
Introduction
Both Ca2+ and Mg2+ are critically involved in essentially every single step of cell proliferation, with cancerous cell growth representing a harmful form of deregulated proliferation. Interestingly, cancerous tissue acts as a Mg2+ trap at the expense of plasma or surrounding tissues, suggesting significant uptake by cancer cells [1-3]. The precise mechanisms by which divalent cations regulate cell proliferation remain to be elucidated, but recent evidence indicates critical involvement of the Ca2+- and Mg2+-transporting transient receptor potential melastatin-related ion channels TRPM7 and TRPM6 [4-6]. TRPM7 and TRPM6 are channel kinases possessing both a Ca2+/Mg2+-permeable ion channel pore and a carboxyl-terminal atypical α-kinase [4,7,8]. The two channels are highly analogous in many aspects, such as channel selectivity [9,10], current-voltage relationship, as well as modulation by acidic pH [7,9,10], extracellular divalent cations [9,10], and PIP2[11,12]. However, recently, they were shown to be differentially regulated by the channel blocker waixenicin A [13], 2-aminoethoxydiphenyl borate (2-APB) [9,14], intracellular halides [15], as well as intracellular Mg2+ and ATP [14].
While TRPM7 is ubiquitously expressed and the native TRPM7-like current MagNuM (Mg2+-nucleotide-regulated metal ion current [7]) is measurable in virtually all cell types examined [4,16], native currents of the more limitedly expressed TRPM6 have not yet been reported. TRPM7 channels are indispensable for Mg2+ homeostasis at both cellular and whole organism levels [17,18]. In contrast, TRPM6 is believed to be responsible for systemic Mg2+ regulation by mediating Mg2+ (re)absorption, as a mutation in the TRPM6 gene leads to an autosomal recessive form of familial hypomagnesemia with secondary hypocalcemia [19,20]. Consistent with its physiological function, TRPM6 expression appears to be relatively tissue-restricted, predominantly in the absorptive epithelia in the colon and kidney [6,19,20]. However, it is unknown whether TRPM6 is aberrantly expressed in other tissues or cells, possibly in a pathophysiological context.
Neuroblastoma, arising in tissues of the sympathetic nervous system such as sympathetic ganglia and the adrenal medulla, is the most common extracranial solid tumor of childhood and accounts for around 15% of all pediatric cancer deaths [21-23]. A factor that is strongly associated with advanced high-risk neuroblastoma and predicts poor outcome is amplification and concomitant high expression of MYCN [24,25], an oncogene encoding N-Myc, a nuclear phosphoprotein in the Myc family of helix-loop-helix transcription factors [26]. Enhanced expression of the oncoprotein N-Myc regulates a large number of genes, disrupting the cell cycle exit and terminal differentiation of neuroblasts and hence promoting neuroblastoma pathogenesis [27,28]. Given the role of Mg2+ in cell proliferation, the present study carefully examined two Mg2+-transporting channel kinases, TRPM7 and TRPM6, in neuroblastoma cells. Our results reveal that TRPM7 is essential for neuroblastoma proliferation and the additional expression of TRPM6 modulates the phenotype of native MagNuM currents, favoring divalent cation transport mediated by TRPM6 and TRPM7 and promoting neuroblastoma cell proliferation.
Results
N-Myc regulates expression of TRPM6 and TRPM7 in human SHEP-21N neuroblastoma
We set out to examine the expression level of the Mg2+-influx channels TRPM6 and TRPM7 in the largest publicly available neuroblastoma expression profiling dataset, the Kocak-649 cohort [29]. On the basis of their MYCN gene copy number status, these tumor samples can be divided into MYCN-amplified (n=93) or non-MYCN-amplified (n=550) tumors. We first examined the expression level of N-Myc in these two groups and found that the expression level is, as expected, significantly higher in the MYCN-amplified samples (p = 1.5 x 10-51, Fig. 1A). Interestingly, TRPM7 mRNA expression was found in all tumor samples, and was significantly correlated with both MYCN amplification (p = 2.3 x 10-7, Fig. 1B) and mRNA expression (p = 3.5 x 10-3 in a 2logPearson test). No significant correlations were found for TRPM6 (Fig. 1C), most likely due to the small numbers of tumors with significant TRPM6 expression (average TRPM6 expression was 8-times lower than that of TRPM7). In order to investigate the role of N-Myc in TRPM7/TRPM6 regulation, we chose the SHEP-21N cell line, a clone derived from the SHEP-2 neuroblastoma, in which N-Myc is constitutively expressed but can be experimentally repressed [30]. These cells contain a MYCN trans-gene under the control of a tetracycline-responsive repressor element, so that tetracycline exposure turns off N-Myc expression. Quantitative RT-PCR (qRT-PCR) analysis showed that SHEP-21N cells without N-Myc expression had basal expression of TRPM7 and TRPM6, which was considerably enhanced by N-Myc up-regulation (Figs. 1D-F). Induction of TRPM6 and TRPM7 expression by N-Myc was significant for both channel kinases, but higher for TRPM6 (2.1-fold for TRPM6 and 1.5-fold for TRPM7). This suggests that N-Myc up-regulates both TRPM genes and concomitantly increases the ratio of TRPM6 over TRPM7.
