
INTRODUCTION
Wounding results in increased vascular permeability, a process that is markedly enhanced if a wound becomes infected. In response to infection, the mammalian host mobilizes an army of immunoglobulins, complement, white blood cells and cytokines. To allow this army to engage the enemy, the vascular system at the site of infection must open its gates. This process has been studied in detail and many of the biochemical mechanisms have been identified[1].
Interestingly, it has been said that tumors resemble “unhealed wounds”[2]. Accordingly, it is known that the vasculature of tumors is different from that of normal cells, and much effort has gone into exploiting this difference through therapeutic agents like Avastin[3-5]. A particularly important phenomenon related to this vascular distinction is referred to as Enhanced Permeability and Retention (EPR)[6]. EPR has been identified in many experimental tumor systems and is believed to result from the aberrant tumor vasculature combined with a lack of functional lymphatics in solid tumors. Because of its selectivity for large molecules, EPR has been exploited for therapeutic purposes by using macromolecular drugs or nanoparticles within an appropriate size range[7-10]. One notable example is Doxil, a liposomal formulation of doxorubicin, which has been approved for the treatment of human cancers.
We wondered whether generating an inflammatory environment within a tumor could enhance the EPR effect, just as infection within a wound dramatically enhances its vascular permeability. To test this approach, we determined whether inflammatory and vascular-active agents could improve selective nanoparticle accumulation within tumors and whether such accumulation would lead to improved therapeutic results in animal models.
Results
Bacterial infection enhances antibody accumulation in experimental tumors
The research described in this work was stimulated by unexpected observations made through the investigation of C. novyi-NT, an attenuated anaerobic bacterial strain that can infect experimental tumors[11]. This infection often leads to eradication of the internal hypoxic regions of tumors but leaves the oxygenated rim of the tumors intact. C. novyi-NT secretes an enzyme called liposomase at high levels in the infected tumors[12, 13]. We hypothesized that a radiolabeled anti-liposomase antibody would synergize with C. novyi-NT by binding to liposomase secreted by the bacteria, thereby eradicating the oxygenated tumor rim through β-particle irradiation. A monoclonal antibody against liposomase was generated and used to evaluate this hypothesis (see Methods).
Mice bearing subcutaneous CT26 tumors were intraveneously injected with C. novyi-NT spores together with the radiolabeled anti-liposomase antibody or with a similarly labeled IgG control antibody. The anti-liposomase antibody was highly enriched in the tumors infected with C. novyi-NT but not in uninfected tumors (Fig. 1A). Surprisingly, however, the radiolabeled IgG control antibody was also enriched in the C. novyi-NT infected tumors, albeit to a lesser extent (Fig. 1A). Biodistribution analyses showed that the level of radioactivity in the tumor was four-fold higher than that in most normal tissues (Fig. 1B).
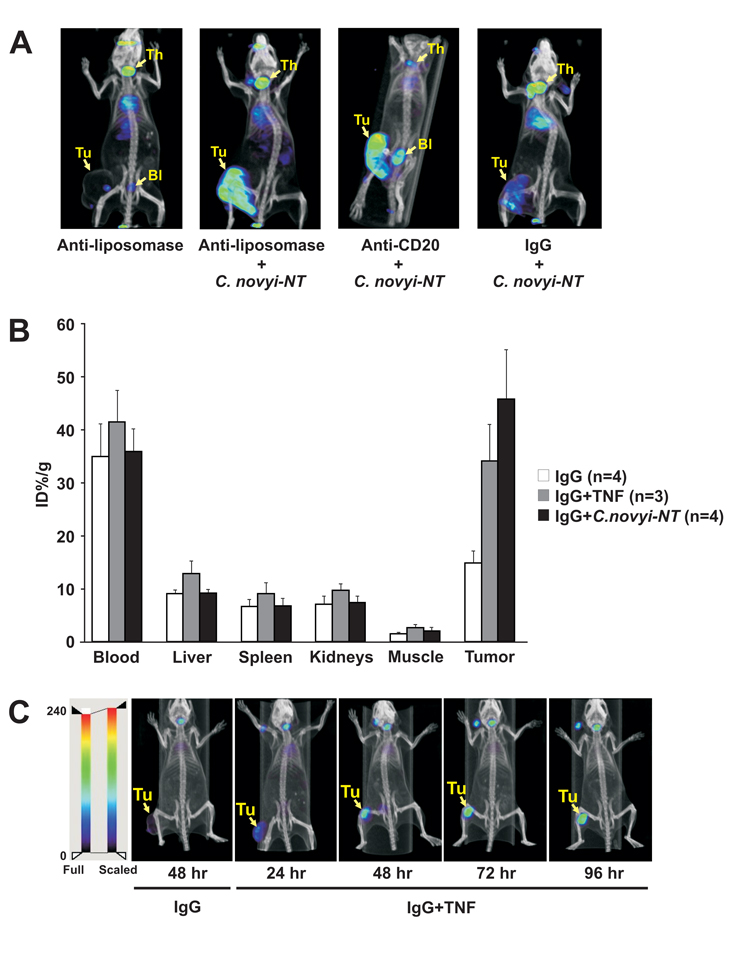
Figure 1: Inflammatory responses enhance tumor-selective accumulation of radiolabeled antibodies. (A) BALB/c mice bearing subcutaneous CT26 tumors were administered C. novyi-NT spores plus 125I-labeled liposomase antibody, CD20 antibody, or an IgG control antibody by tail vein injection. The animals were imaged by SPECT/CT 24 hours thereafter. Tumor (Tu), thyroid (Th) and bladder (Bl) are indicated. (B, C) Tumor-bearing mice were administered 125I-labeled IgG plus C. novyi-NT spores or TNF-α by tail vein injection. For biodistribution analysis (B), mice were sacrificed 48 hours later and percent injected dose per gram of tissue (ID%/g) was determined. Means and s.d. are shown. For imaging study (C), SPECT/CT images were taken at the indicated time points after the injections. Tumor (Tu) is indicated.
