
INTRODUCTION
Generally, jawed vertebrates, such as Osteichthyes and mammals, have developed an elaborate adaptive immune system with diversified B-cell and T-cell antigen receptors (BCRs and TCRs) [1, 2]. Yet, jawless vertebrates, such as cyclostomes, have variable lymphocyte receptors (VLRs) with somatically rearranged LRR ectodomains [3, 4]. Therefore, the adaptive immunity may be traced to an early stage of vertebrate evolution (Figure 1). A recent study found that primordial recombination-activating gene (protoRAG) in amphioxus could correctly splice mRNA and conduct actual transposition in human cells [5]. Cao et al. reported the structure of a VLR-like gene (similar to VLR-c type) and preliminarily detected innate immune function of this gene in amphioxus [6]. However, antigen receptors with naturally adaptive immune activity have not been found in amphioxus. Therefore, the assumption is that cephalochordates strongly rely on the innate immune system according to our current knowledge [1, 7].
Amphioxus, the only modern representative of the subphylum Cephalochordata, has long been considered the extant invertebrate most closely related to the proximate invertebrate ancestor of vertebrates [7-9]. Amphioxus is a model organism widely used in comparative genomics, developmental homology of vertebrates and comparative immunology studies [10]. Generally, model amphioxus primarily includes three genera (Branchiostoma, Epigonichthys and Asymmetron), with most species (over 28 of ~31 species) in Branchiostoma [7], which has a wide distribution from low to high latitudes [11]. For the tropical amphioxus Asymmetron, two recognized species are primarily distributed along low latitudes [12], whereas only one Epigonichthys species is recognized [7]. Yue et al. reported that many fast-evolving genes between Branchiostoma and Asymmetron were associated with innate immunity [7]. However, two Branchiostoma species (B. floridae and B. belcheri) separated from the common ancestor ~120 million years ago (Mya) (Figure 1) [7]. Moreover, Branchiostoma has a wide distribution, indicating that Branchiostoma species may have experienced more diverse and complex environments than the other two genera in amphioxus. Thus, fast-evolving genes have evolved functional divergence between Branchiostoma and Asymmetron, which would be different from that of the interspecific divergence of fast-evolving genes within Branchiostoma. Within the completed Branchiostoma genome (floridae and belcheri) sequence [1, 9], amphioxus exhibits complexity and diversity of the gene repertoire for innate immunity [1]. However, evolutionary characteristics of innate immunity in the speciation of amphioxus remain largely unknown, impeding our understanding of the evolution of innate immune-related genes in amphioxus.
Previous studies showed that the emergence of mmunoglobulin-based adaptive immunity accompanied the origin of jawed vertebrates (gnathostomes) [2] (Figure 1). However, the adaptive immune system of cartilaginous fish (Chondrichthyes, the original jawed vertebrates) lacks key transcription factors, such as the cluster of differentiation 4 (CD4), RAR related orphan receptor C (RORC), and most CD4-lineage-specific cytokines and cytokine receptors, suggesting that adaptive immunity is more complete in bony vertebrates with the acquisition of helper and regulatory functions of T-cells that recognize MHC class II molecules [2]. Osteichthyes superclass, the oldest group in bony vertebrates (Figure 1), have developed more elaborate adaptive immune systems [2]. This animal group is composed of lobe-finned fish (Sarcopterygii) and ray-finned fish (Actinopterygii). The African coelacanth (Latimeria chalumnae) is a rare existing in Coelacanthimorpha in Sarcopterygians [13] (Figure 1). Because of a wide distribution and high adaptability and diversity, ray-finned fish are important experimental materials in studies of genetics and immunity [14-16]. The zebrafish (Danio rerio) and pufferfish (Takifugu rubripes) are two kinds of fish that are well-known model animals with sequenced genomes that are the subjects of many comparative studies conducted to explore genome evolution, gene function, and development and to discover immune- and disease-related genetic resources [17-20].
Here, we ask scientific questions: Within Branchiostoma, are innate immune-related genes fast-evolving? Next, whether this situation exists within Osteichthyes? Finally, what difference of immune-related genes in Osteichthyes can be obtained based on functional enrichments of fast-evolving gene sets? First, we performed comparative analyses between B. floridae and B. belcheri (Bf vs Bb) to identify fast-evolving gene sets (including particularly fast-evolving genes, PFEGs, and moderately fast-evolving genes, MFEGs); second, enrichment of functional terms (Gene ontology, GO) of PFEGs and MFEGs was determined to explore their potential functions; finally, to detect pathways that contained fast-evolving genes, we determined significantly enriched pathways (as defined in Kyoto Encyclopedia of Genes and Genomes, KEGG) for MFEGs. Furthermore, two parallel analyses for fast-evolving gene sets were conducted for Danio rerio vs. Latimeria chalumnae (Dr vs Lc) and D. rerio vs. Takifugu rubripes (Dr vs Tr) (these animals are in basal or advanced evolutionary nodes of Crossopterygii or Actinopterygii in Osteichthyes), respectively. To observe whether PFEGs (enriched KEGG pathways associated with metabolism and energy production) possessed innate immune activity in amphioxus, we used quantitative real-time PCR (qRT-PCR) to detect the expression responses of four genes in skin, intestine or muscle of B. belcheri challenged with lipopolysaccharide (LPS). The results of this study will provide some broader insights into the evolution of immune-related genes in cephalochordates and Osteichthyes.
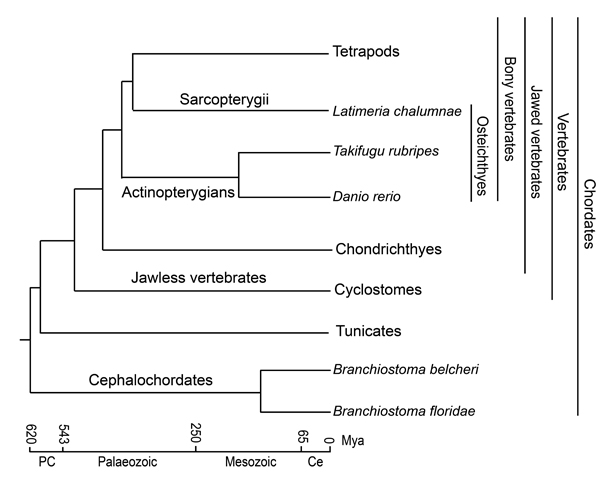
Figure 1: Phylogenetic tree and evolutionary time frame for chordates. Adapted from Yue et al. and Venkatesh et al. [2, 7].