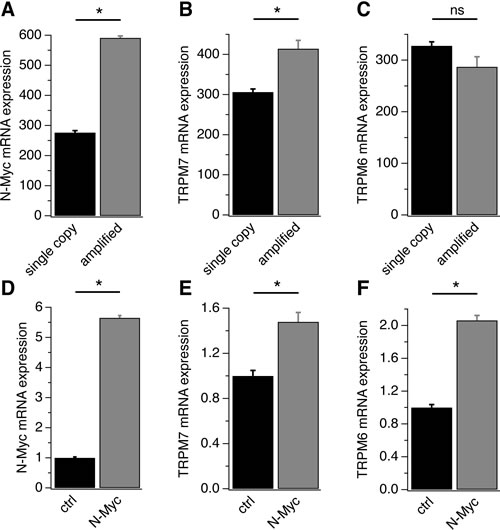
Figure 1: TRPM6 and TRPM7 correlation with MYCN in neuroblastoma. A-C, N-Myc, TRPM7 and TRPM6 mRNA expression correlation with MYCN amplification in the Kocak-649 cohort. Microarray analysis of N-Myc, TRPM7 and TRPM6 mRNA expression in Kocak-649 (GSE45547), the largest neuroblastoma cohort in the public domain. The graphs present N-Myc (A), TRPM7 (B) and TRPM6 (C) expression in tumors without (n=550) and with (n=93) MYCN amplification. Y-axes represent sample ranks in a non-parametric Kruskal-Wallis t test; actual mean ± s.e.m. expression values were: 17,839 ± 1,137 (MYCN), 104.9 ± 3.7 (TRPM6), 807.0 ± 321.1 (TRPM7). Both N-Myc and TRPM7 expression are significantly higher in tumors with MYCN amplification (p=1.5 • 10-51 and p=2.3 • 10-7 respectively; Kruskal-Wallis t test). D-F, qRT-PCR analysis of N-Myc (D), TRPM7 (E) and TRPM6 (F) expression levels in the SHEP-21N cell line where MYCN transgene is controlled by tetracycline, i.e., N-Myc expression is repressed in the presence of tetracycline (control), but removal of tetracycline induces N-Myc expression (N-Myc). The graphs show normalized N-Myc, TRPM7 and TRPM6 expression in SHEP-21N cells with (N-Myc, n = 9) or without (control, n = 9) N-Myc expression. *, p<0.01.
Endogenous MagNuM currents in SHEP-21N are mediated by TRPM6 and TRPM7
Heterologous expression of heteromeric TRPM7/TRPM6 produces an outwardly rectifying current whose current-voltage relationship is indistinguishable from homomeric TRPM7 or TRPM6 currents [9,14]. Patch-clamp recordings in SHEP-21N cells also revealed such currents (Figs. 2A, B). Importantly, SHEP-21N cells exhibited significantly larger current amplitudes when they expressed N-Myc (Figs. 2A, B), consistent with the observed N-Myc-induced increases in TRPM7 and TRPM6 mRNA levels (Figs. 1E, F). Employing specific small interfering RNA (siRNA) against TRPM7 and TRPM6 (Figs. 3A, B), we found that MagNuM currents were significantly suppressed upon TRPM6 knockdown, and both knockdown of TRPM7 or TRPM7/TRPM6 combined nearly abolished any endogenous MagNuM currents (Figs. 3C-F). These results demonstrate that N-Myc expression produces significantly larger overall MagNuM currents and suggest that, independent of N-Myc levels, endogenous MagNuM currents in SHEP-21N cells are largely composed of TRPM7 with a lesser, yet significant, contribution of TRPM6.
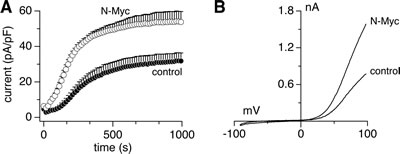
Figure 2: Endogenous MagNuM currents are increased by N-Myc upregulation. Currents were measured in divalent-free internal solution with 5 mM EGTA and 5 mM EDTA. A, whole-cell current development in SHEP-21N cells expressing (N-Myc, n = 11) or not (control, n = 10). B, corresponding representative ramp currents (I-V curves) extracted at 1,000 s from both groups.
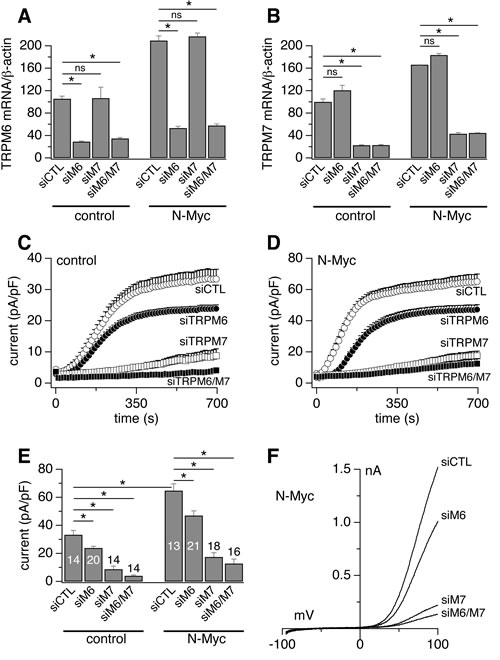
Figure 3: Endogenous MagNuM currents are mediated by TRPM7 and TRPM6. Currents were measured in divalent-free internal solution with 5 mM EGTA and 5 mM EDTA. A-B, siRNAs efficacy measured by qRT-PCR analysis of TRPM6 (A) and TRPM7 (B) expression levels in the SHEP-21N cell line under both control and N-Myc upregulation conditions (n = 9). C, MagNuM currents in control SHEP-21N cells treated with negative non-silencing (siCTL) or specific siRNA sequences against TRPM7, TRPM6, TRPM7&TRPM6. D, current measurement in N-Myc-expressing SHEP-21N cells treated with negative (siCTL) or specific siRNA sequences against TRPM7, TRPM6, TRPM7&TRPM6. E, statistical summary of current amplitudes at 700 s as in c-d (*, p<0.01). The number of patched cells is indicated in graph. F, typical current traces evoked by voltage ramps (I-V curves) in N-Myc-expressing SHEP-21N cells as in D.
