
INTRODUCTION
Surgery is the most effective treatment for localized gastrointestinal cancer, and is often curative. Advanced stage cancers, however, are difficult to control with surgery alone. In these cases, surgery with the addition of multidisciplinary treatment strategies, such as combined chemoradiotherapy and molecular targeted therapy, can be beneficial. For example, combination chemotherapy with cetuximab for unresectable colorectal cancer (CRC) liver metastases refractory to conventional chemotherapy, increased resection rates and improved patient outcomes [1]. More recently, the emergence of immune checkpoint inhibitors has brought about a paradigm shift in cancer treatment. These have had dramatic effects in several advanced solid cancers [2–4], and accumulating evidence suggests promising outcomes in advanced gastrointestinal cancers [5]. Importantly, in some patients, immune checkpoint inhibitors can provide a cure for metastatic cancer, which is beyond the ability of conventional surgical treatment.
Immunotherapy, however, is only effective in a small proportion of patients, and current methods cannot identify which tumor is likely to respond. Predictive biomarkers are therefore needed to assist oncologists identify candidates for whom this therapeutic approach is most likely to succeed. Recent progress in genomic analysis using next-generation sequencing (NGS) technology has enabled comprehensive detection of mutations and mutation burden in cancer tissues. A hypermutated tumor is defined as a tumor with an increased mutation burden (a high rate of somatic mutation). The threshold above which tumors are considered hypermutated, however, depends on the sequencing methodology and type of cancer (Table 1). Importantly, the clinical significance of identifying hypermutated tumors has recently been demonstrated by several studies showing tumor mutation burden correlates with the generation of neo-antigens (mutated proteins) and a clinical response to immunotherapy [6, 7] (Figure 1). The causes of hypermutation vary between cancer types (Table 2). Ultraviolet (UV) light is the cause of many mutations in melanoma [8], while tobacco smoke causes the mutations that accumulate in non-small cell lung cancer [6]. A leading cause of the mutations found in several gastrointestinal cancers, such as colorectal, gastric, and hepato-pancreato-biliary cancer, is dysfunction in the mismatch repair (MMR) system. Indeed, CRC patients with MMR deficiency, who would be expected to develop a hypermutated phenotype, exhibited excellent outcomes after anti-PD-1 therapy [5]. This highlights the clinical significance of identifying hypermutated tumors for immunotherapy treatment.
Table 1: Definitions of hypermutated tumors reported in the literature
Cancer |
Sequence method |
Mutation rate |
Reference |
|---|---|---|---|
Colorectal |
WES |
>12 per Mb |
[16] |
Stomach |
WES |
>11.4 per MB |
[50] |
Stomach |
WES and WGS |
20.5 per Mb |
[113] |
Biliary tract |
WES and transcriptome sequencing |
>11.13 per Mb |
[87] |
Endometrium |
WES |
>18 per Mb |
[114] |
Melanoma |
WES |
>100 per exome |
[10] |
Lung |
WES |
≧178 nonsynonymous mutation per tumor |
[6] |
Glioblastoma |
WES |
>100 per tumor exome |
[115] |
Glioma |
Targeted NGS |
>20 per 1.4 Mb |
[116] |
WES, whole-exome sequencing; WGS, whole-genome sequencing; NGS, next-generation sequencing.
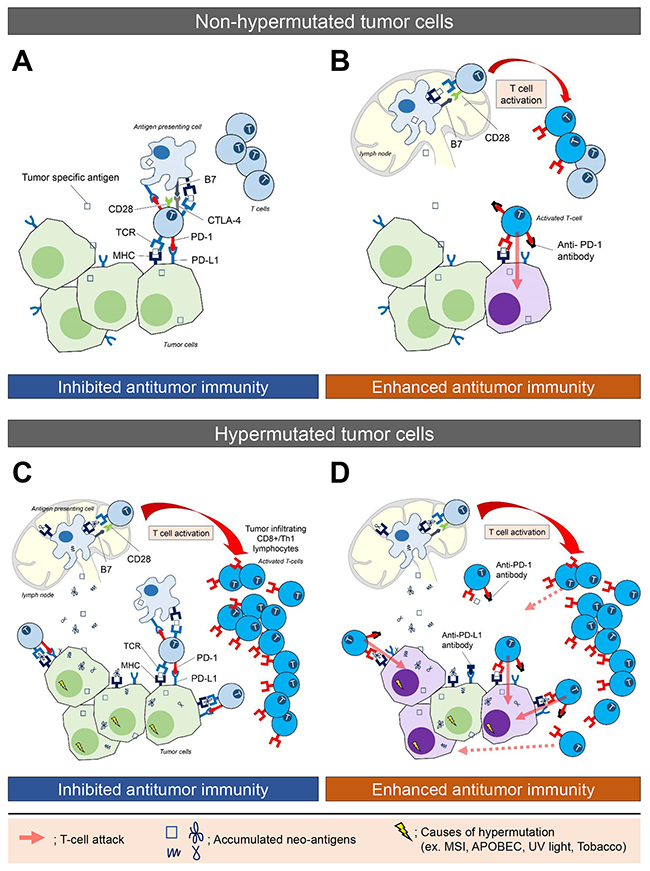
Figure 1: The immune microenvironment in non-hypermutated and hypermutated tumors, and enhanced immune activity following blockade of the PD-L1/PD-1 interaction. (A) Tumor-specific antigens are processed and presented by cancer cells and antigen presenting cells (APCs). Upregulated expression of checkpoint molecules, including PD-1 and CTLA-4 on T cells and PD-L1 on tumor cells and APCs, delivers inhibitory signals that suppress T cell activation, and produce an immunosuppressive microenvironment. (B) Blockade of the PD-L1/PD-1 interaction by an anti-PD-1 antibody enhances immune activity, which leads to T cells attacking and killing tumor cells. The attacked tumor cells are shown as purple cells. (C) Hypermutated cancer cells, derived by various mutagenic processes, generate numerous neo-antigens (mutated proteins) that are processed and presented by cancer cells and APCs. This stimulates T cell activation, leading to an infiltration of cytotoxic (CD8+) T-lymphocytes. Checkpoint molecules inhibit antitumor activity. (D) Blockade of the PD-L1/PD-1 interaction by an anti-PD-1 antibody enhances immune activation, whereby the infiltrating cytotoxic (CD8+) T-lymphocytes and activated Th1 cells attack tumor cells presenting the tumor specific antigen. This is thought to explain why hypermutated tumors demonstrate a significant durable efficacy to immune checkpoint therapy.
Table 2: The various causes of hypermutation in different cancers
Cause of hypermutation |
Organ |
Reference |
|---|---|---|
UV light |
Skin cancer |
[8] |
Tobacco smoke |
Lung cancer |
[6] |
MSI |
Gastrointestinal cancer |
[16] |
APOBEC |
Breast cancer |
[15] |
POLE, POLD1 |
Colorectal cancer |
UV, ultraviolet; MSI, microsatellite instability; APOBEC, apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like; POLE, DNA polymerase E; POLD1, DNA polymerase D1.
In this article, we review the current understanding of hypermutation and MMR deficiency in gastrointestinal cancer from the perspective of surgical oncology. We discuss how new knowledge of the cancer genome can be best used to improve the treatments available for patients with gastrointestinal cancer.
HYPERMUTATION IN CANCER AND ITS CAUSES
Mutations in oncogenes and tumor suppressor genes are the main mechanisms for cancer development. Increased spontaneous or environmentally enhanced mutagenesis has been correlated with increased mutation load and cancer risk [9]. Importantly, hypermutated cancer cells are believed to create numerous neo-antigens, which promote infiltration of cytotoxic (CD8+) T-lymphocytes and activated Th1 cells to the tumor microenvironment (Figure 1). Furthermore, studies have indicated an association between elevated mutation burden and response to checkpoint blockade immunotherapy in certain solid cancers [5, 6, 10, 11].