RESULTS
Sequence annotation, putative orthologous genes and test for selection pressure
Statistics for the annotation information of the gene sets used in this study is shown in Supplementary Table 1. We obtained 15,008, 12,645 and 14,475 orthologous gene pairs in Bf vs Bb, Dr vs Lc and Dr vs Tr, respectively. After trimming and gap filtration, the alignment coverage of each of the orthologous gene pairs in these three comparable groups was ≥ 80%, and the range of the alignment lengths was from 138 to 22,551 bp in Bf vs Bb, 129 to 21,705 bp in Dr vs Lc and 141 to 19,671 bp in Dr vs Tr. Table 1 shows the values of Ka, Ks and Ka/Ks across all genes for the three comparable groups. For the Bf vs Bb, we removed 4,393 invalid genes and the remaining genes were used in further analyses based on Ka/Ks values. Regarding the Dr vs Lc and Dr vs Tr sets of orthologous genes that we used, more than 50% of orthologous gene pairs had values of Ks > 1 (genome-wide average Ks > 3, Table 1). Thus, we only considered the Ka values as the criteria to scale evolutionary rates in Dr vs Lc and Dr vs Tr. Independent of Ka/Ks or Ka values that we used when identifying the PFEGs, the PFEG sets showed a 2.5- to 3.5-fold higher evolutionary rates than that of the genome-wide average (Table 1). We found a 2.1- to 3.2-fold higher evolutionary rate in MFEG sets than that of the genome-wide average under more relaxed criterion.
Analysis of fast-evolving genes
The top 20 fast-evolving genes in three comparable groups are presented in Table 2. In Bf vs Bb, the most fast-evolving genes encoded an iron-sulfur cluster assembly scaffold protein (ISCU). Additionally, seven fast-evolving genes encoded typical immune-related proteins, including Toll-like receptor 4 (TLR4), interleukin-17 receptor D (IL17RD), complement component C1q receptor (CD93), complement factor H-related protein 1 (CFHR1), interferon regulatory factor 4 (IRF4), TNF receptor-associated factor 6 (TRAF6), NACHT, LRR and PYD domains-containing protein 3 (NLRP3). The most fast-evolving genes in Dr vs Lc encoded mucin-1 (MUCIN1). A large number of fast-evolving genes encoded typical immune-related proteins were detected, such as macrophage receptor with collagenous structure (MARCO), interleukin 2 receptor, beta (IL2RB), hematopoietic death receptor (HDR), and so on. In Dr vs Tr, the most fast-evolving genes encoded an interleukin 13 receptor, alpha 1 (IL13RA1), followed by lymphotoxin alpha (LTA). Moreover, three fast-evolving genes encoded cytokine receptor family member b2, b1, b6 (CRFB2, 1, 6); six fast-evolving genes encoded interleukin-related proteins, including interleukin 20 receptor, alpha (IL20RA), interleukin 21 (IL21), interleukin 15, like (IL15L), interleukin-1 family member A (IL1FMA), interleukin 2 receptor, beta (IL2RB), interleukin 12a (IL12A). Notably, CD79b molecule, immunoglobulin-associated beta (CD79B) involving B-cell antigen receptor function, was identified.
Analysis of functional classes of fast-evolving genes
We found that 130 and 138 GO categories were significantly enriched for PFEG sets in Bf vs Bb based on the values of Ka and Ka/Ks, respectively (Table 1, Supplementary Tables 2 and 3); for the MFEG sets, 227 (top 10% Ka) and 166 (top 15% Ka/Ks) terms were significantly enriched. Most GO terms (> 80%) enriched in the PFEG set were included in those enriched in the MFEG sets (Supplementary Tables 2 and 4). Additionally, for MFEG sets in Bf vs Bb, more than 90% of GO terms identified by Ka/Ks overlapped with terms identified by Ka (Supplementary Tables 4 and 5). We found that 21 and 23 GO terms were overrepresented in the PFEG and MFEG sets of Dr vs Lc, respectively (Table 1, Supplementary Tables 6 and 7). For Dr vs Tr, 32 and 61 GO categories were significantly enriched in the PFEG and MFEG sets, respectively (Table 1, Supplementary Tables 8 and 9). GO terms enriched in the MFEG set included most of GO terms (> 70%) enriched in the PFEG set in both Dr vs Lc and Dr vs Tr.
Table 1: Means of genome-wide Ka, Ks, and Ka/Ks values and statistics of GO and KEGG terms for three comparable groups.
All genes |
Particularly fast-evolving genes (top 5% identified by Ka) |
Fast-evolving genes (top 10% identified by Ka) |
Particularly fast-evolving genes (top 5% identified by Ka/Ks) |
Fast-evolving genes (top 15% identified by Ka/Ks) |
|||||||
Bf vs Bb |
Dr vs Lc |
Dr vs Tr |
Bf vs Bb |
Dr vs Lc |
Dr vs Tr |
Bf vs Bb |
Dr vs Lc |
Dr vs Tr |
Bf vs Bb |
Bf vs Bb |
|
Ka |
0.3033 |
0.2826 |
0.2042 |
1.0338 |
0.6965 |
0.5756 |
1.0174 |
0.6341 |
0.5003 |
1.0307 |
1.0174 |
Ks |
0.8714 |
3.2931 |
3.3977 |
0.8963 |
0.9459 |
0.8936 |
0.9459 |
||||
Ka/Ks |
0.3162 |
1.1584 |
1.0804 |
1.1586 |
1.0804 |
||||||
T# |
130 |
21 |
32 |
227 |
23 |
61 |
138 |
166 |
|||
N* |
87 |
22 |
25 |
147 |
20 |
40 |
92 |
112 |
|||
N/T (%) |
66.92 |
95.65 |
78.13 |
64.76 |
95.24 |
65.57 |
66.67 |
67.47 |
|||
K& |
20 |
31 |
37 |
19 |
|||||||
# T: Total number of GO terms for which the particular gene sets are significantly enriched; *N: Number of immune-related GO for which the particular gene sets are significantly enriched; &K: Number of KEGG terms for which the particular gene sets are significantly enriched. Branchiostoma floridae vs B. belcheri (Bf vs Bb); Danio rerio vs. Latimeria chalumnae (Dr vs Lc); D. rerio vs. Takifugu rubripes (Dr vs Tr).