N-Myc expression shapes the phenotype of native TRPM7/TRPM6 currents
Heteromeric TRPM7/TRPM6 channels behave differently from both homomeric TRPM7 and homomeric TRPM6. In heterologous expression systems, 2-APB suppresses TRPM7 currents, potentiates TRPM6 currents, and leaves heteromeric TRPM7/TRPM6 largely unaffected, indicating that incorporation of TRPM6 renders the TRPM7/TRPM6 heteromer less sensitive to 2-APB [9,14]. Indeed, N-Myc induction attenuated the inhibitory effects of 2-APB on the MagNuM currents in SHEP-21N cells (Figs. 4A, B). Since homomeric TRPM6 channels are strongly suppressed by free intracellular Mg2+, heteromeric TRPM7/TRPM6 channels exhibit increased sensitivity of Mg2+-mediated suppression relative to TRPM7. This has been shown to be due to cross-phosphorylation of TRPM7 by the TRPM6 kinase domain [14]. Consistent with this mechanism, the dose-response curves for intracellular free Mg2+ in SHEP-21N cells revealed that N-Myc expression and the resulting up-regulation of TRPM6 considerably enhanced the sensitivity of native currents to Mg2+, as indicated by a decrease in IC50 from 425 µM to 236 µM (Figs. 4D-F). Finally, and physiologically most relevant, intracellular Mg·ATP strongly suppresses heterologously expressed TRPM7 channels [7], but has no inhibitory effect on either TRPM6 or TRPM7/TRPM6 currents [14]. Accordingly, N-Myc expression in SHEP-21N cells significantly reduced Mg·ATP-mediated suppression of endogenous MagNuM currents (Fig. 4C). Thus N-Myc expression caused changes in the behavior of native MagNuM currents in SHEP-21N cells with respect to 2-APB, intracellular free Mg2+ and Mg·ATP that resemble the channel phenotype of overexpressed TRPM7/TRPM6 heteromers [14] and consequently would favor enhanced constitutive activity of these channels at rest.
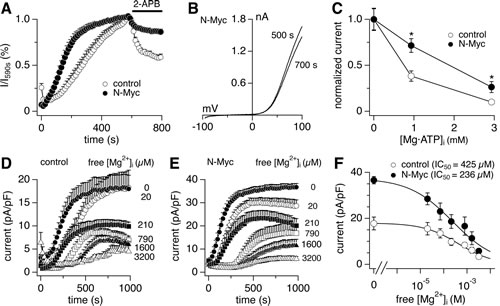
Figure 4: N-Myc expression shapes the phenotype of native TRPM7/TRPM6 currents. Whole-cell MagNuM currents were measured in SHEP-21N cells treated with tetracycline (control) or not (N-Myc) and peak outward currents at +80 mV were analyzed. A, inhibition of MagNuM current by 200 µM 2-APB (control, n = 10; N-Myc, n = 7), normalized to current amplitude just prior to 2-APB application. B, representative currents evoked by voltage ramps (I-V curves) were derived from N-Myc-expressing cells before (500 s) and after (700 s) 2-APB application. C, ATP sensitivity of currents (n = 7-13 cells for each point; *, p<0.01). Mg·ATP concentrations were fixed at 1 or 3 mM with an internal free Mg2+ concentration of 264 µM. Currents were normalized against the control condition containing no Mg·ATP internally. D, E, current measurements in control and N-Myc-expressing cells perfused with intracellular solutions of defined free Mg2+ concentrations as labeled in the graph (control, n = 5-6 for each Mg2+ concentration; N-Myc, n = 5-7 for each Mg2+ concentration). Free Mg2+ concentration in internal solution was clamped to the indicated levels with 10 mM EGTA. F, peak current amplitudes derived from D and E as a function of free Mg2+ concentration. Dose-response fit rendered IC50 values of 425 µM and 236 µM for control and N-Myc-expressing cells, respectively.
Suppression of TRPM7/TRPM6 inhibits proliferation of neuroblastoma
We performed knockdown experiments using siRNA as described above to investigate the relative role of both TRPM6 and TRPM7 channels in SHEP-21N cell proliferation. N-Myc induction following tetracycline withdrawal promoted cell proliferation compared to tetracycline-treated SHEP-21N cells without N-Myc expression (Figs. 5A, B). In either group, the additional siRNA-mediated knockdown of TRPM7, TRPM6 or TRPM7/TRPM6 expression markedly inhibited SHEP-21N cell proliferation (Figs. 5A, B). Notably, knockdown of TRPM6 alone strongly inhibited cell proliferation, an effect that was comparable to that seen with TRPM7 suppression (Figs. 5A, B). Given that TRPM6 knockdown only causes moderate reduction in total MagNuM currents recorded under divalent-free intracellular solutions (Figs. 3C, D), this indicates that TRPM6 may play a vital role in promoting cell proliferation under physiological conditions by enhancing the constitutive activity of MagNuM currents.
