice were challenged topically with 10μl of 0.2% DNFB on the left ear, whereas 10μl of acetone/olive oil (4:1) was painted on the right ear. Ear thicknesses were measured by comparing challenged (left) and unchallenged (right) ears using a thickness gauge (Digimatic caliper, Mitutoyo, Japan) in a blinded manner. And, ear thickness increases were calculated by subtracting pre-challenge (0 hour) from post-challenge measurements (24 hours, 48 hours, 72 hours, 96 hours).
LC replenishment under skin inflammation
For the depletion of LCs, mice were exposed to ultraviolet (UV) for 15 minutes (wavelength 254 nm, voltage 8W, source: 38 cm). The replenishment of the LC network was assessed 1 week after UV treatment.
Statistical analysis
Data were presented as mean ± standard deviation (SD). Statistics analyses were performed with GraphPad Prism software (GraphPad, San Diego, CA, USA) using a two-tailed Student t test. Differences were considered to be statistically significant when P < 0.05.
ACKNOWLEDGMENTS
The authors thank Matthew Weiland for maintaining mouse colonies. This study is supported by the Henry Ford Health System Research Grant for the Immunology Program (T71016 to LZ) and National Science Foundation of China (81602751).
CONFLICTS OF INTEREST
The authors have declared no conflicting interests.
REFERENCES
1. Valladeau J, Ravel O, Dezutter-Dambuyant C, Moore K, Kleijmeer M, Liu Y, Duvert-Frances V, Vincent C, Schmitt D, Davoust J, Caux C, Lebecque S, Saeland S. Langerin, a novel C-type lectin specific to Langerhans cells, is an endocytic receptor that induces the formation of birbeck granules. Immunity. 2000; 12:71–81.
2. Hoeffel G, Wang Y, Greter M, See P, Teo P, Malleret B, Leboeuf M, Low D, Oller G, Almeida F, Choy SH, Grisotto M, Renia L, et al. Adult Langerhans cells derive predominantly from embryonic fetal liver monocytes with a minor contribution of yolk sac-derived macrophages. J Exp Med. 2012; 209:1167–1181.
3. Ghigo C, Mondor I, Jorquera A, Nowak J, Wienert S, Zahner SP, Clausen BE, Luche H, Malissen B, Klauschen F, Bajenoff M. Multicolor fate mapping of Langerhans cell homeostasis. J Exp Med. 2013; 210:1657–1664.
4. Sere K, Baek JH, Ober-Blobaum J, Muller-Newen G, Tacke F, Yokota Y, Zenke M, Hieronymus T. Two distinct types of Langerhans cells populate the skin during steady state and inflammation. Immunity. 2012; 37:905–916.
5. Mutyambizi K, Berger CL, Edelson RL. The balance between immunity and tolerance: the role of Langerhans cells. Cell Mol Life Sci. 2009; 66:831–840.
6. Igyarto BZ, Jenison MC, Dudda JC, Roers A, Müller W, Koni PA, Campbell DJ, Shlomchik MJ, Kaplan DH. Langerhans cells suppress contact hypersensitivity responses via cognate CD4 interaction and langerhans cell-derived IL-10. J Immunol. 2009; 183:5085–93.
7. Elentner A, Finke D, Schmuth M, Chappaz S, Ebner S, Malissen B, Kissenpfennig A, Romani N, Dubrac S. Langerhans cells are critical in the development of atopic dermatitis-like inflammation and symptoms in mice. J Cell Mol Med. 2009; 13:2658–2672.
8. Cunningham AL, Abendroth A, Jones C, Nasr N, Turville S. Viruses and Langerhans cells. Immunol Cell Biol. 2010; 88:416–423.
9. Zhang X, Gu J, Yu FS, Zhou L, Mi QS. TGF-beta1-induced transcription factor networks in Langerhans cell development and maintenance. Allergy. 2016; 71:758–764.
10. Borkowski TA, Letterio JJ, Farr AG, Udey MC. A role for endogenous transforming growth factor beta 1 in Langerhans cell biology: the skin of transforming growth factor beta 1 null mice is devoid of epidermal Langerhans cells. J Exp Med. 1996; 184:2417–2422.
11. Kaplan DH, Li MO, Jenison MC, Shlomchik WD, Flavell RA, Shlomchik MJ. Autocrine/paracrine TGFbeta1 is required for the development of epidermal Langerhans cells. J Exp Med. 2007; 204:2545–2552.
12. Bobr A, Igyarto BZ, Haley KM, Li MO, Flavell RA, Kaplan DH. Autocrine/paracrine TGF-beta1 inhibits Langerhans cell migration. Proc Natl Acad Sci USA. 2012; 109:10492–10497.