Among enriched GO terms for Bf vs Bb, most were associated with the neural/immune category. For example, 66.92% of GO terms identified by the top 5% Ka and 66.67% by the top 5% Ka/Ks values were included in the list of neural/immune-related GO terms [21]. Additionally, 64.76% and 67.47% of GO terms identified by the top 10% Ka and the top 15% Ka/Ks values, respectively, were detected in this curated collection [21]. The nervous and immune systems are well known to interact with each other; the function of the immune system is modulated by the nervous system, which in turn influences nervous system [22-24]. In the analysis of Dr vs Lc, the enrichment of neural/immune-related functional GO terms (both innate and adaptive) by fast-evolving genes was more obvious than that of Bf vs Bb and Dr vs Tr (Table 1). A relatively fast evolution of immune-related genes was also observed in other fish, including the zebrafish and grass carp (Ctenopharyngodon idellus) [25], large yellow croaker (Larimichthys crocea) and Atlantic cod (Gadus morhua) [14, 26]. Many of the enriched GO terms for fast-evolving genes identified by Bf vs Bb were related to transmembrane cellular function, signal activity and receptor activity associated with response to stimuli, indicating potential roles in innate immunity. The most compelling GO categories were those involved with the mitochondrial membrane, such as “mitochondrial part”, “mitochondrial inner membrane”, “mitochondrial membrane”, “mitochondrial envelope”, “mitochondrial respiratory chain” and “mitochondrial membrane part” (Supplementary Tables 2-5). Many immune-related genes are located in mitochondrial membranes and mitochondria and are part of the respiratory chains that are key in the innate immune system [27, 28]. Additionally, several well-known immune genes of amphioxus were also detected in fast-evolving gene sets, such as the NOD-like receptor (NLR) genes, fork-head transcription factor (FOX) gene and IkB kinase gene. Furthermore, the enriched GO categories for fast-evolving genes in Dr vs Lc and Dr vs Tr were primarily related to immune/defense response, cytokines, receptor activity and cell death. In particular, the GO terms involved in adaptive immunity were detected in the comparable groups within bony fish and included terms (i.e. “MHC protein complex”) which functioned in the recognition of cell surfaces for T-cells (Supplementary Tables 7 and 8).
Analysis of pathways of fast-evolving genes
Fast-evolving genes in Bf vs Bb, the top 10% Ka and 15% Ka/Ks values were classified significantly into 20 and 19 KEGG pathways, respectively (Table 1). The results showed some immune- and disease-related pathways, such as the proteasome, Jak-STAT signaling pathway, the peroxisome, bacterial invasion of epithelial cells and NOD-like receptor signaling pathway (Figure 2). Some well-known pathways involved in energy production and amino acid metabolism were notable. For Dr vs Lc and Dr vs Tr, the significant results of enriched KEGG pathways for the MFEG set had 31 and 37 terms (Table 1), respectively. When the top 20 KEGG categories were plotted based on descending order of the significance level, we found that almost all pathways were related to a response to immune stimulation (Figure 3). These pathways primarily involved cell surface receptors in innate immunity, infections and immune diseases, cytokines, apoptosis and adaptor molecules in immune signaling pathways. Particularly, we detected “T-cell receptor signaling pathway”, “intestinal immune network for IgA production” and “hematopoietic cell lineage”, indicating that some of the genes associated with adaptive immunity were fast evolving in Dr vs Tr.
Table 2: Top 20 most fast-evolving genes in each of three comparable groups.
Gene description (B. floridae versus B. belcheri ) |
Ka/Ks |
Gene name |
Iron-sulfur cluster assembly scaffold protein IscU |
1.5847 |
ISCU |
Ultraviolet-B receptor UVR8 |
1.5294 |
UVR8 |
Toll-like receptor 4 |
1.4546 |
TLR4 |
Interleukin-17 receptor D |
1.4422 |
IL17D |
DNA-directed RNA polymerase II subunit RPB7 |
1.4417 |
POLR2G |
Vacuolar ATPase assembly integral membrane protein VMA21 |
1.4351 |
VMA21 |
Complement component C1q receptor |
1.4237 |
CD93 |
E3 ubiquitin-protein ligase TRIM71 |
1.4069 |
TRIM71 |
Transforming growth factor-beta-induced protein |
1.3847 |
TGFBI |
Complement factor H-related protein 1 |
1.3837 |
CFHR1 |
UBA-like domain-containing protein 2 |
1.3790 |
UBALD2 |
Interferon regulatory factor 4 |
1.3760 |
IRF4 |
Thymosin beta-10 |
1.3751 |
TMSB10 |
Polycomb group RING finger protein 1 |
1.3662 |
PCGF1 |
Astrocytic phosphoprotein PEA-15 |
1.3645 |
PEA15 |
Cytochrome c2 |
1.3574 |
C2 |
TNF receptor-associated factor 6 |
1.3521 |
TRAF6 |
Transmembrane protein 203 |
1.3421 |
TMEM203 |
Sperm-associated antigen 7 |
1.3412 |
SPAG7 |
NACHT, LRR and PYD domains-containing protein 3 |
1.3376 |
NLRP3 |
Gene description (D. rerio versus L. chalumnae) |
Ka |
Gene name |
Mucin-1 |
0.9894 |
MUCIN1 |
Chromogranin B |
0.9799 |
CHGB |
Macrophage receptor with collagenous structure |
0.9249 |
MARCO |
Podocalyxin-like 2 |
0.8992 |
PODXL2 |
Meis homeobox 1 b |
0.8945 |
MEIS1 |
Eph receptor A3 |
0.8915 |
EPHA3 |
MACRO domain containing 1 |
0.8908 |
MACROD1 |
Notch 2 |
0.8753 |
NOTCH 2 |
Complement factor H like 4 |
0.8690 |
CFHL4 |
Spalt-like transcription factor 1a |
0.8690 |
SALL1A |
Myelin transcription factor 1a |
0.8648 |
MYT1A |
CD79a molecule, immunoglobulin-associated alpha |
0.8630 |
CD79a |
Mitogen-activated protein kinase kinase kinase 5 |
0.8609 |
MAP3K5 |
Interleukin 2 receptor, beta |
0.8536 |
IL2RB |
Hematopoietic death receptor |
0.8523 |
HDR |
NLR family, CARD domain containing 3 |
0.8498 |
NLRC3 |
Calcitonin/calcitonin-related polypeptide, alpha |
0.8456 |
CALCA |
Caspase 8 associated protein 2 |
0.8445 |
CASP8AP2 |
MAP7 domain containing 2a |
0.8443 |
MAP7D2A |
Nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, zeta |
0.8405 |
NFKBIZ |
Gene description (D. rerio versus T. rubripes) |
Ka |
Gene name |
Interleukin 13 receptor, alpha 1 |
1.1271 |
IL13RA1 |
lymphotoxin alpha (TNF superfamily, member 1) |
0.9460 |
LTA |
Telomeric repeat binding factor a |
0.8847 |
TERFA |
Secreted phosphoprotein 1 |
0.8533 |
SPP1 |
Cytokine receptor family member b2 |
0.8403 |
CRFB2 |
Neurotrophic tyrosine kinase, receptor, type 2a |
0.8241 |
NTRK2A |
Serine/threonine/tyrosine interacting-like 1 |
0.8119 |
STYXL1 |
Interferon-induced protein with tetratricopeptide repeats 15 |
0.8108 |
IFIT15 |
Interleukin 20 receptor, alpha |
0.8070 |
IL20RA |
CD79b molecule, immunoglobulin-associated beta |
0.8065 |
CD79B |
Polymerase (DNA directed) kappa |
0.8056 |
POLK |
Interleukin 21 |
0.8042 |
IL21 |
Cytokine receptor family member b1 |
0.7987 |
CRFB1 |
Interleukin 15, like |
0.7981 |
IL15L |
Interleukin-1 family member A |
0.7902 |
IL1FMA |
Interleukin 2 receptor, beta |
0.7902 |
IL2RB |
Interleukin 12a |
0.7884 |
IL12A |
Collagen, type IV, alpha 3 |
0.7844 |
COI4A3 |
Threonine synthase-like 2 |
0.7801 |
THNSL2 |
Cytokine receptor family member b6 |
0.7744 |
CRFB6 |
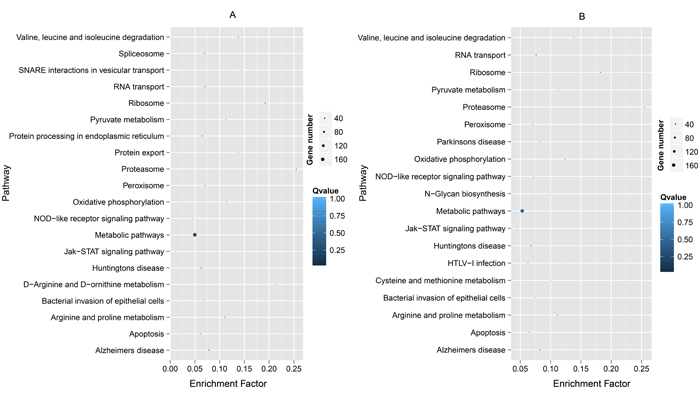
Figure 2: Top 20 KEGG pathways enriched by the top 10% Ka (A) and the top 15% Ka/Ks (B) for Branchiostoma belcheri vs B. floridae in descending order of the P-values. A low P-value indicates a high level of enrichment.