Since TRPM6 and TRPM7 are permeable to both Ca2+ and Mg2+ and both divalent cations can regulate cell proliferation, we assessed the role of Ca2+ and Mg2+ in SHEP-21N proliferation assays. We first established a dose-response relationship of Ca2+ and Mg2+ by incubating SHEP-21N cells in media with Ca2+ and Mg2+ concentrations adjusted in the range of 0.4 mM to 10 mM (while the congeneric divalent remained at 0.4 mM). Increasing Mg2+ up to 10 mM had no effect on SHEP-21N cell proliferation regardless of N-Myc expression (Fig. 5C). Increasing Ca2+ levels up to 1.8 mM had no significant effect on N-Myc-expressing cells, but yielded a slight increase in cell numbers in cells without N-Myc expression (Fig. 5D). At higher concentrations of Ca2+, cell proliferation started to decline in both cell populations. For this reason, we assessed possible compensatory effects of divalent supplementation on cell proliferation in cells in which TRPM6, TRPM7, or both, were knocked down with siRNA using concentrations of 10 mM Mg2+ and 1.8 mM Ca2+ (Fig. 5B). As illustrated in Fig. 5B, 10 mM Mg2+ had no effect on cell proliferation, indicating the low abundance or absence of alternative Mg2+ uptake mechanisms in these cells. Indeed, qRT-PCR analysis of SLC41A1, a Mg2+ transporter that has been found to rescue TRPM7-deficient DT40 cells from growth arrest [17,31], revealed that its mRNA levels were 123/24-fold less expressed than those of TRPM7/TRPM6, respectively (Figs. 5E and 1E, F). Importantly, SLC41A1 mRNA levels did not change significantly upon N-Myc induction. Interestingly, Ca2+ supplementation to 1.8 mM partially rescued the growth-arrest phenotype by causing a slight increase in cell numbers under all experimental conditions, except in N-Myc-expressing SHEP-21N cells in which TRPM6 and TRPM7 were left uninhibited and which were already strongly proliferating. However, neither cation was able to decisively rescue the growth suppression imposed by the knockdown of either channel. Thus, TRPM7, particularly in combination with TRPM6, appears to represent the major Ca2+ and/or Mg2+ accumulation mechanisms in SHEP-21N cells.
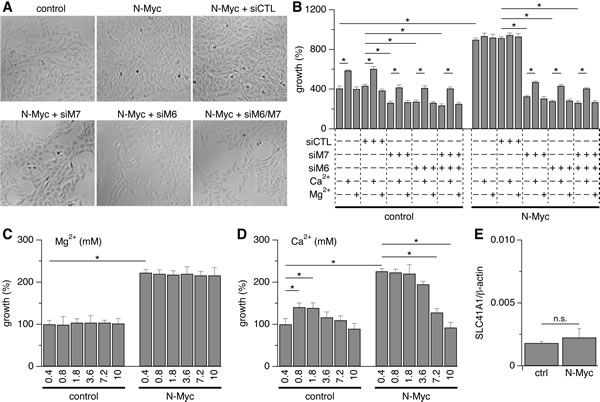
Figure 5: siRNA, Ca2+ and Mg2+ supplementation and TRPM7/M6-dependent cell proliferation. A, representative images of SHEP-21N cells treated with siRNA as indicated. Tetracycline was added (control) or not (N-Myc) to repress or induce N-Myc expression, respectively. siCTL = negative control siRNA; siM6 = siRNA against TRPM6; siM7 = siRNA against TRPM7. B, statistical analysis of cell growth in SHEP-21N cells treated with the indicated siRNA (n = 6; *, p<0.01). Cells were transfected with siRNA, seeded, treated or not treated with Mg2+ or Ca2+ on day 1, and analyzed on day 3 after seeding. C-D, analysis of cell growth in SHEP-21N cells treated for 2 days with the indicated Mg2+ (C) or Ca2+ (D) concentrations (n = 6). E, qRT-PCR analysis of SLC41A1 transcripts in both control and N-Myc-expressing SHEP21N cells (n = 6; n.s., not significant).
Discussion
While TRPM6 was initially discovered in the absorptive epithelia in the kidney and intestine [5,6], the present study now demonstrates that the protein is also expressed in SHEP-21N neuroblastoma. TRPM6 expression in SHEP-2 and SHEP-21N is not serendipitous, as data mining of Kocak-649 and other public neuroblastoma cohort expression profiles showed very similar expression levels and patterns: TRPM7 mRNA expression is ubiquitous and TRPM6 mRNA is much lower, and present in only ~10% of samples (results not shown). Remarkably, SHEP-21N cells expressed TRPM6 at a sufficiently high level that endogenous TRPM6-dependent currents could be measured for the first time. The central observation made in the present study was that the enhanced proliferation phenotype mediated by N-Myc expression (Figs. 1 and 5A, B) was paralleled by increased TRPM6 and TRPM7 expression levels (Figs. 1E, F), as well as by enlarged endogenous MagNuM currents (Figs. 2A, B). The phenotypic characteristics of the resulting currents are consistent with heteromeric TRPM6/TRPM7 channels (Fig. 4). Molecular suppression of either channel through siRNA (Figs. 3C-F and 5A, B) completely suppressed both the N-Myc-enhanced MagNuM currents and the growth phenotype (Figs. 3 and 4).
Even though both TRPM6 and TRPM7 also possess kinase activity in addition to their channel function, TRPM6 has no known phosphorylation substrates other than TRPM7 [32] and the currently known TRPM7 substrates, annexin A1 and myosin IIA heavy chain [33,34], are not likely to determine cell proliferation. This indicates that enhanced ion transport activities are in part responsible for the growth phenotype observed in N-Myc-expressing SHEP-21N cells. Our results are therefore consistent with the hypothesis that N-Myc enhances TRPM6 expression, which in turn promotes Ca2+ and Mg2+ transport across the plasma membrane by enhancing the overall activity of TRPM6/TRPM7 heteromeric channel assemblies, and thereby promotes cell proliferation.