To further confirm that the accumulation in the tumors was not antibody-specific, we repeated the experiment with another antibody generated against human CD20, a B-cell antigen. The partially humanized version of this antibody, Rituximab, has been marketed for the treatment of B cell lymphoma and chronic lymphocytic leukemia[14, 15]. Systemically administered anti-CD20 antibody was also enriched in the tumor if the animal was simultaneously injected with C. novyi-NT spores (Fig. 1A).
Bacterial infection and pro-inflammatory cytokine both enhance tumor-selective accumulation of macromolecular drug formulations
We reasoned that the inflammatory response to the bacterial infection led to an increased vascular permeability, resulting in the preferential antibody accumulation at the infected tumor site. We therefore sought to identify a pro-inflammatory cytokine that might mimic the effects of C. novyi-NT. Among those considered, Tumor Necrosis Factor-α (TNF-α) was of particular interest as this cytokine has been identified as the serum factor responsible for endotoxin-induced vascular permeabilization[16, 17]. Furthermore, a similar hemorrhagic necrosis in tumors is observed following systemic administration of either TNF-α or C. novyi-NT spores[11, 16]. Based on these parallels, we repeated the protocol described above, substituting systemically-administered TNF-α for C. novyi-NT spores. When CT26 tumor-bearing mice were injected with murine TNF-α and radiolabeled murine IgG, significant IgG accumulation was observed in the tumors but not in the normal tissues (Fig. 1B and C). A time course study revealed that the IgG tumor accumulation progressed slowly and peaked between 72 and 96 hours after injection (Fig. 1C).
The effect of vascular-active agents on tumor vasculature will henceforth be referred to as Enhanced EPR (E2PR). Sterically stabilized liposomal nanoparticles (SSLs) of ~100 nm in diameter have been shown to be susceptible to the EPR effect[8]. To evaluate whether such liposomes were susceptible to E2PR, we fabricated radioactive liposomes using a Bolton-Hunter (BH) reagent-based iodination strategy[18]. Iodinated BH reagent labels proteins by forming amide bonds with free amino groups such as those present on arginine[19]. SSLs were loaded with arginine at low pH and then the loaded SSLs were incubated with 125I-labeled BH reagent. The 125I-BH reagent passed through the lipid bilayer but was unable to exit after covalent binding to the arginine because of the latter’s positive charge. We were thus able to achieve a very high concentration of radioactivity within the SSLs while avoiding prolonged exposure to the radioactivity during the preparation.
125I-labeled SSLs were intravenously injected into tumor-bearing mice in combination with either C. novyi-NT or TNF-α. Both C. novyi-NT and TNF-α treatments significantly augmented the selective retention of 125I within tumors (Fig. 2). Furthermore, the radioactivity in normal tissues was markedly lower compared to the animals treated with 125I-labeled SSLs without TNF-α or C. novyi-NT (Fig. 2A). Thus, the tumor-to-blood ratio of radioactivity following TNF-α treatment was as high as 22-fold, far higher than achieved with radiolabeled IgG (compare Fig. 2A to Fig. 1B). SPECT/CT also revealed that the kinetics of tumor accumulation was different with radiolabeled SSLs than with IgG: SSL accumulation peaked at 24 hours, 48 - 72 hours earlier than IgG.