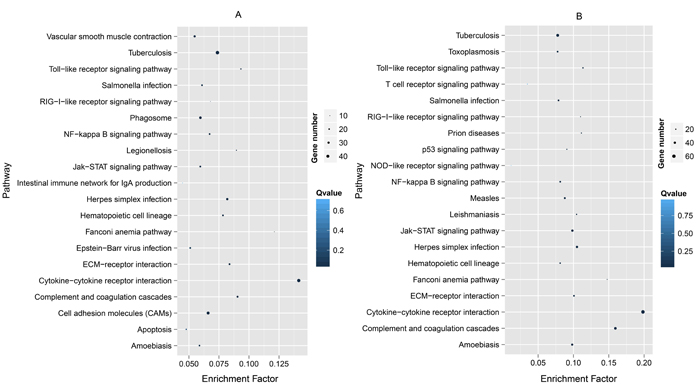
Figure 3: Top 20 KEGG pathways enriched by the top 10% Ka for Latimeria chalumnae vs Danio rerio (A) and for Takifugu rubripes vs D. rerio (B) in descending order of the P-values.
Expression of HMGCL, SAT2, CYBS and MDH2 challenged with lipopolysaccharide
To determine the innate immune activity of the fast-evolving genes in metabolism or energy production-related pathways, we measured the expression of four of these genes in the muscle, intestine or skin of B. belcheri challenged by a LPS. The expression responses of four types of genes to LPS stimulation were determined over a 72 hour time course: mitochondrial hydroxymethylglutaryl-CoA lyase (HMGCL) in skin, diamine acetyltransferase 2 (SAT2) in the intestine, and succinate dehydrogenase cytochrome B small subunit precursor (CYBS) and mitochondrial malate dehydrogenase (MDH2) in muscle (Figure 4). Upon a LPS simulation, only one expression peak was detected for HMGCL at 6 hours post infection (hpi) with a 4.4-fold increase compared with the control, and the level of expression was significantly higher than that from 12 to 72 hpi. For SAT2, no significant increase was detected at any hpi. In this experiment, we found that CYBS only showed significantly higher expressions at 24 and 48 hpi than that in the control. Regarding the expression profile for MDH2, we detected an initial, significant up-regulation at 2 hpi, then reached a single expression peak in muscle at 24 hpi when the expression was 4.3-fold higher than that of the control. At the two later time points, the expression of MDH2 continued to be up-regulated, compared with that of the control.
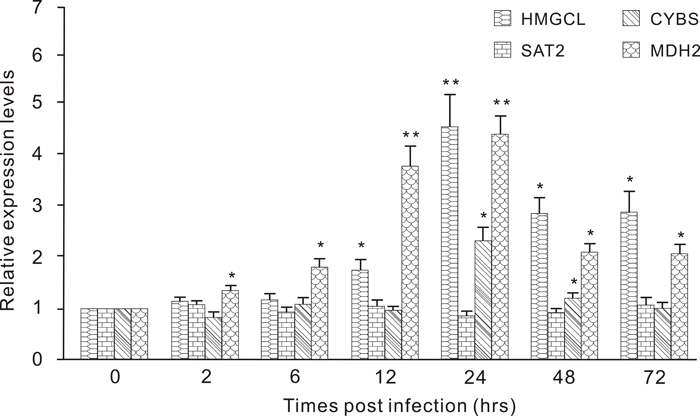
Figure 4: Expression profiles of HMGCL, SAT2, CYBS and MDH2 in the different tissues (skin, intestine and muscle) of Branchiostoma belcheri challenged with LPS. HMGCL in the skin, SAT2 in the intestine, and CYBS and MDH2 in the muscle Expression level at each time point is shown as the mean ± SD (n = 3). “**” highly significant difference compared with the control, P < 0.01; “*”significant difference, P < 0.05.