The expression patterns of TRPM6 and TRPM7 in MYCN-amplified neuroblastoma cell lines and SHEP-21N cells suggests that their expression might not necessarily relate to N-Myc expression, as some MYCN-amplified cell lines had low TRPM6 expression levels (data not shown). Nevertheless, in SHEP-21N cells, N-Myc is able to elevate basal TRPM6/7 expression upon induction. In addition, in the largest publicly available neuroblastoma expression profiling dataset, the Kocak-649 cohort [29], TRPM7 mRNA expression is found in all tumor samples, and is significantly correlated to N-Myc amplification. No significant correlations were found for TRPM6, most likely due to the small numbers of tumors with significant TRPM6 expression. Future investigations might reveal oncogene-dependent increases of TRPM6 expression in other cells that already express some basal levels of TRPM6.
We previously demonstrated in heterologous expression systems that TRPM7/TRPM6 channel heteromers, when co-expressed at a stoichiometric ratio of ~1:1, exhibit unique phenotypes that are characterized by insensitivity to 2-APB, lack of modulation by Mg·ATP and increased sensitivity to Mg2+ inhibition compared with homomeric TRPM7 [14]. The insensitivity of heteromeric TRPM7/TRPM6 channels to cytosolic ATP leaves the channel function largely unaffected by cellular energy status and augmented Mg2+ inhibition indicates an enhanced negative feedback loop, a common regulatory mechanism for many ion channels, such as CRAC and TRPV5/TRPV6 [35,36]. These distinctive features of heteromeric TRPM7/TRPM6 channels are primarily controlled by cross-phosphorylation of TRPM7 by the TRPM6 kinase domain [32] and will determine the functional activity of the channel complex as a Ca2+ and Mg2+ influx conduit under physiological circumstances. Consistent with the findings in TRPM7/TRPM6 overexpression systems, the native currents in SHEP-21N cells expressing N-Myc were less inhibited by 2-APB, less suppressed by Mg·ATP, and more sensitive to Mg2+ inhibition compared to the currents in cells without N-Myc expression (Fig. 4). Thus, N-Myc shaped the MagNuM phenotype towards that of heteromeric TRPM7/TRPM6 by increasing the relative amount of TRPM6.
It is noteworthy that the extent to which the MagNuM phenotype was altered in native SHEP-21N cells is slightly less pronounced than that previously observed in a TRPM7/TRPM6 overexpression system, where the ratio of TRPM6/TRPM7 was ~1 [14,32], suggesting that the ratio of TRPM6 over TRPM7 within channel complexes in SHEP-21N cells is lower than 1. This would be consistent with the observed mRNA levels (Figs. 1E, F) and also be consistent with the observation that TRPM6 suppression downregulated the MagNuM currents by 29%, whereas knockdown of TRPM7 reduced current amplitudes by 74% (Fig. 4). Nevertheless, the impact of TRPM6 knockdown on cell proliferation was as severe as that of TRPM7, suggesting that relatively small increases in TRPM6 expression can significantly alter the MagNuM current behavior under physiological conditions. Since TRPM6/TRPM7 heteromers are less sensitive to Mg·ATP and slightly more sensitive to Mg2+ inhibition (Fig. 4; [14]), TRPM6-containing heteromeric channels in SHEP-21N would likely be less suppressed under physiological conditions, as the most dominant physiological regulator of MagNuM is Mg·ATP [7]. As a result, these heteromeric channels would be more effective in supporting proliferation than TRPM7 homomers.
TRPM7 and TRPM6 conduct divalent cations with preferential transport of Mg2+ over Ca2+. Consistent with this notion is that TRPM6 plays a well-documented role in Mg2+ absorption/reabsorption in epithelia of colon and kidney [6,19,20] and TRPM7 has been found to be critical for cellular Mg2+ homeostasis [17,18]and a critical factor for cell proliferation [13,37,38]. The presence and regulation of TRPM6 in SHEP-21N cells thus would suggest that this channel may also serve a role in the pathological context of neuroblastoma by regulating cellular Mg2+ levels in conjunction with TRPM7. However, the role of Ca2+ transport through TRPM6 and/or TRPM7 may not be insignificant, particularly in cells in which Ca2+ is an important regulator of cell proliferation. Thus, it has been suggested that Ca2+ influx through TRPM7 may be critical for fibroblast proliferation [39] and a recent study on prostate cancer cells proposed that enhanced Ca2+ influx through TRPM7 determined their proliferation rate [40].
Our Ca2+ and Mg2+ supplementation experiments revealed that increasing extracellular Mg2+ from 0.4 to 10 mM had no effect on proliferation regardless of N-Myc expression in SHEP-21N cells (Fig. 5), whereas Ca2+ had a slight facilitatory effect up to 1.8 mM. Higher concentrations of Ca2+ reduced cell proliferation. Extracellular Mg2+ can be taken up by mammalian cells through Mg2+-permeable channels, including TRPM7 and TRPM6 [5], and/or Mg2+ transporter, such as SLC41A1 [41]. Genetic ablation of TRPM7 in DT40 cells induces growth arrest, and supplementation with high extracellular Mg2+ (10 mM), but not Ca2+, rescues the growth arrest [42], a compensatory effect partly mediated by the Mg2+ transporter SLC41A1 that is natively expressed in DT40 cells [31]. However, Mg2+ supplementation was completely ineffective in overcoming the growth arrest of SHEP-21N cells in which either TRPM6, TRPM7, or both were knocked down with siRNA (Fig. 5B), suggesting that TRPM6/TRPM7 channels, rather than SLC41A1, constitute the major Mg2+ uptake mechanism in these cells. An alternative explanation could be that Mg2+ may not be the limiting factor for cell proliferation in these cells and the more relevant cation might be Ca2+. Indeed, supplementation of Ca2+ (1.8 mM) in the extracellular solution slightly enhanced proliferation, although not in N-Myc-expressing SHEP-21N cells. The reason for this could be that these SHEP-21N cells already proliferated at maximal rates. It remains to be determined whether the facilitatory effect of Ca2+ is mediated by Ca2+ influx through TRPM6/TRPM7 channels or represents an alternative Ca2+ influx pathway that offsets a Mg2+-dependent growth impairment.