Like EPR, the effect of E2PR is particle size-dependent. In contrast to 125I-labeled SSLs, tumor retention of 125I-labeled arginine (the substrate of 125I labeling in SSLs) is not affected by TNF-α. However, at the other end of the size spectrum, 125I-labeled C. novyi-NT spores (~1 µm in diameter[20]) are highly enriched in tumors only when combined with TNF-α (data not shown). Thus, E2PR appears to reflect a more substantial vascular disruption than EPR: while EPR favors accumulation of nanoparticles in the range around 100 nm[8], E2PR extends that range to >1 µm.
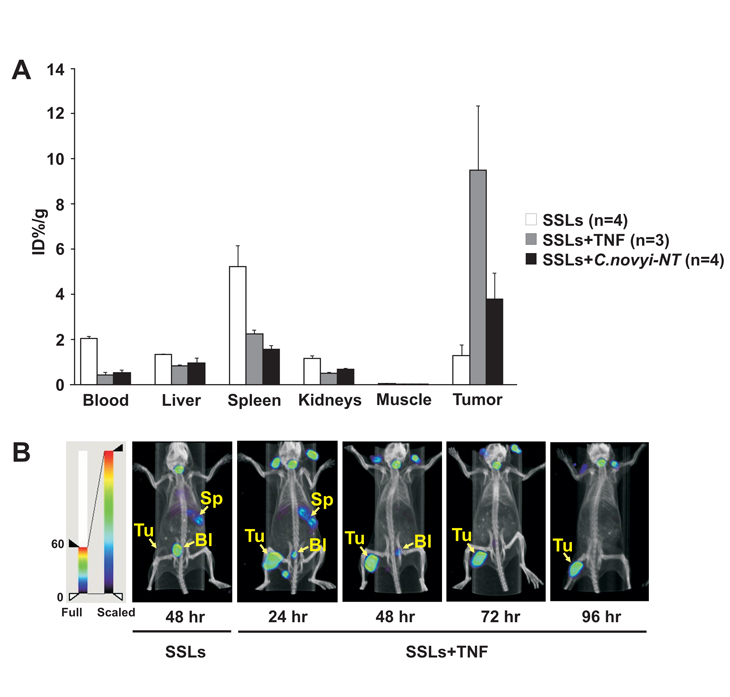
Figure 2: Inflammatory responses enhance tumor-selective accumulation of radiolabeled SSLs. BALB/c mice bearing subcutaneous CT26 tumors were administered 125I-labeled SSLs plus C. novyi-NT spores or TNF-α by tail vein injection. For biodistribution (A), mice were sacrificed 48 hours later and percent injected dose per gram of tissue (ID%/g) was determined. Means and s.d. are shown. For imaging analysis (B), SPECT/CT images were taken at the indicated time points after the injections. Tumor (Tu), bladder (Bl) and spleen (Sp) are indicated.
To determine whether the accumulation was dependent on the volume of the tumor, we injected TNF-α plus 125I-labeled IgG or 125I-labeled SSLs into animals with a small subcutaneous tumor on one flank and a large tumor on the other flank. SPECT/CT showed retention of radioactivity in both tumors (examples in Supplementary Fig. S1). We also tested the relative timing of injection of TNF-α and 125I-labeled SSLs. Though TNF-α and SSLs were administered jointly in the experiments recorded above, we found that similar results were obtained when TNF-α was administered within12 hours after SSLs. Conversely, E2PR was not observed when TNF-α was administered 6 hours prior to SSL administration (data not shown).
Microtubule-interacting agents are also able to disrupt the tumor vasculature[21]. We therefore determined whether such agents could induce E2PR. Combretastatin A4P (CA4P) and vinorelbine are microtubule-interacting agents with completely different structures and modes of interaction with microtubules[22, 23]. Injection of either resulted in E2PR, though not as impressively as TNF-α (Supplementary Fig. S2).
TNF-α and macromolecular drug formulations synergize in the treatment of experimental tumors
We next investigated whether the E2PR could be translated into therapeutic gain. Mice bearing fully developed CT26 tumors were treated by simultaneous i.v. injections of TNF-α plus Doxil (10 mg/kg) or radiolabeled IgG. 131I rather than 125I was chosen for radiolabeling in light of the type of ionizing radiation required for a radiotherapeutic effect. Although treatment with Doxil or 131I-labeled IgG in the absence of TNF-α retarded tumor growth and prolonged animal survival, the tumors always grew back (Fig. 3A and B). When combined with TNF-α, however, a single administration of these agents led to complete tumor regression in all animals and long-term cures in more than 75% of them. When a lower dose (25 µg/kg) of TNF-α was used, none of the treated animals were cured, although prolonged survival was observed. It is important to note that humans tolerate multiple injections (3 infusions/week) of a dose comparable to the highest dose of TNF-α we used[24]. We also tested SSLs containing 131I, generated using the chemical trapping approach described above. While 131I-labeled SSLs alone retarded tumor growth, complete tumor regression and cures were only observed when they were used in combination with TNF-α (Fig. 3C).