DISCUSSION
In the absence of an adaptive immune system, amphioxus primarily depends on a diverse and complex innate immune system to provide protection against viral and microbial pathogens in seawater [1, 7]. Huang et al. found that many classical innate immune-related gene repertoires experienced significant expansion in B. floridae, including cell surface receptors (Toll-like receptors, TLRs; NLRs), scavenger receptors (SRCRs), caspases, adaptors and components of the complement system [1]. Some domains have undergone strongly positive selection in expansion, such as leucine-rich repeats (LRRs) [1]. Under gene expansion and domain shuffling, the innate immune system that developed in amphioxus was even shaped by altering the topology of protein-protein networks [1, 29]. Therefore, the high proportion of fast-evolving genes involved in innate immunity observed in our results was not unexpected. In particular, based on this exploration, improvement in the immune system likely relied on rapid variation in the sequences and copies of innate immune-related genes; perhaps, this fast evolution of sequences was an important factor in the expansion of genes, as reported in ticks (Ixodes scapularis) [30, 31]. Among the top 20 fast-evolving genes, we found many typical immune-related genes in innate immune system, i.e. cytokines, pattern recognition receptor (PRRs), complement and signal transducers. Some well-known pathways with annotated information were involved in metabolism and energy production in enriched KEGG terms of Bf vs Bb, such as “metabolic pathway” and “oxidative phosphorylation”. This result was not completely unexpected because several neural/immune GO terms were also annotated to this information, such as “metabolic process” and “oxidation-reduction process” (Figure 2). Additionally, many studies showed that several metabolism-related genes and pathways played key roles in innate immunity [32-34]. In particular, genes associated with metabolism and energy production usually experienced fast evolution among groups (ecotypes) living in habitats with extreme environmental differences (e.g., fish living at high and low elevations, facing severe cold and heat, and inhabiting marine and fresh water) [35, 36]. Nevertheless, amphioxus are generally found half-buried in sand (with much less movement than jawed vertebrates) between 5 and 10 meters below sea level, with a distribution in moderate temperature ranges (from shallow temperate to tropical seas) [37]. Thus, the enriched pathways with annotation information involved in metabolism and energy most likely participated in innate immunity. Additionally, three PFEGs (HMGCL, CYBS and MDH2) enriched in metabolic and energy production-related pathways showed an expression response to LPS, further supporting our contention. However, most innate immune activity of these genes commonly appeared at 24 hpi, suggesting possible indirect roles in innate immunity. SAT2 did not show a response in the intestine challenged with LPS, indicating that some members associated with these pathways might indeed function primarily to meet metabolism and energy requirements. However, this relation requires further confirmation in more challenged tissues with more post-infection time points, number of genes and types of stimuli. Overall, many fast-evolving genes were related to innate immunity within Branchiostoma. Our study provided evolutionary characteristics of the interspecific divergence of immune-related genes within the same amphioxus genus. Moreover, previous studies have reported that many fast evolving genes were involved in innate immunity between Branchiostoma and Asymmetron [7], therefore we proposed that this phenomenon may be common in cephalochordates.
In addition, in our parallel analyses of Dr vs Lc and Dr vs Tr, several GO terms that were primarily enriched involved cytokines, immune processes, defense, receptor signaling pathways and apoptosis. Both within Actinopterygii (Dr vs Tr) and between Actinopterygii and Crossopterygii (Dr vs Lc), most of enriched GO terms were related to immunity. Additionally, we also detected many typical innate immune-related genes among the top 20 fast-evolving genes in these two comparable groups, of which genes involving interleukin function were found much more frequently and the other genes associated with complement, PRRs, cytokines, signal transducers and caspases were also well-know typical innate immune components. However, Osteichthyes have developed a more elaborate adaptive immune system, which includes major histocompatibility complex (MHC), immunoglobulins (IGs), costimulatory molecules and relevant cytokines [2, 38]. In this study, we detected some genes involving adaptive immunity, including CD79b molecule, immunoglobulin-associated beta (CD79B), and lymphotoxin alpha (LTA), in the list of the top 20 fast-evolving genes. In addition, many cytokines and adaptors play key roles in both innate and adaptive immunity [38, 39]. Therefore, the enriched GO terms only indicated that many fast-evolving genes in Osteichthyes were involved in immunity, and further analyses will be required to confirm whether these genes are related to innate or adaptive immunity. Notably, most KEGG terms enriched by fast-evolving genes in Dr vs Lc were highly similar to those in Dr vs Tr, and some of these are well known to participate in innate immunity, including “Toll-like receptor signaling pathway”, “Complement and coagulation cascades”, “NOD-like receptor signaling pathway”, and “RIG-I-like receptor signaling pathway”. Thus, genes involved in innate immunity had a high proportion of the fast-evolving genes, which showed a pattern of parallel evolution between Osteichthyes and cephalochordates. A highly open environment provided opportunities for aquatic pathogens and other microorganisms in natural aquatic systems to spread rapidly and widely [40, 41], and within variable microenvironments, populations of microorganisms could evolve rapidly [42]. Therefore, it was possible explanation that many of the genes involved in innate immunity were fast evolving.
Compared with cephalochordates, some lineage-specific genes, GO and KEGG terms involved in adaptive immunity were fast-evolving in Osteichthyes, such as genes encoding CD79B, the “MHC protein complex”, “T-cell receptor signaling pathway”, and “Intestinal immune network for IgA production” (Figure 3). Indeed, many components associated with adaptive immunity (i.e. immunoglobulin genes, adaptive immunity relevant cytokines and MHC genes) experienced a certain level of expansion in Osteichthyes [43, 44], which suggested that the functions of fast-evolving genes involved in immunity were more diverse in Osteichthyes than those in cephalochordates. Therefore, we hypothesized that improved ability of Osteichthyes to defend against pathogens may be due to a diverse and complex immune system and also an increase in the diversity of immune-related gene libraries with fast evolution (i.e., rapid evolution of adaptive immune-related genes played a key role). It is noteworthy that within Osteichthyes (between Crossopterygii and Actinopterygii), we found that the ratio of number of immune-related GO to total number of GO terms for which the particular gene sets are significantly enriched in Dr vs Lc was notably higher (~95%) than that of Dr vs Tr (Table 1), indicating that immune-related genes tend to rapidly evolve in different intensity for different groups within Osteichthyes. In addition to those immune-related genes in cephalochordates, Osteichthyes also possessed many other fast-evolving genes involved in activity, proliferation of and immunity with B- and T- cells, immunoglobulin function, regulation of cytokines, leukocyte function, regulation of CD4 and CD8, and lymphocyte function. Moreover, in previously paralleled analysis in mammals, Yue et al. found that the GO terms enriched by fast-evolving genes in the placental–marsupial comparison (mouse vs. opossum) were highly and diversely involved in adaptive immunity (see Supplementary Tables 2 and 3 in [7]). Thus, compared with cephalochordates, Osteichthyes developed some fast-evolving genes associated with adaptive immunity to combat pathogens, but the diversity and complexity may be much less than that of mammals. Given that amphioxus and Osteichthyes are key models for investigating the evolution of chordate immune systems [8, 9, 38], the results of this study demonstrated fast evolution of immune-related genes in cephalochordates and Osteichthyes, and provided some useful information for understanding the evolutionary characteristics of immune-related genes in chordates.
MATERIALS AND METHODS
Downloading and annotation of protein-coding gene sets
The protein-coding sequences (CDSs) and corresponding protein sequences of each sampled species were downloaded from online databases (Supplementary Table 10). Considering alternative splicing in which multiple CDSs corresponded to a single gene, the longest CDS of each of the reference genes was extracted as a unique representative for the corresponding gene. The Basic Local Alignment Search Tool (BLAST) finds regions of local similarity between sequences. The software compares nucleotide or protein sequences to target databases to infer functional and evolutionary relationships between sequences (https://blast.ncbi.nlm.nih.gov/Blast.cgi). BLAST tool (ftp://ftp.ncbi.nlm.nih.gov/blast/) with a default E-value (10e-5) was used to annotate genes to the NCBI nonredundant (NR) protein, GO and KEGG databases.