The data presented in this study suggest that TRPM7 channels have a significant impact on cell proliferation of neuroblastoma, regardless of whether or not they express N-Myc, whereas additional TRPM6 expression further synergizes with TRPM7 in augmenting the proliferative activity. The relevance of TRPM7 for cell proliferation is also evident in other cancers, as genetic suppression of TRPM7 can inhibit the proliferation of human head and neck squamous carcinoma (HNSCC) [37] and ascending aortic vascular smooth muscle cells (VSMC) [38]. Pharmacological inhibition of TRPM7 by the channel blocker waixenicin A has also been shown to inhibit cell proliferation of rat basophilic and human Jurkat T-cell leukemia cells [13]. This makes Ca2+/Mg2+-transporting channels attractive candidates as pharmacological targets for potential therapeutic drugs. Collectively, genetic or pharmacological suppression of TRPM7/TRPM6 channels inhibits cell proliferation, underscoring the importance of these channels in promoting proliferation and pointing towards a potential therapeutic avenue for the treatment of neuroblastoma.
Materials and Methods
Cell culture and microarray analysis
Neuroblastoma cell line and culture conditions were as in [28,43]. N-Myc expression in SHEP-21N [28,30] was inhibited by the addition of tetracycline (100 ng/ml) and incubation for at least 48 hours. The Agilent-020382 Human Custom Microarray 44k data for Kocak-649 was described in [29]. Expression data for this dataset were retrieved from the public Gene Expression Omnibus (GEO) dataset on the NCBI website (GEO ID: GSE45547). The TranscriptView genomic analysis and visualization tool was used to confirm the anti-sense position in a (posterior) exon of the probe-sets. The Agilent probe-sets MYCN 24_P94402 (MYCN), 24_P410463 (TRPM6), and 23_P88470 (TRPM7), that fulfilled these criteria, and in addition showed the highest sensitivity for detection of the target gene, were selected. Alternative correct probe-sets did not yield conflicting results. All analyses were performed using R2 (http://r2.amc.nl).
Electrophysiology and solutions
Whole-cell patch-clamp experiments were performed at room temperature (20-25 °C). High-resolution whole-cell currents were recorded by EPC-9 (HEKA, Bellmore, NY) and Patchmaster v2.4 (HEKA). Unless otherwise stated, all voltages were corrected for a liquid junction potential of 10 mV. Patch pipettes pulled from borosilicate glass had resistances of 2.5-3.5 MΩ when filled with internal solutions. Cells were held at 0 mV holding potential, with voltage ramps of 50 ms duration spanning the voltage range of -100 mV to +100 mV delivered at a rate of 0.5 Hz following the establishment of whole-cell configuration. The currents were filtered at 2.9 kHz and digitized at 10 kHz. SHEP-21N cells plated on coverslips were bathed in Mg2+-free external solution containing (in mM) 140 NaCl, 2.8 KCl, 1 CaCl2, 10 HEPES, 11 Glucose, pH 7.4 adjusted with NaOH. The divalent-free internal recording solution was composed of (in mM): 140 Cs-glutamate, 8 NaCl, 5 Cs-EDTA, 5 Cs-EGTA, 10 Cs-HEPES, pH 7.2 adjusted with CsOH. When needed, internal free Mg2+ was clamped by 10 mM EGTA to various levels, as calculated by WebMaxC (http://maxchelator.stanford.edu).
RNA extraction and quantitative real-time PCR (qRT-PCR)
Quantitative real-time PCR (qRT-PCR) was used to examine the mRNA expression levels of TRPM6 and TRPM7 using β-actin for normalization. Total RNA (1 µg) was extracted from SHEP-21N cells treated with or without tetracycline using the RNeasy Mini kit (Qiagen, Valencia, CA). Conversion of mRNA to cDNA was achieved using random priming by ABI’s High Capacity cDNA RT Kit with RNase Inhibitor (Applied Biosystems, Life Technologies, Foster City, CA). The qRT-PCR was performed using the ABI HT7900 FAST Real-Time PCR System (Applied Biosystems) and ABI POWER SYBRGreen (Applied Biosystems) according to the manufacturers’ protocols. Gene-specific primer pairs of human MYCN (QT00201404) human TRPM6 (QT00043456), TRPM7 (QT00082425) and β-actin (QT01680476) were purchased from Qiagen.
RNA interference and cell proliferation assay
The small interfering RNA (siRNA) duplexes specific against human TRPM6 (4392420) and TRPM7 (4390824), and a non-silencing RNA sequence (4390843) as a negative control were transiently transfected with RNAiMAX (Invitrogen) according to the manufacturer’s protocols. siRNA transfection and cell seeding were carried out simultaneously, according to the reverse transfection protocol of RNAiMAX. Briefly, siRNA at a final concentration of 20 nM, diluted in Opti-MEM (Invitrogen), was mixed with RNAiMAX in 6-well plates, followed by seeding of SHEP-21N cells at 5x104 cells/well in 6-well plates,. On day 2 after siRNA transfection, micrographs of SHEP-21N cells were taken at ×10 magnification. Currents were measured and cell numbers counted on day 3 after transfection. To count the cell number, SHEP-21N cells were trypsinized to single cells and stained with trypan blue, followed by cell counting with a hemocytometer. Each well was counted four times. The experiments were repeated as indicated in the figure legends.