Hypermutation in cancer can be caused by a variety of mechanisms, including exogenous mutagens and endogenous processes. Exposure to exogenous mutagens, such as UV light in melanoma or tobacco smoke in lung cancer [12], can cause an accumulation of mutations in affected cells. Endogenous mutagenic processes can also affect the number of mutations [13]. They include activation-induced deaminase in chronic lymphocytic leukemia and B-cell lymphomas [14], and excessive APOBEC (apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like) activity in breast and gastric cancers [15]. One of the leading causes of hypermutation in gastrointestinal cancer is defective DNA MMR systems, which results in microsatellite instability (MSI) [16].
DETERMINING MSI STATUS AND MMR DEFICIENCY
MSI is a well-established tumorigenesis pathway that refers to the hypermutable state of cells. It is caused by a dysfunction of the MMR system, which results in a reduction in the length of highly repeated DNA sequences termed microsatellites. The MMR system corrects nucleotide mismatches that occur during replication. Microsatellites are simple repeat sequences of one to six base pairs (also known as short tandem DNA repeats) that are prone to DNA replication errors, resulting in MSI [17]. In sporadic CRCs, MMR deficiency most commonly occurs through epigenetic inactivation from hypermethylation of the MLH1 gene [16]. A germline mutation inactivating one of the MMR genes (MLH1, MSH2, MSH6, and PMS2) may lead to a hereditary form, termed hereditary non-polyposis colon cancer (HNPCC) [18].
MSI status is commonly determined by polymerase chain reaction amplification. DNA is isolated from microdissected tumor and normal tissues to compare the length of microsatellite alleles. The National Cancer Institute (NCI) recommends a microsatellite panel (NCI panel) consisting of two mononucleotide repeats (BAT25 and BAT26) and three dinucleotide repeats (D5S346, D2S123, and D17S250) [17]. Using the NCI panel, microsatellite instability-high (MSI-H) tumors are defined as having instability in two or more markers, and tumors with low or stable MSI have instability in one or no markers. Tumor MMR status is determined by immunohistochemical analysis of the proteins encoded by genes involved in DNA MMR. MSI-H tumors are found not only in colorectal cancer patients, but also in other gastrointestinal cancer patients, with the frequency of their occurrence differing between each cancer type (Table 3).
Table 3: The frequency of microsatellite instability (MSI-H) in each gastrointestinal cancer
Cancer |
Frequency of MSI-H |
Reference |
|---|---|---|
Colorectal cancer |
12-17 % |
|
Gastric cancer |
8-37 % |
|
Hepatocellular carcinoma |
0-18 % |
|
Pancreatic cancer |
0-13 % |
|
Intrahepatic cholangiocarcinoma |
10 % |
[90] |
Gallbladder cancer |
0-42 % |
|
Ampullary carcinoma |
0-22 % |
Recently, several groups have described NGS as a suitable testing platform for MSI [19–21]. The advantage of NGS technology is that it allows massively parallel sequencing capable of producing millions of sequences at once [22]. This usually translates to more efficient genetic sequencing compared to traditional genetic testing platforms, which is beneficial when testing large batches of tumor samples [20].
MSI IN PATIENTS WITH CRC
MSI is often reported in patients with CRC, and is detected in about 15% of all CRCs [17, 23–25]. Of these, 3% are associated with Lynch syndrome, also known as HNPCC, and the remaining 12% are sporadic, caused by acquired hypermethylation of the MLH1 promoter [25]. Hypermutated CRC is highly correlated with MSI. The Cancer Genome Atlas (TCGA) Network has reported that 16% of CRCs are hypermutated (defined as cancers with mutation rates of >12 per 106 bases), with three-quarters of these having high MSI [16]. One of the most frequent genetic alterations in CRCs with MSI is the oncogenic BRAF V600E mutation. The TCGA study has also revealed that hypermutated CRCs had fewer APC, KRAS, and TP53 mutations compared with the non-hypermutated CRCs. In contrast, mutations in transforming growth factor (TGF)-β signaling genes and BRAF were dramatically elevated in the hypermutated tumors.
Regardless of the origin (hereditary or sporadic) or type of mutation, MSI-H CRCs share some distinct histopathological cancer features. For example, these tumors tend to arise in the proximal or right side, and are more common in females [26]. This is particularly true in sporadic MSI-H cancers where over 90% are located in the proximal colon [27, 28]. It has been reported that CRCs with MSI-H, especially tumors with BRAF mutations, show increased proliferative activities [29]. Histologically, MSI-H CRCs tend to be poorly differentiated, are often mucinous, and sometimes contain signet ring cells and undifferentiated medullary carcinoma. Pathological characteristics are associated with the presence of lymphocytic infiltration [30, 31], and a Crohn’s-like lymphocytic reaction. Indeed, excessive lymphocyte infiltration was classically used as a pathological screening criterion for MSI by hematoxylin and eosin staining.
Clinically, MSI-H CRC develops a large size tumor with high levels of cell growth but less metastasis [30, 32]. MSI-H CRC patients are reported to have a good prognosis [33] and it has been suggested they respond differently to chemotherapy than microsatellite stable (MSS) tumor patients. For example, MSI-H patients have been reported to be less likely to respond to fluoropyrimidine and 5-fluorouracil (5-FU) [33, 34]. This finding remains controversial, however, as several reports have indicated there is no significant clinical benefit in using MSI status to guide treatment decisions on the use of 5-FU for CRC [35]. Furthermore, 5-FU, alone or in combination with other drugs, has been the standard of care for first-line treatment in Stage III, Stage IV, and high-risk Stage II CRC since the late 1950s.
MSI tumors strongly express various immunological checkpoint proteins, such as PD-1, PD-L1, CTLA-4, LAG-3 and IDO [36–39], and, as such, are the focus of many current clinical trials. Their inhibitory signals prevent elimination of neoplastic cells by counteracting the active immune microenvironment of the MSI tumor [5, 36]. Recently, it has been reported that CRC patients with MSI showed significantly better progression-free and overall survival than patients without MSI. Therefore, the MSI status and hypermutated phenotype may be a predictive marker for immuno-modulating agents [25].
In addition to dysfunction in the MMR system, mutations in DNA polymerase D1 (POLD1) and DNA polymerase E (POLE) genes have been described as another cause of hypermutated CRC [40, 41]. In addition to somatic mutations, germline mutations in these genes have been identified in familial CRC. POLD1 synthesizes the lagging strand and POLE1 synthesizes the leading strand in a bidirectional replication fork [42]. CRC with mutations in the exonuclease domain of POLE1 is associated with a high number of mutations, multiple tumor neo-epitopes, and extensive T lymphocyte infiltration. Taken together, a hypermutated phenotype, not only with MSI-H, but also with a POLE mutation, may be a useful predictive marker for CRC.