Construction of putative orthologs
We identified putative orthologs for each of the three comparable groups using Proteinortho (v5.15) [45], a reciprocal best hits based method that is suitable for large-scale analysis. If multiple genes from a species are detected, they are assigned into a co-orthologous group; next, we retained the first member as a unique representative of each co-orthologous group for further analyses. We extracted corresponding CDS sequences according to the output results of Proteinortho using custom Perl script. Then, nucleotide sequence of each orthologs CDS was aligned by PRANK with the “-codon” parameter [46], and aligned sequences with ambiguous alignments were removed. A manual check was also conducted to correct potential errors. Trimmed alignments that contained fewer than 20 codons were removed.
Tests for selection pressure and selection of fast-evolving genes
For the ortholog sets obtained, we calculated the nonsynonymous substitution (Ka), synonymous substitution (Ks) and Ka/Ks values using the KaKs_calculator based on a maximum-likelihood (ML) method [47]. Then, invalid orthologous pairs (with Ka = 0, Ks = 0 or Ks > 1) were removed to avoid substitution saturation (mutations are in steady state) at the synonymous substitutions when analyses were further conducted based on Ka/Ks values [7]; genes with values of Ks > 1 were retained when the fast-evolving gene sets were defined by Ka [7]. Generally, a higher Ka or Ka/Ks value indicates a faster evolutionary rate [7, 48]. For Bf vs Bb, we defined genes with the top 5% Ka and Ka/Ks values as PFEGs according to the method of Yue et al. [7]. To ensure the robustness of this analysis and reliable results, we examined alternative Ka (top 10%) and Ka/Ks cutoffs (top 15%) (MFEGs) to determine whether enriched GO terms were sensitive to the different thresholds.
Additionally, when the number of genes with Ks > 1 (saturated values) was more than 40% in all orthologous pairs, which suggested that the evolutionary scale was too large between two species and that the saturated Ks values biased the estimated Ka/Ks ratio, we were required to mask out the Ka/Ks ratio [7]. Ka values are recommended as a superior proxy for the measurement of evolutionary rates in sorting genes, particularly under this condition; thus, we defined the fast-evolving genes based on Ka measurements according to previous methods [7, 48]. For Dr vs Lc and Dr vs Tr, two PFEG sets were identified using the top 5% Ka criteria. Alternatively, we also defined two MFEG (top 10% Ka) sets for the Dr vs Lc and Dr vs Tr. Our analyses reflected the divergence degree of orthologous pairs between two species and did not discriminate whether a given gene had a faster evolutionary rate in one species than in another.
Enrichment for functional classes and pathways of fast-evolving genes
Enrichment for functional terms of the defined fast-evolving gene sets was implemented in the Blast2GO pipeline based on Fisher’s exact test [49]. Multiple test correction was conducted with the Benjamini and Hochberg (BH) methods using the p.adjust module in the R statistical software package to control the false discovery rate (FDR). The set of significantly enriched GO categories (FDR values <0.05) was further filtered using GO Trimming software (v2.0) to remove redundant categories [50]. Immune-related GO terms were identified according to the method of Yue et al. [7, 21]. Namely, GO terms enriched for in the defined PFEG and MFEG sets were included in a list of neural/immune-related GO terms [21], which were considered as the target GO categories. Additionally, enriched analyses for KEGG pathways of the PFEG and MFEG sets were performed using KOBAS 2.0 [51], and the pathways with P-values < 0.05 were considered as significantly enriched targets.
Collection of experimental tissues of amphioxus
To explore whether genes in the metabolic and energy production-related KEGG pathways enriched in fast-evolving gene sets had innate immune activity, we selected the following four genes with different evolutionary rates in the sets that only contained genes with Ka/Ks > 1: HMGCL (higher rate, Ka/Ks = 1.38, in “Metabolic pathways”, ko01100), SAT2 (high rate, Ka/Ks = 1.14, in “Arginine and proline metabolism”, ko00330), CYBS (moderate rate, Ka/Ks = 1.09, in “Oxidative phosphorylation”, ko00190) and MDH2 (low rate, Ka/Ks = 1.04, in “Metabolic pathways”, ko01100), according to annotated information. Then, we further manually confirmed the genes using online BLAST in NCBI. Furthermore, we detected expression profiles of the four target genes in the transcriptomic database of different normal tissues of B. belcheri (http://wcy.pkusulab.com/). HMGCL showed the highest expression in the skin, SAT2 in the intestine, and CYBS and MDH2 showed specific expression in muscle. Therefore, adult individuals were fed and treated to empty contents of intestine before sampling, following the methods of Zhang et al. [52]. Then, we collected three tissues (intestine, skin and muscle) of B. belcheri which were challenged with 1 mg/ml LPS according to the method of previous studies [53]. For each tissue, samples with three biological replications were collected at seven timing points after LPS challenge (0, 2, 6, 12, 24, 48 and 72 h; 10 μl/individual). Each sample group contained approximate 12 adult individuals. The samples collected at 0 h were only injected with PBS (used to dissolve LPS powder to 1 mg/ml) and used as the control. All samples were stored in TRIzol reagent (Invitrogen, Carlsbad, CA) at -20°C until use.
RNA extraction and cDNA synthesis
Total RNA was isolated from each sample according to a previous protocol [54]. Residual genomic DNA was removed with an RNase-free DNase kit (Qiagen, Germany) following the manufacturer’s protocols. The RNA concentration and purity were determined by examining the OD260/D280 ratio with expected values between 1.8 and 2.0 using a BioPhotometer Plus (Eppendorf, Germany). Single-stranded synthesis was conducted using a RervertAid First Strand cDNA Synthesis kit (Thermo Scientific, USA). The cDNA (100 ng/μl) was diluted with RNase-free water for use in further analyses.
Quantitative real-time PCR and statistical analyses
qRT-PCR was performed in a 20 μl reaction volume using the ABI 7300 real-time PCR system (Applied Biosystems, USA) with the SYBR Premix Ex Taq (Takara, Japan) according to manufacturer’s protocol. The reaction volume contained 1 μl (approximately 100 ng) of cDNA, 1 μl of each sense and anti-sense primer (10μM), 10 μl of 2 × SYBR Green Premix and 7 μl of ddH2O. The following PCR running program was used: 95 °C for 60 s, followed by 40 cycles of 95 °C for 10 s, 57 °C for 30 s and 72 °C for 30 s. The PCR reaction for each of three biological replicates was implemented according to the procedure described above, and each reaction was performed in triplicate. Melting curves were used to analyze the specificity of amplifications. Specific primers for all the genes used in this study were designed using Beacon Designer 7 (Supplementary Table 11), and the ribosomal protein S20 gene (S20) was used as the endogenous control [52]. The relative expression level of each gene was analyzed according to the 2-ΔΔCTmethod [55]. Data are presented as the means ± standard deviation (SD). Statistical analysis, one-way ANOVA, was performed using the IBM SPSS statistical software package 22.0, with significance declared at P < 0.05.