13. Kel JM, Girard-Madoux MJ, Reizis B, Clausen BE. TGF-beta is required to maintain the pool of immature Langerhans cells in the epidermis. J Immunol. 2010; 185:3248–3255.
14. Mohammed J, Beura LK. Stromal cells control the epithelial residence of dcs and memory t cells by regulated activation of TGF-beta. 2016; 17:414–421.
15. Xu YP, Shi Y, Cui ZZ, Jiang HH, Li L, Wang XF, Zhou L, Mi QS. TGFbeta/Smad3 signal pathway is not required for epidermal Langerhans cell development. J Invest Dermatol. 2012; 132:2106–2109.
16. Li G, Gao XH, Mi QS. TGF-β1-Smad signaling pathways are not required for epidermal LC homeostasis. Oncotarget. 2016; 7:15290–91. https://doi.org/10.18632/oncotarget.8167.
17. Subramaniam M, Harris SA, Oursler MJ, Rasmussen K, Riggs BL, Spelsberg TC. Identification of a novel TGF-beta-regulated gene encoding a putative zinc finger protein in human osteoblasts. Nucleic Acids Res. 1995; 23:4907–4912.
18. Fautsch MP, Vrabel A, Rickard D, Subramaniam M, Spelsberg TC, Wieben ED. Characterization of the mouse TGFbeta-inducible early gene (TIEG): conservation of exon and transcriptional regulatory sequences with evidence of additional transcripts. Mamm Genome. 1998; 9:838–842.
19. Spittau B, Krieglstein K. Klf10 and Klf11 as mediators of TGF-beta superfamily signaling. Cell Tissue Res. 2012; 347:65–72.
20. Yajima S, Lammers CH, Lee SH, Hara Y, Mizuno K, Mouradian MM. Cloning and characterization of murine glial cell-derived neurotrophic factor inducible transcription factor (MGIF). J Neurosci. 1997; 17:8657–8666.
21. Hsu CF, Sui CL, Wu WC, Wang JJ, Yang DH, Chen YC, Yu WC, Chang HS. Klf10 induces cell apoptosis through modulation of BI-1 expression and Ca2+ homeostasis in estrogen-responding adenocarcinoma cells. Int J Biochem Cell Biol. 2011; 43:666–673.
22. Subramaniam M, Hawse JR, Rajamannan NM, Ingle JN, Spelsberg TC. Functional role of KLF10 in multiple disease processes. Biofactors. 2010; 36:8–18.
23. Papadakis KA, Krempski J, Svingen P, Xiong Y, Sarmento OF, Lomberk GA, Urrutia RA, Faubion WA. Kruppel-like factor KLF10 deficiency predisposes to colitis through colonic macrophage dysregulation. Am J Physiol Gastrointest Liver Physiol. 2015; 309:G900–909.
24. Jörgl A, Platzer B, Taschner S, Heinz LX, Höcher B, Reisner PM, Göbel F, Strobl H. Human Langerhans-cell activation triggered in vitro by conditionally expressed mkk6 is counterregulated by the downstream effector RelB. Blood. 2007; 109:185.
25. Leduc C, Sobilo L, Toumi H, Mondon P, Lespessailles E, Ossant F, Kurfurst R, Pichon C. TGF-beta-induced early gene-1 overexpression promotes oxidative stress protection and actin cytoskeleton rearrangement in human skin fibroblasts. Biochim Biophys Acta. 2016; 1860:1071–1078.
26. Bae CJ, Song KD, Lee SC, Jung JH, Yun CH, Lee CK, Lee HK, Hwang DY, Lee WK. Phthalic anhydride-induced skin inflammation is augmented in KLF10-deficient mice. J Dermatol Sci. 2013; 71:221–224.
27. Toews GB, Bergstresser PR, Streilein JW. Epidermal Langerhans cell density determines whether contact hypersensitivity or unresponsiveness follows skin painting with DNFB. J Immunol. 1980; 124:445–453.
28. Kaplan DH, Jenison MC, Saeland S, Shlomchik WD, Shlomchik MJ. Epidermal Langerhans cell-deficient mice develop enhanced contact hypersensitivity. Immunity. 2005; 23:611–620.
29. Subramaniam M, Gorny G, Johnsen SA, Monroe DG, Evans GL, Fraser DG, Rickard DJ, Rasmussen K, van Deursen JM, Turner RT, Oursler MJ, Spelsberg TC. TIEG1 null mouse-derived osteoblasts are defective in mineralization and in support of osteoclast differentiation in vitro. Mol Cell Biol. 2005; 25:1191–1199.