MSI IN PATIENTS WITH GASTRIC CANCER
The MSI-H phenotype in gastric cancer is predominantly caused by epigenetic hypermethylation of MLH1 rather than germline mutations in an MMR gene [39, 43–45]. The incidence of MSI-H in gastric cancers varies from 8-37% [43, 46–49]. TCGA has defined four molecular subgroups of gastric cancer by unsupervised clustering, and one of the groups, comprising 22% of all cases, was enriched for MSI and showed elevated mutation rates and hypermethylation [50]. Gastric cancer with MSI-H is reported to display distinct clinical and molecular features compared to MSS gastric cancer [46, 51–53]. They are usually associated with female sex, older age [54, 55], antral location, intestinal type [49], smaller risk in lymph node metastasis [49, 55], shallower tumor invasion [49], earlier stage, and a better prognosis [52, 53, 55], most of which are characteristics similar to CRC MSI-H patients, as described above. Association with tumor necrosis, expanding growth pattern, and tumor-infiltrating lymphocytes (TILs) are also reported [56]. Although reports are inconsistent [47, 56–58], MSI in gastric cancer may be considered a favorable prognostic indicator [52, 58] for both early [53, 59–61] and advanced [46, 49] stages.
Conflicting results have been reported in MSI-H gastric cancers regarding response to adjuvant 5-FU-based chemotherapy. No difference in overall survival between MSI-H and MSS was observed in a study of 240 patients [39]. However, a more recent study found disease-free survival was improved in the MSI-Low/MSS group treated with 5-FU-based chemotherapy [62]. The disparity between these studies might be due to the high morphological, phenotypic, and molecular heterogeneity of gastric cancer [56]. Recently, significant correlations have been found between defective MMR systems and immune system activity, suggesting that this group of patients might be optimal candidates for immunotherapies including anti-PD-1/PD-L1 antibodies [63–65].
Mutational analysis of MSI-H gastric cancers revealed 37 significantly mutated genes, including TP53, KRAS, PIK3A, and AR1D1A [50]. Genes in the TGF-β pathway were predicted to be key drivers in MSI. Indeed, TGFBR2, ACVR2A, SMAD4, and ELF3 are frequently mutated, suggesting an important role in gastric cancer biology [66, 67].
MSI IN PATIENTS WITH HEPATO CELLULAR CARCINOMA (HCC) AND PANCREATIC CANCER
MSI is seldom observed in HCC and pancreatic carcinoma. MSI-H occurrence in HCC ranges from 0-18% [68–72], and alterations in MMR genes are not implicated in its pathogenesis [73–75]. As such, the biological and clinicopathological significance of MSI in HCC remains to be determined [74]. In a small-sized study, the histology and the prognosis of patients with MSI-H HCC were worse than those with non-MSI-H HCC [71]. MSI-H tumors tended to exhibit a large, unique nodule without a capsule, corresponding to a more aggressive tumor [71]. Progression of the primary tumor leading to liver failure is the general cause of death in HCC patients, rather than tumor metastasis. Considering MSI-H HCC patients exhibit a more aggressive primary tumor than those with non-MSI-H, it is to be expected that MSI-H HCC patients have a worse prognosis. In contrast, in other cancer types such as CRC, gastric cancer and other gastrointestinal cancers, prognosis is usually determined by metastatic disease rather than primary tumor characteristics. As such, MSI-H patients with these cancer types often exhibit a better prognosis than non-MSI-H patients, likely due to the lower frequency of metastasis in MSI-H cancer.
MSI is rarely found in sporadic pancreatic ductal adenocarcinoma, occurring in less than 1% of cases based on molecular MSI testing [76]. Studies that used the NCI panel to define MSI-H found it occurred in 0-13% of pancreatic carcinoma [77–80]. A recent study of 385 pancreatic cancers subjected to whole genome or exome sequencing reported four (1%) cases with MSI [81]. MSI is found in both medullary [77, 82, 83] and non-medullary [79, 84, 85] subtypes of pancreatic carcinoma. Previous studies on MSI as a prognostic predictor of pancreatic cancer are limited. Some reports suggest that MSI-H pancreatic cancer might have a comparatively better prognosis than non-MSI-H cancer [77, 86], but larger studies are needed to confirm this finding.
MSI IN PATIENTS WITH BILIARY TRACT CANCER (BTC)
Published literature on the mutational profile and MMR deficiency in BTCs is limited, but there are reports that some BTCs have a significantly high mutation burden [87, 88]. Most studies evaluating MSI in intrahepatic cholangiocarcinoma come from analyses of patients in Southeast Asia, particularly Thailand, where liver cholangiocarcinoma represents one of the most common cancers and is believed to be associated with liver fluke (Opisthorchis viverrini) infection [89, 90]. In these studies, the frequency of MSI-H in intrahepatic cholangiocarcinoma was 10% [90]. Liver-fluke-associated intrahepatic cholangiocarcinoma shows a higher somatic mutation burden compared with non-parasite-associated BTC [91].
Recent studies from Japan have investigated mutational signatures of intrahepatic cholangiocarcinoma believed to be caused from exposure to organic solvents, mainly haloalkanes such as 1,2-dichloropropane and/or dichloromethane [92–94]. The number of single-nucleotide variants in cholangiocarcinoma resected from printing workers exposed to organic solvents was significantly higher than in control common cholangiocarcinoma tissues, with somatic mutations at an average of 44.8/Mb [92]. This suggests 1,2-dichloropropane is an exogenous mutagen that results in hypermutated cholangiocarcinoma.
Previous reports have found a correlation between MSI and gallbladder carcinoma. The prevalence of MSI varied from 0% to 42%, and averaged 5% overall [90, 95–99]. There was no significant difference in tumor stage or overall survival between patients with and without MSI [100], and no association between MSI status and tumor grade, or the presence of extracellular mucin or TILs [100]. Expression of long interspersed nuclear element-1 (LINE-1), a surrogate marker of global methylation status, was lacking in MSI gallbladder carcinomas, suggesting the loss of MMR proteins was due to changes in methylation [100]. MSI was found, not only in cancer regions, but also in severe chronic cholecystitis [97], and areas of both intestinal metaplasia and dysplasia [98]. This suggests that MMR deficiency in tumor development may be associated with prolonged inflammation and may occur very early in gallbladder cancer development [90, 97, 98].
Several studies have correlated ampullary carcinoma with MMR deficiency [90, 101–106], while others have reported that MSI could not be identified in ampullary carcinoma [96, 107]. The reported frequency of MSI-H in ampullary carcinomas is 0-22%. Ampullary and colorectal carcinomas share a significant overlap in phenotypic and molecular characteristics [106]. Ampullary carcinoma with MSI-H often demonstrates better prognosis [101, 105] with increased TILs [104, 105], poor differentiation with “medullary”-like histology, and intestinal type morphology [104, 105, 108].
Most recently, a relatively large-scale study by Nakamura et al. [87] has molecularly characterized 260 BTCs and uncovered a spectrum of genomic alterations. Fourteen cases were classified as hypermutated, with mutation rates of >11.13/Mb. Of these, five harbored inactivating (nonsense, frameshift or splice-site) mutations in mismatch-repair complex components. Transcriptome sequencing and hierarchical clustering of gene expression levels classified BTC into four molecular subgroups that had prognostic implications. The hypermutated cases were significantly enriched in the worst prognosis group, with increased expression of immune checkpoint molecules and enrichment for the genes involved in cytokine activity and anti-apoptosis. In total, 45% of cases showed increased expression of immune checkpoint molecules, which suggests that this subgroup may be a good target population for immunotherapy [87, 88, 109].
FUTURE PERSPECTIVE AND CONCLUSION
While immunotherapy and targeted therapies continue to advance the treatment of cancer, surgery still plays a vital role in the treatment of gastrointestinal malignancies, especially in the management of early-stage, localized disease. However, despite surgery with curative intent in patients with advanced disease, many experience tumor recurrence that leads to poor outcome [110]. The genomic profile of the primary and metastatic tumor provides critical information for guiding decisions about treatment. This review has outlined how hypermutation may play a pivotal role as a cancer biomarker able to identify cancer subtypes most likely to respond to treatment, and therefore predict the clinical benefit of immunotherapy, especially in advanced cancers where surgery is usually not indicated.