ACKNOWLEDGMENTs AND FUNDING
This work was supported by the program B for Outstanding PhD candidate of Nanjing University (Grants No. 201702B075) and the 973 project of Ministry of Science and Technology of China (Grants No. 2013CB835300) and the Natural Science Foundation of China (41272008).
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
REFERENCES
1. Huang S, Yuan S, Guo L, Yu Y, Li J, Wu T, Liu T, Yang M, Wu K, Liu H, Ge J, Yu Y, Huang H, et al. Genomic analysis of the immune gene repertoire of amphioxus reveals extraordinary innate complexity and diversity. Genome Res. 2008; 18:1112-1126.
2. Venkatesh B, Lee AP, Ravi V, Maurya AK, Lian MM, Swann JB, Ohta Y, Flajnik MF, Sutoh Y, Kasahara M, Hoon S, Gangu V, Roy SW, et al. Elephant shark genome provides unique insights into gnathostome evolution. Nature. 2014; 505:174-179.
3. Han BW, Herrin BR, Cooper MD, Wilson IA. Antigen recognition by variable lymphocyte receptors. Science. 2008; 321:1834–37.
4. Nagawa F, Kishishita N, Shimizu K, Hirose S, Miyoshi M, Nezu J, Nishimura T, Nishizumi H, Takahashi Y, Hashimoto S, Takeuchi M, Miyajima A, Takemori T, et al. Antigen-receptor genes of the agnathan lamprey are assembled by a process involving copy choice. Nat Immunol. 2007; 8:206-13.
5. Huang S, Tao X, Yuan S, Zhang Y, Li P, Beilinson HA, Zhang Y, Yu W, Pontarotti P, Escriva H, Le Petillon Y, Liu X, Chen S, et al. Discovery of an Active RAG Transposon Illuminates the Origins of V(D)J Recombination. Cell. 2016; 166:102–114.
6. Cao DD, Liao X, Cheng W, Jiang YL, Wang WJ, Li Q, Chen JY, Chen Y, Zhou CZ. Structure of a variable lymphocyte receptor-like protein from the amphioxus Branchiostoma floridae. Sci Rep. 2016; 6:19951.
7. Yue JX, Yu JK, Putnam NH, Holland LZ. The transcriptome of an amphioxus, Asymmetron lucayanum, from the Bahamas: A window into chordate evolution. Genome Biol Evol. 2014; 6:2681-2696.
8. Huang S, Chen Z, Yan X, Yu T, Huang G, Yan Q, Pontarotti PA, Zhao H, Li J, Yang P, Wang R, Li R, Tao X, et al. Decelerated genome evolution in modern vertebrates revealed by analysis of multiple lancelet genomes. Nat Commun. 2014; 5:5896-5896.
9. Putnam NH, Butts T, Ferrier DE, Furlong RF, Hellsten U, Kawashima T, Robinson-Rechavi M, Shoguchi E, Terry A, Yu JK, Benito-Gutiérrez EL, Dubchak I, Garcia-Fernàndez J, et al. The amphioxus genome and the evolution of the chordate karyotype. Nature. 2008; 453:1064-1071.
10. Wang Y, Zhang S. EF1α is a useful internal reference for studies of gene expression regulation in amphioxus Branchiostoma japonicum. Fish Shellfish Immunol. 2012; 32:1068-1073.
11. Chen Y, Cheung SG, Kong RYC, Shin PKS. Morphological and molecular comparisons of dominant amphioxus populations in the China Seas. Mar Biol. 2007; 153:189-198.
12. Kon T, Nohara M, Nishida M, Sterrer W, Nishikawa T. Hidden ancient diversification in the circumtropical lancelet Asymmetron lucayanum complex. Mar Biol. 2006; 149:875-883.
13. Amemiya CT, Alfoldi J, Lee AP, Fan S, Philippe H, MacCallum I, Braasch I, Manousaki T, Schneider I, Rohner N, Organ C, Chalopin D, Smith JJ, et al. The African coelacanth genome provides insights into tetrapod evolution. Nature. 2013; 496:311-316.
14. Wu C, Zhang D, Kan M, Lv Z, Zhu A, Su Y, Zhou D, Zhang J, Zhang Z, Xu M, Jiang L, Guo B, Wang T, et al. The draft genome of the large yellow croaker reveals well-developed innate immunity. Nat Commun. 2014; 5:5227–5227.
15. Askary A, Smeeton J, Paul S, Schindler S, Braasch I, Ellis NA, Postlethwait J, Miller CT, Crump JG. Ancient origin of lubricated joints in bony vertebrates. eLife. 2016; 5:e16415.
16. Krasnov A, Kileng Ø, Skugor S, Jørgensen SM, Afanasyev S, Timmerhaus G, Sommer AI, Jensen I. Genomic analysis of the host response to nervous necrosis virus in Atlantic cod ( Gadus morhua ) brain. Mol Immunol. 2013; 54:443-452.
17. Howe K, Clark MD, Torroja CF, Torrance J, Berthelot C, Muffato M, Collins JE, Humphray S, McLaren K, Matthews L, McLaren S, Sealy I, Caccamo M, et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature. 2013; 496:498-503.
18. Kettleborough RN, Busch-Nentwich EM, Harvey SA, Dooley CM, de Bruijn E, van Eeden F, Sealy I, White RJ, Herd C, Nijman IJ, Fényes F, Mehroke S, Scahill C, et al. A systematic genome-wide analysis of zebrafish protein-coding gene function. Nature. 2013; 496:494-7.
19. Hikima JI, Mihara K, Maekawa S, Wang HC, Aoki T, Kono T, Sakai M. Functional characterization of recombinant interleukin (IL)-17A/F1 in the Japanese pufferfish ( Takifugu rubripes ). Fish Shellfish Immunol. 2016; 53:79-80.
20. Morimoto T, Biswas G, Kono T, Sakai M, Hikima JI. Immune responses in the Japanese pufferfish (Takifugu rubripes) head kidney cells stimulated with particulate silica. Fish Shellfish Immunol. 2016; 49:84-90.
21. Geifman N, Monsonego A, Rubin E. The Neural/Immune Gene Ontology: clipping the Gene Ontology for neurological and immunological systems. BMC Bioinformatics. 2010; 11:458.