Statistical analysis
Currents were analyzed with FitMaster v2.11 (HEKA) and Igor Pro (Wavemetrics, Portland, OR). Peak outward currents at +80 mV were extracted for statistical analysis. Current amplitudes were normalized to cell size upon break-in as current density (pA/pF). Dose-response curves were calculated using the function f(x) = (Ymax*(1/(1+(IC50/x)n))), where Ymax is the maximal normalized current, IC50 is the concentration at which inhibition is half maximal, x is the concentration, and n is the Hill coefficient. All data are given as mean ± standard error of mean (s.e.m.). Appropriate ANOVA, Student’s, or Kruskal-Wallis t tests, and 2logPearson correlation were performed to assess statistical significance. P values <0.05 were considered as statistically significant.
Acknowledgements
The authors would like to thank Lori Tsue and Chris Maggio for their excellent technical assistance. Funding: The study was supported by NIH Grants U54 CA143727 (R.P.) and P01 GM078195 (A.F.). Author contribution: Z.Z., M.F. design, execution, interpretation of data and manuscript preparation. J.H., D.G., H.Y. execution and interpretation of data. R.P., A.F. conception, design, interpretation of data and manuscript preparation.
Competing Interests
All authors declare no conflict of interest.
References
1. Castiglioni S, Maier JAM. Magnesium and cancer: a dangerous liason. Magnes. Res. 2011; 24:S92–100.
2. Wolf FI, Trapani V, Cittadini A. Magnesium and the control of cell proliferation: looking for a needle in a haystack. Magnes. Res. 2008; 21:83–91.
3. Wolf FI, Cittadini ARM, Maier JAM. Magnesium and tumors: ally or foe? Cancer Treat. Rev. 2009; 35:378–82.
4. Penner R, Fleig A. The Mg2+ and Mg(2+)-nucleotide-regulated channel-kinase TRPM7. Handb Exp Pharmacol. 2007; 313–28.
5. Schlingmann KP, Waldegger S, Konrad M, Chubanov V, Gudermann T. TRPM6 and TRPM7--Gatekeepers of human magnesium metabolism. Biochim Biophys Acta. 2007; 1772:813–21.
6. Van der Wijst J, Hoenderop JGJ, Bindels RJM. Epithelial Mg2+ channel TRPM6: insight into the molecular regulation. Magnes Res. 2009; 22:127–32.
7. Nadler MJ, Hermosura MC, Inabe K, Perraud AL, Zhu Q, Stokes AJ, Kurosaki T, Kinet JP, Penner R, Scharenberg AM, Fleig A. LTRPC7 is a Mg.ATP-regulated divalent cation channel required for cell viability. Nature. 2001; 411:590–5.
8. Runnels LW. TRPM6 and TRPM7: A Mul-TRP-PLIK-cation of channel functions. Curr Pharm Biotechnol. 2011; 12:42–53.
9. Li M, Jiang J, Yue L. Functional characterization of homo- and heteromeric channel kinases TRPM6 and TRPM7. J Gen Physiol. 2006; 127:525–37.
10. Monteilh-Zoller MK, Hermosura MC, Nadler MJ, Scharenberg AM, Penner R, Fleig A. TRPM7 provides an ion channel mechanism for cellular entry of trace metal ions. J Gen Physiol. 2003; 121:49–60.
11. Xie J, Sun B, Du J, Yang W, Chen H-C, Overton JD, Runnels LW, Yue L. Phosphatidylinositol 4,5-bisphosphate (PIP2) controls magnesium gatekeeper TRPM6 activity. Sci. Rep. 2011; 1:146.
12. Runnels LW, Yue L, Clapham DE. The TRPM7 channel is inactivated by PIP2 hydrolysis. Nat. Cell Biol. 2002; 4:329–36.
13. Zierler S, Yao G, Zhang Z, Kuo WC, Porzgen P, Penner R, Horgen FD, Fleig A. Waixenicin A inhibits cell proliferation through magnesium-dependent block of transient receptor potential melastatin 7 (TRPM7) channels. J. Biol. Chem. 2011; 286:39328–35.
14. Zhang Z, Yu H, Huang J, Faouzi M, Schmitz C, Penner R, Fleig A. The TRPM6 kinase domain determines the Mg·ATP sensitivity of TRPM7/M6 heteromeric ion channels. J. Biol. Chem. 2014; 289:5217–27.
15. Yu H, Zhang Z, Lis A, Penner R, Fleig A. TRPM7 is regulated by halides through its kinase domain. Cell. Mol. Life Sci. 2013; 70:2757–71.
16. Bates-Withers C, Sah R, Clapham DE. TRPM7, the Mg2+ inhibited channel and kinase. Adv. Exp. Med. Biol. 2011; 704:173–83.
17. Schmitz C, Perraud A-L, Johnson CO, Inabe K, Smith MK, Penner R, Kurosaki T, Fleig A, Scharenberg AM. Regulation of vertebrate cellular Mg2+ homeostasis by TRPM7. Cell. 2003; 114:191–200.
18. Ryazanova LV, Rondon LJ, Zierler S, Hu Z, Galli J, Yamaguchi TP, Mazur A, Fleig A, Ryazanov AG. TRPM7 is essential for Mg2+ homeostasis in mammals. Nat. Commun. 2010; 1:109.
19. Schlingmann KP, Weber S, Peters M, Niemann Nejsum L, Vitzthum H, Klingel K, Kratz M, Haddad E, Ristoff E, Dinour D, Syrrou M, Nielsen S, Sassen M, et al. Hypomagnesemia with secondary hypocalcemia is caused by mutations in TRPM6, a new member of the TRPM gene family. Nat Genet. 2002; 31:166–70.