BTCs are heterogeneous cancers with an increasing incidence worldwide. They are often refractory to standard chemotherapy regimens and exhibit a poor prognosis. Recent studies have revealed that BTCs are rich in actionable genetic aberrations [111]. As such, it is now possible to identify unique molecular subsets of BTC, such as hypermutated phenotypes, that can be effectively treated with a personalized medicine approach, which will hopefully lead to an improved prognosis [111].
Detection of hypermutation in cancers may not only predict the clinical benefit of immune checkpoint inhibitors, but has the potential to also provide better surgical strategies for the patients with hypermutated tumors. Of particular benefit may be the treatment of locally advanced hypermutated tumors. Hypermutated tumors tend to show expansive growth in a localized region and have less metastasis. As such, surgical resection, rather than the currently indicated neoadjuvant chemotherapy, may prove more beneficial. Furthermore, MSI-H tumors tend to show resistance to 5-FU-based chemotherapy compared to non-MSI-H tumors. Therefore, a combination of surgery and immune checkpoint inhibitors would be an attractive approach for cancer treatment. This approach, however, needs further investigation since studies of combined immunotherapy and surgery are lacking. Immune checkpoint inhibitors are now being incorporated in various clinical trials in the neoadjuvant setting (NCT02957968, NCT02735239, NCT03003637, NCT02918162) [112], the adjuvant setting (NCT02775812, NCT02641093), or both (NCT02296684). These studies will provide valuable evidence of any clinical benefit from immune checkpoint inhibitor use combined with surgery.
Abbreviations
5-FU, 5-fluorouracil; APOBEC, apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like; BTC, biliary tract cancer; CD, cluster of differentiation; CRC, colorectal cancer; CTLA-4, cytotoxic T-lymphocyte-associated antigen 4; HCC, hepatocellular carcinoma; HNPCC, hereditary non-polyposis colon cancer; IDO, indoleamine 2,3-dioxygenase; LAG-3, lymphocyte activation gene-3; LINE-1, long interspersed nuclear element-1; MMR, mismatch repair; MSI, microsatellite instability; MSI-H, microsatellite instability-high; MSS, microsatellite stable; NCI, The National Cancer Institute; NGS, next-generation sequencing; PD-1, programmed death-1; PD-L1, programmed death ligand 1; POLD1, DNA polymerase D1; POLE, DNA polymerase E; TCGA, The Cancer Genome Atlas; TGF, transforming growth factor; Th1, T helper cell type 1; TILs, tumor-infiltrating lymphocytes; UV, ultraviolet.
Author contributions
K. Yuza, M. Nagahashi, S. Watanabe and K. Takabe wrote the manuscript; T. Wakai designed the aim of the editorial.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
The authors would like to acknowledge funding from the Japan Society for the Promotion of Science (JSPS) Grant-in-Aid for Scientific Research, Grant Numbers 17K10579 for K. Yuza, 15H05676 and 15K15471 for M. Nagahashi, and 15H04927 and 16K15610 for T. Wakai. M. Nagahashi is also supported by Takeda Science Foundation, and Tsukada Medical Foundation. K. Takabe is supported by NIH/NCI grant R01CA160688 and Susan G. Komen Investigator Initiated Research Grant IIR12222224.
REFERENCES
1. Adam R, Aloia T, Levi F, Wicherts DA, de Haas RJ, Paule B, Bralet MP, Bouchahda M, Machover D, Ducreux M, Castagne V, Azoulay D, Castaing D. Hepatic resection after rescue cetuximab treatment for colorectal liver metastases previously refractory to conventional systemic therapy. J Clin Oncol. 2007; 25:4593–602. https://doi.org/10.1200/jco.2007.10.8126.
2. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010; 363:711–23. https://doi.org/10.1056/NEJMoa1003466.
3. Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E, Waterhouse D, Ready N, Gainor J, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015; 373:123–35. https://doi.org/10.1056/NEJMoa1504627.
4. Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, Tykodi SS, Sosman JA, Procopio G, Plimack ER, Castellano D, Choueiri TK, Gurney H, et al, and CheckMate 025 Investigators. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015; 373:1803–13. https://doi.org/10.1056/NEJMoa1510665.
5. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, Biedrzycki B, Donehower RC, Zaheer A, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015; 372:2509–20. https://doi.org/10.1056/NEJMoa1500596.
6. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015; 348:124–8. https://doi.org/10.1126/science.aaa1348.
7. McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT, Watkins TB, Shafi S, Murugaesu N, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016; 351:1463–9. https://doi.org/10.1126/science.aaf1490.
8. Mar VJ, Wong SQ, Li J, Scolyer RA, McLean C, Papenfuss AT, Tothill RW, Kakavand H, Mann GJ, Thompson JF, Behren A, Cebon JS, Wolfe R, et al. BRAF/NRAS wild-type melanomas have a high mutation load correlating with histologic and molecular signatures of UV damage. Clin Cancer Res. 2013; 19:4589–98. https://doi.org/10.1158/1078-0432.ccr-13-0398.
9. Roberts SA, Gordenin DA. Hypermutation in human cancer genomes: footprints and mechanisms. Nat Rev Cancer. 2014; 14:786–800. https://doi.org/10.1038/nrc3816.
10. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, Hollmann TJ, Bruggeman C, Kannan K, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014; 371:2189–99. https://doi.org/10.1056/NEJMoa1406498.
11. Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Geukes Foppen MH, Goldinger SM, Utikal J, Hassel JC, Weide B, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015; 350:207–11. https://doi.org/10.1126/science.aad0095.
12. Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, Kiezun A, Hammerman PS, McKenna A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013; 499:214–8. https://doi.org/10.1038/nature12213.
13. Schlesner M, Eils R. Hypermutation takes the driver’s seat. Genome Med. 2015; 7:31. https://doi.org/10.1186/s13073-015-0159-x.
14. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, Boyault S, Burkhardt B, Butler AP, et al, and Australian Pancreatic Cancer Genome Initiative, and ICGC Breast Cancer Consortium, and ICGC MMML-Seq Consortium, and ICGC PedBrain. Signatures of mutational processes in human cancer. Nature. 2013; 500:415–21. https://doi.org/10.1038/nature12477.
15. Roberts SA, Lawrence MS, Klimczak LJ, Grimm SA, Fargo D, Stojanov P, Kiezun A, Kryukov GV, Carter SL, Saksena G, Harris S, Shah RR, Resnick MA, et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat Genet. 2013; 45:970–6. https://doi.org/10.1038/ng.2702.
16. Muzny DM, Bainbridge MN, Chang K, Dinh HH, Drummond JA, Fowler G, Kovar CL, Lewis LR, Morgan MB, Newsham IF, Reid JG, Santibanez J, Shinbrot E, et al, and Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487:330–7. https://doi.org/10.1038/nature11252.
17. Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, Srivastava S. A national cancer institute workshop on microsatellite instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998; 58:5248–57.
18. Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Ruschoff J, Fishel R, Lindor NM, Burgart LJ, Hamelin R, Hamilton SR, Hiatt RA, Jass J, et al. Revised bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004; 96:261–8.
19. Salipante SJ, Scroggins SM, Hampel HL, Turner EH, Pritchard CC. Microsatellite instability detection by next generation sequencing. Clin Chem. 2014; 60:1192–9. https://doi.org/10.1373/clinchem.2014.223677.
20. Gan C, Love C, Beshay V, Macrae F, Fox S, Waring P, Taylor G. Applicability of next generation sequencing technology in microsatellite instability testing. Genes (Basel). 2015; 6:46–59. https://doi.org/10.3390/genes6010046.