22. Amor S, Woodroofe MN. Innate and adaptive immune responses in neurodegeneration and repair. Immunology. 2014; 141:287–91.
23. Wraith DC, Nicholson LB. The adaptive immune system in diseases of the central nervous system. J Clin Invest. 2012; 122:1172-9.
24. Ransohoff RM, Brown MA. Innate immunity in the central nervous system. J Clin Invest. 2012; 122:1164-1171.
25. Wang Y, Lu Y, Zhang Y, Ning Z, Li Y, Zhao Q, Lu H, Huang R, Xia X, Feng Q, Liang X, Liu K, Zhang L, et al. The draft genome of the grass carp (Ctenopharyngodon idellus) provides insights into its evolution and vegetarian adaptation. Nat Genet. 2015; 47:625-631.
26. Star B, Nederbragt AJ, Jentoft S, Grimholt U, Malmstrøm M, Gregers TF, Rounge TB, Paulsen J, Solbakken MH, Sharma A, Wetten OF, Lanzén A, Winer R, et al. The genome sequence of Atlantic cod reveals a unique immune system. Nature. 2011; 477:207-10.
27. West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, Bestwick M, Duguay BA, Raimundo N, MacDuff DA, Kaech SM, Smiley JR, Means RE, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015; 520:553-557.
28. West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nat Rev Immunol. 2011; 11:389-402.
29. Zhang Q, Zmasek CM, Dishaw LJ, Mueller MG, Ye Y, Litman GW, Godzik A. Novel genes dramatically alter regulatory network topology in amphioxus. Genome Biol. 2008; 9:R123.
30. Dai SX, Zhang AD, Huang JF. Evolution, expansion and expression of the Kunitz/BPTI gene family associated with long-term blood feeding in Ixodes Scapularis. BMC Evol Biol. 2012; 12:4.
31. Wang Y, Zhu S. The defensin gene family expansion in the tick Ixodes scapularis. Dev Comp Immunol. 2011; 35:1128-1134.
32. Turner ML, Cronin JG, Noleto PG, Sheldon IM. Glucose availability and AMP-activated protein kinase link energy metabolism and innate immunity in the bovine endometrium. PLoS ONE. 2016; 11:e0151416.
33. Nizet V, Johnson RS. Interdependence of hypoxic and innate immune responses. Nat Rev Immunol. 2009; 9:609-17.
34. McGettrick AF, O’Neill LA. How metabolism generates signals during innate immunity and inflammation. J Biol Chem. 2013; 288:22893-8.
35. Wang Y, Shen Y, Feng C, Zhao K, Song Z, Zhang Y, Yang L, He S. Mitogenomic perspectives on the origin of Tibetan loaches and their adaptation to high altitude. Sci Rep. 2016; 6:29690.
36. Jones FC, Grabherr MG, Chan YF, Russell P, Mauceli E, Johnson J, Swofford R, Pirun M, Zody MC, White S, Birney E, Searle S, Schmutz J, et al, and Broad Institute Genome Sequencing Platform & Whole Genome Assembly Team. The genomic basis of adaptive evolution in threespine sticklebacks. Nature. 2012; 484:55-61.
37. Fang Y. The ecological habit and resource protection of the amphioxus. Dongwuxue Zazhi. 1987; 22:44-48.
38. Zhu LY, Nie L, Zhu G, Xiang LX, Shao JZ. Advances in research of fish immune-relevant genes: a comparative overview of innate and adaptive immunity in teleosts. Dev Comp Immunol. 2013; 39:39-62.
39. Fischer U, Koppang EO, Nakanishi T. Teleost T and NK cell immunity. Fish Shellfish Immunol. 2013; 35:197-206.
40. McCallum H, Harvell D, Dobson A. Rates of spread of marine pathogens. Ecol Lett. 2003; 6:1062–1067.
41. Hellberg ME, Burton RS, Neigel JE, Palumbi SR. Genetic assessment of connectivity among marine populations. Bull Mar Sci. 2002; 70:273-290.
42. Boto L, Martínez JL. Ecological and temporal constraints in the evolution of bacterial genomes. Genes (Basel). 2011; 2:804-828.
43. Lutfalla G, Roest Crollius H, Stange-Thomann N, Jaillon O, Mogensen K, Monneron D. Comparative genomic analysis reveals independent expansion of a lineage-specific gene family in vertebrates: The class II cytokine receptors and their ligands in mammals and fish. BMC Genomics. 2003; 4:29.
44. Dixon B, van Erp SH, Rodrigues PN, Egberts E, Stet RJ. Fish major histocompatibility complex genes: an expansion. Dev Comp Immunol. 1995; 19:109-133.
45. Lechner M, Findeiss S, Steiner L, Marz M, Stadler PF, Prohaska SJ. Proteinortho : Detection of (Co-)orthologs in large-scale analysis. BMC Bioinformatics. 2011; 12:124.
46. Löytynoja A, Goldman N. An algorithm for progressive multiple alignment of sequences with insertions. Proc Natl Acad Sci U S A. 2005; 102:10557-10562.
47. Zhang Z, Li J, Zhao XQ, Wang J, Wong GK, Yu J. KaKs_Calculator: calculating Ka and Ks through model selection and model averaging. Genomics Proteomics Bioinformatics. 2006; 4:259–63.
48. Wang D, Liu F, Wang L, Huang S, Yu J. Nonsynonymous substitution rate (Ka) is a relatively consistent parameter for defining fast-evolving and slow-evolving protein-coding genes. Biol Direct. 2011; 6:13-13.
49. Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005; 21:3674-3676.
50. Jantzen SG, Sutherland BJ, Minkley DR, Koop BF. GO Trimming: systematically reducing redundancy in large Gene Ontology datasets. BMC Res Notes. 2011; 4:267–267.
51. Xie C, Mao X, Huang J, Ding Y, Wu J, Dong S, Kong L, Gao G, Li CY, Wei L. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011; 39:W316-22.
52. Zhang QL, Zhu QH, Liao X, Wang XQ, Chen T, Xu HT, Wang J, Yuan ML, Chen JY. Selection of reliable reference genes for normalization of quantitative RT-PCR from different developmental stages and tissues in amphioxus. Sci Rep. 2016; 6:37549.
53. Yuan S, Yu Y, Huang S, Liu T, Wu T, Dong M, Chen S, Yu Y, Xu A. Bbt-TNFR1 and Bbt-TNFR2, two tumor necrosis factor receptors from Chinese amphioxus involve in host defense. Mol Immunol. 2007; 44:756-62.
54. Yu JK, Holland LZ. Extraction of RNA from amphioxus embryos or adult amphioxus tissue. Cold Spring Harbor Protocols. 2009; 2009: pdb.prot5288.
55. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001; 25:402-408.