20. Walder RY, Landau D, Meyer P, Shalev H, Tsolia M, Borochowitz Z, Boettger MB, Beck GE, Englehardt RK, Carmi R, Sheffield VC. Mutation of TRPM6 causes familial hypomagnesemia with secondary hypocalcemia. Nat Genet. 2002; 31:171–4.
21. Maris JM. Recent advances in neuroblastoma. N. Engl. J. Med. 2010; 362:2202–11.
22. Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet. 2007; 369:2106–20.
23. Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat. Rev. Cancer. 2003; 3:203–16.
24. Brodeur GM, Seeger RC, Schwab M, Varmus HE, Bishop JM. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science. 1984; 224:1121–4.
25. Seeger RC, Brodeur GM, Sather H, Dalton A, Siegel SE, Wong KY, Hammond D. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N. Engl. J. Med. 1985; 313:1111–6.
26. Schwab M, Varmus HE, Bishop JM, Grzeschik KH, Naylor SL, Sakaguchi AY, Brodeur G, Trent J. Chromosome localization in normal human cells and neuroblastomas of a gene related to c-myc. Nature. 1984; 308:288–91.
27. Grimmer MR, Weiss WA. Childhood tumors of the nervous system as disorders of normal development. Curr. Opin. Pediatr. 2006; 18:634–8.
28. Boon K, Caron HN, van Asperen R, Valentijn L, Hermus MC, van Sluis P, Roobeek I, Weis I, Voûte PA, Schwab M, Versteeg R. N-myc enhances the expression of a large set of genes functioning in ribosome biogenesis and protein synthesis. EMBO J. 2001; 20:1383–93.
29. Kocak H, Ackermann S, Hero B, Kahlert Y, Oberthuer A, Juraeva D, Roels F, Theissen J, Westermann F, Deubzer H, Ehemann V, Brors B, Odenthal M, et al. Hox-C9 activates the intrinsic pathway of apoptosis and is associated with spontaneous regression in neuroblastoma. Cell Death Dis. 2013; 4:e586.
30. Lutz W, Stöhr M, Schürmann J, Wenzel A, Löhr A, Schwab M. Conditional expression of N-myc in human neuroblastoma cells increases expression of alpha-prothymosin and ornithine decarboxylase and accelerates progression into S-phase early after mitogenic stimulation of quiescent cells. Oncogene. 1996; 13:803–12.
31. Mandt T, Song Y, Scharenberg AM, Sahni J. SLC41A1 Mg2+ transport is regulated via Mg2+-dependent endosomal recycling through its N-terminal cytoplasmic domain. Biochem. J. 2011; 439:129–39.
32. Schmitz C, Dorovkov MV, Zhao X, Davenport BJ, Ryazanov AG, Perraud AL. The channel kinases TRPM6 and TRPM7 are functionally nonredundant. J Biol Chem. 2005; 280:37763–71.
33. Dorovkov MV, Ryazanov AG. Phosphorylation of annexin I by TRPM7 channel-kinase. J. Biol. Chem. 2004; 279:50643–6.
34. Clark K, Middelbeek J, Lasonder E, Dulyaninova NG, Morrice NA, Ryazanov AG, Bresnick AR, Figdor CG, van Leeuwen FN. TRPM7 regulates myosin IIA filament stability and protein localization by heavy chain phosphorylation. J. Mol. Biol. 2008; 378:790–803.
35. Prakriya M. The molecular physiology of CRAC channels. Immunol. Rev. 2009; 231:88–98.
36. Den Dekker E, Hoenderop JGJ, Nilius B, Bindels RJM. The epithelial calcium channels, TRPV5 & TRPV6: from identification towards regulation. Cell Calcium. 2003; 33:497–507.
37. Jiang J, Li MH, Inoue K, Chu XP, Seeds J, Xiong ZG. Transient receptor potential melastatin 7-like current in human head and neck carcinoma cells: role in cell proliferation. Cancer Res. 2007; 67:10929–38.
38. Zhang Z, Wang M, Fan X-H, Chen J-H, Guan Y-Y, Tang Y-B. Upregulation of TRPM7 channels by angiotensin II triggers phenotypic switching of vascular smooth muscle cells of ascending aorta. Circ. Res. 2012; 111:1137–46.
39. Du J, Xie J, Zhang Z, Tsujikawa H, Fusco D, Silverman D, Liang B, Yue L. TRPM7-mediated Ca2+ signals confer fibrogenesis in human atrial fibrillation. Circ. Res. 2010; 106:992–1003.
40. Sun Y, Selvaraj S, Varma A, Derry S, Sahmoun AE, Singh BB. Increase in serum Ca2+/Mg2+ ratio promotes proliferation of prostate cancer cells by activating TRPM7 channels. J. Biol. Chem. 2013; 288:255–63.
41. Kolisek M, Launay P, Beck A, Sponder G, Serafini N, Brenkus M, Froschauer EM, Martens H, Fleig A, Schweigel M. SLC41A1 is a novel mammalian Mg2+ carrier. J. Biol. Chem. 2008; 283:16235–47.
42. Sahni J, Scharenberg AM. TRPM7 ion channels are required for sustained phosphoinositide 3-kinase signaling in lymphocytes. Cell Metab. 2008; 8:84–93.
43. Duijkers FAM, Gaal J, Meijerink JPP, Admiraal P, Pieters R, de Krijger RR, van Noesel MM. Anaplastic lymphoma kinase (ALK) inhibitor response in neuroblastoma is highly correlated with ALK mutation status, ALK mRNA and protein levels. Cell. Oncol. 2011; 34:409–17.