21. Nowak JA, Yurgelun MB, Bruce JL, Rojas-Rudilla V, Hall DL, Shivdasani P, Garcia EP, Agoston AT, Srivastava A, Ogino S, Kuo FC, Lindeman NI, Dong F. Detection of mismatch repair deficiency and microsatellite instability in colorectal adenocarcinoma by targeted next-generation sequencing. J Mol Diagn. 2017; 19:84–91. https://doi.org/10.1016/j.jmoldx.2016.07.010.
22. Liu L, Li Y, Li S, Hu N, He Y, Pong R, Lin D, Lu L, Law M. Comparison of next-generation sequencing systems. J Biomed Biotechnol. 2012; 2012:251364. https://doi.org/10.1155/2012/251364.
23. Hemminki A, Mecklin JP, Jarvinen H, Aaltonen LA, Joensuu H. Microsatellite instability is a favorable prognostic indicator in patients with colorectal cancer receiving chemotherapy. Gastroenterology. 2000; 119:921–8.
24. Gryfe R, Kim H, Hsieh ET, Aronson MD, Holowaty EJ, Bull SB, Redston M, Gallinger S. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N Engl J Med. 2000; 342:69–77. https://doi.org/10.1056/nejm200001133420201.
25. Copija A, Waniczek D, Witkos A, Walkiewicz K, Nowakowska-Zajdel E. Clinical significance and prognostic relevance of microsatellite instability in sporadic colorectal cancer patients. Int J Mol Sci. 2017; 18. https://doi.org/10.3390/ijms18010107.
26. Thibodeau SN, French AJ, Cunningham JM, Tester D, Burgart LJ, Roche PC, McDonnell SK, Schaid DJ, Vockley CW, Michels VV, Farr GH Jr, O’Connell MJ. Microsatellite instability in colorectal cancer: different mutator phenotypes and the principal involvement of hMLH1. Cancer Research. 1998; 58:1713–8.
27. Thibodeau SN, Bren G, Schaid D. Microsatellite instability in cancer of the proximal colon. Science. 1993; 260:816–9.
28. Kim H, Jen J, Vogelstein B, Hamilton SR. Clinical and pathological characteristics of sporadic colorectal carcinomas with DNA replication errors in microsatellite sequences. Am J Pathol. 1994; 145:148–56.
29. Nagahashi M, Wakai T, Shimada Y, Ichikawa H, Kameyama H, Kobayashi T, Sakata J, Yagi R, Sato N, Kitagawa Y, Uetake H, Yoshida K, Oki E, et al. Genomic landscape of colorectal cancer in Japan: clinical implications of comprehensive genomic sequencing for precision medicine. Genome Med. 2016; 8:136. https://doi.org/10.1186/s13073-016-0387-8.
30. Buckowitz A, Knaebel HP, Benner A, Blaker H, Gebert J, Kienle P, von Knebel Doeberitz M, Kloor M. Microsatellite instability in colorectal cancer is associated with local lymphocyte infiltration and low frequency of distant metastases. Br J Cancer. 2005; 92:1746–53. https://doi.org/10.1038/sj.bjc.6602534.
31. Deschoolmeester V, Baay M, Van Marck E, Weyler J, Vermeulen P, Lardon F, Vermorken JB. Tumor infiltrating lymphocytes: an intriguing player in the survival of colorectal cancer patients. BMC Immunol. 2010; 11:19. https://doi.org/10.1186/1471-2172-11-19.
32. Fujiyoshi K, Yamamoto G, Takenoya T, Takahashi A, Arai Y, Yamada M, Kakuta M, Yamaguchi K, Akagi Y, Nishimura Y, Sakamoto H, Akagi K. Metastatic pattern of Stage IV colorectal cancer with high-frequency microsatellite instability as a prognostic factor. Anticancer Res. 2017; 37:239–47.
33. Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol. 2005; 23:609–18. https://doi.org/10.1200/JCO.2005.01.086.
34. Sargent DJ, Marsoni S, Monges G, Thibodeau SN, Labianca R, Hamilton SR, French AJ, Kabat B, Foster NR, Torri V, Ribic C, Grothey A, Moore M, et al. Defective mismatch repair as a predictive marker for lack of efficacy of fluorouracil-based adjuvant therapy in colon cancer. J Clin Oncol. 2010; 28:3219–26. https://doi.org/10.1200/jco.2009.27.1825.
35. Webber EM, Kauffman TL, O’Connor E, Goddard KA. Systematic review of the predictive effect of MSI status in colorectal cancer patients undergoing 5FU-based chemotherapy. BMC Cancer. 2015; 15:156. https://doi.org/10.1186/s12885-015-1093-4.
36. Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, Blosser RL, Fan H, Wang H, Luber BS, Zhang M, Papadopoulos N, Kinzler KW, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015; 5:43–51. https://doi.org/10.1158/2159-8290.CD-14-0863.
37. Xiao Y, Freeman GJ. The microsatellite instable subset of colorectal cancer is a particularly good candidate for checkpoint blockade immunotherapy. Cancer Discov. 2015; 5:16–8. https://doi.org/10.1158/2159-8290.cd-14-1397.
38. Quiroga D, Lyerly HK, Morse MA. Deficient mismatch repair and the role of immunotherapy in metastatic colorectal cancer. Curr Treat Options Oncol. 2016; 17:41. https://doi.org/10.1007/s11864-016-0414-4.
39. Oki E, Kakeji Y, Zhao Y, Yoshida R, Ando K, Masuda T, Ohgaki K, Morita M, Maehara Y. Chemosensitivity and survival in gastric cancer patients with microsatellite instability. Ann Surg Oncol. 2009; 16:2510–5. https://doi.org/10.1245/s10434-009-0580-8.
40. Palles C, Cazier JB, Howarth KM, Domingo E, Jones AM, Broderick P, Kemp Z, Spain SL, Guarino E, Salguero I, Sherborne A, Chubb D, Carvajal-Carmona LG, et al, and WGS500 Consortium. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet. 2013; 45:136–44. https://doi.org/10.1038/ng.2503.
41. Bourdais R, Rousseau B, Pujals A, Boussion H, Joly C, Guillemin A, Baumgaertner I, Neuzillet C, Tournigand C. Polymerase proofreading domain mutations: new opportunities for immunotherapy in hypermutated colorectal cancer beyond MMR deficiency. Crit Rev Oncol Hematol. 2017; 113:242–8. https://doi.org/10.1016/j.critrevonc.2017.03.027.
42. Lange SS, Takata K, Wood RD. DNA polymerases and cancer. Nat Rev Cancer. 2011; 11:96–110. https://doi.org/10.1038/nrc2998.
43. Leung SY, Yuen ST, Chung LP, Chu KM, Chan AS, Ho JC. hMLH1 promoter methylation and lack of hMLH1 expression in sporadic gastric carcinomas with high-frequency microsatellite instability. Cancer Res. 1999; 59:159–64.
44. Bevilacqua RA, Simpson AJ. Methylation of the hMLH1 promoter but no hMLH1 mutations in sporadic gastric carcinomas with high-level microsatellite instability. Int J Cancer. 2000; 87:200–3.
45. Fleisher AS, Esteller M, Tamura G, Rashid A, Stine OC, Yin J, Zou TT, Abraham JM, Kong D, Nishizuka S, James SP, Wilson KT, Herman JG, et al. Hypermethylation of the hMLH1 gene promoter is associated with microsatellite instability in early human gastric neoplasia. Oncogene. 2001; 20:329–35. https://doi.org/10.1038/sj.onc.1204104.
46. Lee HS, Choi SI, Lee HK, Kim HS, Yang HK, Kang GH, Kim YI, Lee BL, Kim WH. Distinct clinical features and outcomes of gastric cancers with microsatellite instability. Mod Pathol. 2002; 15:632–40. https://doi.org/10.1038/modpathol.3880578.
47. Perez RO, Jacob CE, D’Ottaviano FL, Alvarenga C, Ribeiro AS, Ribeiro U Jr, Bresciani CJ, Zilberstein B, Krieger JE, Habr-Gama A, Gama-Rodrigues JJ. Microsatellite instability in solitary and sporadic gastric cancer. Rev Hosp Clin Fac Med Sao Paulo. 2004; 59:279–85. https://doi.org//S0041-87812004000500010.
48. Kim H, An JY, Noh SH, Shin SK, Lee YC, Kim H. High microsatellite instability predicts good prognosis in intestinal-type gastric cancers. J Gastroenterol Hepatol. 2011; 26:585–92. https://doi.org/10.1111/j.1440-1746.2010.06487.x.
49. Zhu L, Li Z, Wang Y, Zhang C, Liu Y, Qu X. Microsatellite instability and survival in gastric cancer: a systematic review and meta-analysis. Mol Clin Oncol. 2015; 3:699–705. https://doi.org/10.3892/mco.2015.506.
50. Bass AJ, Thorsson V, Shmulevich I, Reynolds SM, Miller M, Bernard B, Hinoue T, Laird PW, Curtis C, Shen H, Weisenberger DJ, Schultz N, Shen R, et al, and Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014; 513:202–9.
51. Yamamoto H, Perez-Piteira J, Yoshida T, Terada M, Itoh F, Imai K, Perucho M. Gastric cancers of the microsatellite mutator phenotype display characteristic genetic and clinical features. Gastroenterology. 1999; 116:1348–57.
52. Falchetti M, Saieva C, Lupi R, Masala G, Rizzolo P, Zanna I, Ceccarelli K, Sera F, Mariani-Costantini R, Nesi G, Palli D, Ottini L. Gastric cancer with high-level microsatellite instability: target gene mutations, clinicopathologic features, and long-term survival. Hum Pathol. 2008; 39:925–32. https://doi.org/10.1016/j.humpath.2007.10.024.
53. Beghelli S, de Manzoni G, Barbi S, Tomezzoli A, Roviello F, Di Gregorio C, Vindigni C, Bortesi L, Parisi A, Saragoni L, Scarpa A, Moore PS. Microsatellite instability in gastric cancer is associated with better prognosis in only stage II cancers. Surgery. 2006; 139:347–56. https://doi.org/10.1016/j.surg.2005.08.021.
54. Sugimoto R, Sugai T, Habano W, Endoh M, Eizuka M, Yamamoto E, Uesugi N, Ishida K, Kawasaki T, Matsumoto T, Suzuki H. Clinicopathological and molecular alterations in early gastric cancers with the microsatellite instability-high phenotype. Int J Cancer. 2016; 138:1689–97. https://doi.org/10.1002/ijc.29916.
55. Mathiak M, Warneke VS, Behrens HM, Haag J, Boger C, Kruger S, Rocken C. Clinicopathologic characteristics of microsatellite instable gastric carcinomas revisited: urgent need for standardization. Appl Immunohistochem Mol Morphol. 2017; 25:12–24. https://doi.org/10.1097/pai.039696R1039696R10264.
56. Kim JY, Shin NR, Kim A, Lee HJ, Park WY, Kim JY, Lee CH, Huh GY, Park DY. Microsatellite instability status in gastric cancer: a reappraisal of its clinical significance and relationship with mucin phenotypes. Korean J Pathol. 2013; 47:28–35. https://doi.org/10.4132/KoreanJPathol.2013.47.1.28.
57. An C, Choi IS, Yao JC, Worah S, Xie K, Mansfield PF, Ajani JA, Rashid A, Hamilton SR, Wu TT. Prognostic significance of CpG island methylator phenotype and microsatellite instability in gastric carcinoma. Clin Cancer Res. 2005; 11:656–63.
58. Richman S. Deficient mismatch repair: read all about it (Review). Int J Oncol. 2015; 47:1189–202. https://doi.org/10.3892/ijo.2015.3119.
59. Fang WL, Chang SC, Lan YT, Huang KH, Chen JH, Lo SS, Hsieh MC, Li AF, Wu CW, Chiou SH. Microsatellite instability is associated with a better prognosis for gastric cancer patients after curative surgery. World J Surg. 2012; 36:2131–8. https://doi.org/10.1007/s00268-012-1652-7.
60. Shigeyasu K, Nagasaka T, Mori Y, Yokomichi N, Kawai T, Fuji T, Kimura K, Umeda Y, Kagawa S, Goel A, Fujiwara T. Clinical significance of MLH1 methylation and CpG island methylator phenotype as prognostic markers in patients with gastric cancer. PLoS One. 2015; 10:e0130409. https://doi.org/10.1371/journal.pone.0130409.
61. Bria E, Pilotto S, Simbolo M, Fassan M, de Manzoni G, Carbognin L, Sperduti I, Brunelli M, Cataldo I, Tomezzoli A, Mafficini A, Turri G, Karachaliou N, et al. Comprehensive molecular portrait using next generation sequencing of resected intestinal-type gastric cancer patients dichotomized according to prognosis. Sci Rep. 2016; 6:22982. https://doi.org/10.1038/srep22982.
62. An JY, Kim H, Cheong JH, Hyung WJ, Kim H, Noh SH. Microsatellite instability in sporadic gastric cancer: its prognostic role and guidance for 5-FU based chemotherapy after R0 resection. Int J Cancer. 2012; 131:505–11. https://doi.org/10.1002/ijc.26399.
63. Giampieri R, Maccaroni E, Mandolesi A, Del Prete M, Andrikou K, Faloppi L, Bittoni A, Bianconi M, Scarpelli M, Bracci R, Scartozzi M, Cascinu S. Mismatch repair deficiency may affect clinical outcome through immune response activation in metastatic gastric cancer patients receiving first-line chemotherapy. Gastric Cancer. 2017; 20:156–63. https://doi.org/10.1007/s10120-016-0594-4.
64. Bockorny B, Pectasides E. The emerging role of immunotherapy in gastric and esophageal adenocarcinoma. Future Oncol. 2016; 12:1833–46. https://doi.org/10.2217/fon-2016-0103.
65. Jou E, Rajdev L. Current and emerging therapies in unresectable and recurrent gastric cancer. World J Gastroenterol. 2016; 22:4812–23. https://doi.org/10.3748/wjg.v22.i20.4812.
66. Nagarajan N, Bertrand D, Hillmer AM, Zang ZJ, Yao F, Jacques PÉ, Teo AS, Cutcutache I, Zhang Z, Lee WH, Sia YY, Gao S, Ariyaratne PN, et al. Whole-genome reconstruction and mutational signatures in gastric cancer. Genome Biol. 2012; 13:R115.
67. Wang K, Yuen ST, Xu J, Lee SP, Yan HH, Shi ST, Siu HC, Deng S, Chu KM, Law S, Chan KH, Chan AS, Tsui WY, et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat Genet. 2014; 46:573–82.
68. Macdonald GA, Greenson JK, Saito K, Cherian SP, Appelman HD, Boland CR. Microsatellite instability and loss of heterozygosity at DNA mismatch repair gene loci occurs during hepatic carcinogenesis. Hepatology. 1998; 28:90–7. https://doi.org/10.1002/hep.510280114.
69. Karachristos A, Liloglou T, Field JK, Deligiorgi E, Kouskouni E, Spandidos DA. Microsatellite instability and p53 mutations in hepatocellular carcinoma. Mol Cell Biol Res Commun. 1999; 2:155–61. https://doi.org/10.1006/mcbr.1999.0170.
70. Kondo Y, Kanai Y, Sakamoto M, Mizokami M, Ueda R, Hirohashi S. Genetic instability and aberrant DNA methylation in chronic hepatitis and cirrhosis--a comprehensive study of loss of heterozygosity and microsatellite instability at 39 loci and DNA hypermethylation on 8 CpG islands in microdissected specimens from patients with hepatocellular carcinoma. Hepatology. 2000; 32:970–9. https://doi.org/10.1053/jhep.2000.19797.
71. Chiappini F, Gross-Goupil M, Saffroy R, Azoulay D, Emile JF, Veillhan LA, Delvart V, Chevalier S, Bismuth H, Debuire B, Lemoine A. Microsatellite instability mutator phenotype in hepatocellular carcinoma in non-alcoholic and non-virally infected normal livers. Carcinogenesis. 2004; 25:541–7. https://doi.org/10.1093/carcin/bgh035.
72. Zhang SH, Cong WM, Xian ZH, Wu MC. Clinicopathological significance of loss of heterozygosity and microsatellite instability in hepatocellular carcinoma in China. World J Gastroenterol. 2005; 11:3034–9.
73. Yamamoto H, Itoh F, Fukushima H, Kaneto H, Sasaki S, Ohmura T, Satoh T, Karino Y, Endo T, Toyota J, Imai K. Infrequent widespread microsatellite instability in hepatocellular carcinomas. Int J Oncol. 2000; 16:543–7.
74. Herath NI, Leggett BA, MacDonald GA. Review of genetic and epigenetic alterations in hepatocarcinogenesis. J Gastroenterol Hepatol. 2006; 21:15–21. https://doi.org/10.1111/j.1440-1746.2005.04043.x.
75. Togni R, Bagla N, Muiesan P, Miquel R, O’Grady J, Heaton N, Knisely AS, Portmann B, Quaglia A. Microsatellite instability in hepatocellular carcinoma in non-cirrhotic liver in patients older than 60 years. Hepatol Res. 2009; 39:266–73. https://doi.org/10.1111/j.1872-034X.2008.00455.x.
76. Scarpa A, Cataldo I, Salvatore L. Microsatellite instability - defective DNA mismatch repair: ESMO biomarker factsheet. 2016.
77. Yamamoto H, Itoh F, Nakamura H, Fukushima H, Sasaki S, Perucho M, Imai K. Genetic and clinical features of human pancreatic ductal adenocarcinomas with widespread microsatellite instability. Cancer Res. 2001; 61:3139–44.
78. Abraham SC, Wu TT, Hruban RH, Lee JH, Yeo CJ, Conlon K, Brennan M, Cameron JL, Klimstra DS. Genetic and immunohistochemical analysis of pancreatic acinar cell carcinoma: frequent allelic loss on chromosome 11p and alterations in the APC/beta-catenin pathway. Am J Pathol. 2002; 160:953–62.
79. Luttges J, Beyser K, Pust S, Paulus A, Ruschoff J, Kloppel G. Pancreatic mucinous noncystic (colloid) carcinomas and intraductal papillary mucinous carcinomas are usually microsatellite stable. Mod Pathol. 2003; 16:537–42. https://doi.org/10.1097/01.mp.0000072748.65178.2f.
80. de Wilde RF, Ottenhof NA, Jansen M, Morsink FH, de Leng WW, Offerhaus GJ, Brosens LA. Analysis of LKB1 mutations and other molecular alterations in pancreatic acinar cell carcinoma. Mod Pathol. 2011; 24:1229–36. https://doi.org/10.1038/modpathol.2011.83.
81. Humphris JL, Patch AM, Nones K, Bailey PJ, Johns AL, McKay S, Chang DK, Miller DK, Pajic M, Kassahn KS, Quinn MC, Bruxner TJ, Christ AN, et al, and Australian Pancreatic Cancer Genome Initiative. Hypermutation in pancreatic cancer. Gastroenterology. 2017; 152:68–74.e2. https://doi.org/10.1053/j.gastro.2016.09.060.
82. Wilentz RE, Goggins M, Redston M, Marcus VA, Adsay NV, Sohn TA, Kadkol SS, Yeo CJ, Choti M, Zahurak M, Johnson K, Tascilar M, Offerhaus GJ, et al. Genetic, immunohistochemical, and clinical features of medullary carcinoma of the pancreas: a newly described and characterized entity. Am J Pathol. 2000; 156:1641–51. https://doi.org/10.1016/s0002-9440(10)65035-3.
83. Laghi L, Beghelli S, Spinelli A, Bianchi P, Basso G, Di Caro G, Brecht A, Celesti G, Turri G, Bersani S, Schumacher G, Rocken C, Grantzdorffer I, et al. Irrelevance of microsatellite instability in the epidemiology of sporadic pancreatic ductal adenocarcinoma. PLoS One. 2012; 7:e46002. https://doi.org/10.1371/journal.pone.0046002.
84. Nakata B, Wang YQ, Yashiro M, Ohira M, Ishikawa T, Nishino H, Seki S, Hirakawa K. Negative hMSH2 protein expression in pancreatic carcinoma may predict a better prognosis of patients. Oncol Rep. 2003; 10:997–1000.
85. Maple JT, Smyrk TC, Boardman LA, Johnson RA, Thibodeau SN, Chari ST, Real FX. Defective DNA mismatch repair in long-term (> or =3 years) survivors with pancreatic cancer. Pancreatology. 2005; 5:220–7; discussion 7-8. https://doi.org/10.1159/000085275.
86. Goggins M, Offerhaus GJ, Hilgers W, Griffin CA, Shekher M, Tang D, Sohn TA, Yeo CJ, Kern SE, Hruban RH. Pancreatic adenocarcinomas with DNA replication errors (RER+) are associated with wild-type K-ras and characteristic histopathology. Poor differentiation, a syncytial growth pattern, and pushing borders suggest RER+. Am J Pathol. 1998; 152:1501–07.
87. Nakamura H, Arai Y, Totoki Y, Shirota T, Elzawahry A, Kato M, Hama N, Hosoda F, Urushidate T, Ohashi S, Hiraoka N, Ojima H, Shimada K, et al. Genomic spectra of biliary tract cancer. Nat Genet. 2015; 47:1003–10. https://doi.org/10.1038/ng.3375.
88. Lee H, Ross JS. The potential role of comprehensive genomic profiling to guide targeted therapy for patients with biliary cancer. Therap Adv Gastroenterol. 2017; 10:507–20. https://doi.org/10.1177/1756283x17698090.
89. Sithithaworn P, Haswell-Elkins MR, Mairiang P, Satarug S, Mairiang E, Vatanasapt V, Elkins DB. Parasite-associated morbidity: liver fluke infection and bile duct cancer in northeast Thailand. Int J Parasitol. 1994; 24:833–43.
90. Silva VW, Askan G, Daniel TD, Lowery M, Klimstra DS, Abou-Alfa GK, Shia J. Biliary carcinomas: pathology and the role of DNA mismatch repair deficiency. Chin Clin Oncol. 2016; 5:62. https://doi.org/10.21037/cco.2016.10.04.
91. Chan-On W, Nairismagi ML, Ong CK, Lim WK, Dima S, Pairojkul C, Lim KH, McPherson JR, Cutcutache I, Heng HL, Ooi L, Chung A, Chow P, et al. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat Genet. 2013; 45:1474–8. https://doi.org/10.1038/ng.2806.
92. Mimaki S, Totsuka Y, Suzuki Y, Nakai C, Goto M, Kojima M, Arakawa H, Takemura S, Tanaka S, Marubashi S, Kinoshita M, Matsuda T, Shibata T, et al. Hypermutation and unique mutational signatures of occupational cholangiocarcinoma in printing workers exposed to haloalkanes. Carcinogenesis. 2016; 37:817–26. https://doi.org/10.1093/carcin/bgw066.
93. Akiba N, Shiizaki K, Matsushima Y, Endo O, Inaba K, Totsuka Y. Influence of GSH S-transferase on the mutagenicity induced by dichloromethane and 1,2-dichloropropane. Mutagenesis. 2017; 32:455–62.
94. Toyooka T, Yanagiba Y, Suda M, Ibuki Y, Wang RS. 1, 2-Dichloropropane generates phosphorylated histone H2AX via cytochrome P450 2E1-mediated metabolism. Toxicol Lett. 2017; 272:60–7.
95. Yoshida T, Sugai T, Habano W, Nakamura S, Uesugi N, Funato O, Saito K. Microsatellite instability in gallbladder carcinoma: two independent genetic pathways of gallbladder carcinogenesis. J Gastroenterol. 2000; 35:768–74.
96. Rashid A, Ueki T, Gao YT, Houlihan PS, Wallace C, Wang BS, Shen MC, Deng J, Hsing AW. K-ras mutation, p53 overexpression, and microsatellite instability in biliary tract cancers: a population-based study in China. Clin Cancer Res. 2002; 8:3156–63.
97. Yanagisawa N, Mikami T, Yamashita K, Okayasu I. Microsatellite instability in chronic cholecystitis is indicative of an early stage in gallbladder carcinogenesis. Am J Clin Pathol. 2003; 120:413–7. https://doi.org/10.1309/byrn-alp8-gn63-dhaj.
98. Roa JC, Roa I, Correa P, Vo Q, Araya JC, Villaseca M, Guzman P, Schneider BG. Microsatellite instability in preneoplastic and neoplastic lesions of the gallbladder. J Gastroenterol. 2005; 40:79–86. https://doi.org/10.1007/s00535-004-1497-4.
99. Nagahashi M, Ajioka Y, Lang I, Szentirmay Z, Kasler M, Nakadaira H, Yokoyama N, Watanabe G, Nishikura K, Wakai T, Shirai Y, Hatakeyama K, Yamamoto M. Genetic changes of p53, K-ras, and microsatellite instability in gallbladder carcinoma in high-incidence areas of Japan and Hungary. World J Gastroenterol. 2008; 14:70–5.
100. Moy AP, Shahid M, Ferrone CR, Borger DR, Zhu AX, Ting D, Deshpande V. Microsatellite instability in gallbladder carcinoma. Virchows Arch. 2015; 466:393–402. https://doi.org/10.1007/s00428-015-1720-0.
101. Achille A, Biasi MO, Zamboni G, Bogina G, Iacono C, Talamini G, Capella G, Scarpa A. Cancers of the papilla of vater: mutator phenotype is associated with good prognosis. Clin Cancer Res. 1997; 3:1841–7.
102. Kim SG, Chan AO, Wu TT, Issa JP, Hamilton SR, Rashid A. Epigenetic and genetic alterations in duodenal carcinomas are distinct from biliary and ampullary carcinomas. Gastroenterology. 2003; 124:1300–10.
103. Park S, Kim SW, Kim SH, Darwish NS, Kim WH. Lack of microsatellite instability in neoplasms of ampulla of vater. Pathol Int. 2003; 53:667–70.
104. Sessa F, Furlan D, Zampatti C, Carnevali I, Franzi F, Capella C. Prognostic factors for ampullary adenocarcinomas: tumor stage, tumor histology, tumor location, immunohistochemistry and microsatellite instability. Virchows Arch. 2007; 451:649–57. https://doi.org/10.1007/s00428-007-0444-1.
105. Ruemmele P, Dietmaier W, Terracciano L, Tornillo L, Bataille F, Kaiser A, Wuensch PH, Heinmoeller E, Homayounfar K, Luettges J, Kloeppel G, Sessa F, Edmonston TB, et al. Histopathologic features and microsatellite instability of cancers of the papilla of vater and their precursor lesions. Am J Surg Pathol. 2009; 33:691–704. https://doi.org/10.1097/PAS.0b013e3181983ef7.
106. Agaram NP, Shia J, Tang LH, Klimstra DS. DNA mismatch repair deficiency in ampullary carcinoma: a morphologic and immunohistochemical study of 54 cases. Am J Clin Pathol. 2010; 133:772–80. https://doi.org/10.1309/ajcpgdde8plldrcc.
107. Suto T, Habano W, Sugai T, Uesugi N, Kanno S, Saito K, Nakamura S. Infrequent microsatellite instability in biliary tract cancer. J Surg Oncol. 2001; 76:121–6.
108. Williams AS, Huang WY. The analysis of microsatellite instability in extracolonic gastrointestinal malignancy. Pathology. 2013; 45:540–52. https://doi.org/10.1097/PAT.0b013e3283653307.
109. Ha H, Nam AR, Bang JH, Park JE, Kim TY, Lee KH, Han SW, Im SA, Kim TY, Bang YJ, Oh DY. Soluble programmed death-ligand 1 (sPDL1) and neutrophil-to-lymphocyte ratio (NLR) predicts survival in advanced biliary tract cancer patients treated with palliative chemotherapy. Oncotarget. 2016; 7:76604–12. https://doi.org/10.18632/oncotarget.12810.
110. Murphy MB. Adjunctive therapy of gastric cancer: moving the field forward. Oncology (Williston Park). 2016; 30:646–7.
111. Jain A, Javle M. Molecular profiling of biliary tract cancer: a target rich disease. J Gastrointest Oncol. 2016; 7:797.
112. Elias AW, Kasi PM, Stauffer JA, Thiel DD, Colibaseanu DT, Mody K, Joseph RW, Bagaria SP. The feasibility and safety of surgery in patients receiving immune checkpoint inhibitors: a retrospective study. Front Oncol. 2017; 7:121. https://doi.org/10.3389/fonc.2017.00121.
113. Li X, Wu WK, Xing R, Wong SH, Liu Y, Fang X, Zhang Y, Wang M, Wang J, Li L, Zhou Y, Tang S, Peng S, et al. Distinct subtypes of gastric cancer defined by molecular characterization include novel mutational signatures with prognostic capability. Cancer Res. 2016; 76:1724–32. https://doi.org/10.1158/0008-5472.can-15-2443.
114. Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, Benz CC, Yau C, Laird PW, Ding L, et al, and Cancer Genome Atlas Research Network. Integrated genomic characterization of endometrial carcinoma. Nature. 2013; 497:67–73. https://doi.org/10.1038/nature12113.
115. Bouffet E, Larouche V, Campbell BB, Merico D, de Borja R, Aronson M, Durno C, Krueger J, Cabric V, Ramaswamy V, Zhukova N, Mason G, Farah R, et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol. 2016; 34:2206–11. https://doi.org/10.1200/jco.2016.66.6552.
116. Hodges TR, Ott M, Xiu J, Gatalica Z, Swensen J, Zhou S, Huse JT, de Groot J, Li S, Overwijk WW, Spetzler D, Heimberger AB. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: implications for immune checkpoint immunotherapy. Neuro-oncol. 2017. https://doi.org/10.1093/neuonc/nox026.