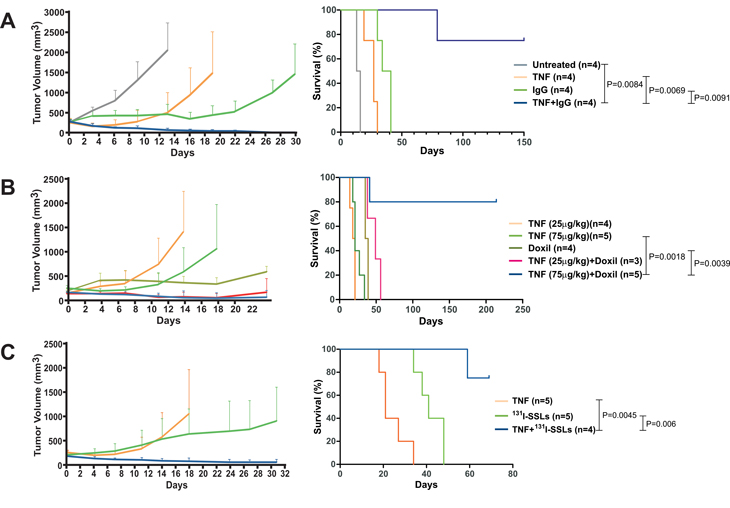
Figure 3: TNF-α enhances the antitumor activity of macromolecular drug formulations. Tumor-bearing mice were treated on day 0 with a single dose of the combinations of TNF-α plus 131I-labeled IgG (A), Doxil (B), or 131I-labeled SSLs (C), respectively. The therapeutic effects on tumor volume and animal survival are shown. Means and s.e.m. are illustrated. The number of animals used in each experimental arm is shown in parentheses. P values between arms are also shown.
Finally, we evaluated the therapeutic potential of E2PR in a murine model of glioblastoma multiforme (GBM). When implanted orthotopically, the brain tumor cell line GL261 forms very aggressive tumors, killing animals within about a month (Fig. 4A). At the histologic level, these tumors are very similar to human GBM, manifesting an infiltrative growth pattern, necrosis and neovascularization[25]. Following stereotactic injection of GL261 cells into the frontal lobe, brain tumors were allowed to grow to substantial size, then 125I-labeled SSLs with or without TNF-α were administered. Tumor accumulation of the radiolabeled SSLs was only observed in TNF-α treated animals (Fig. 4B). Mice with similar tumors were injected with Doxil, either with or without TNF-α. The combination clearly had a therapeutic benefit, prolonging survival up to 103 days even in this highly challenging pre-clinical model (Fig. 4A). Both Doxil and TNF-α showed limited therapeutic benefit when used as single agents, with no animal surviving beyond 50 days following tumor implantation.
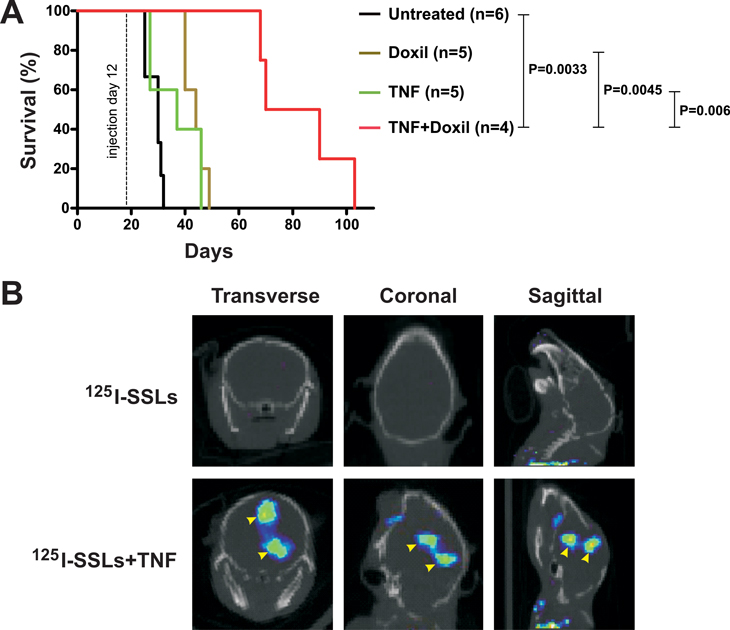
Figure 4: Vascular effect of TNF-α on a brain tumor model. (A) C57BL6 mice bearing orthotopic brain tumors were treated with a single dose of the indicated therapeutic agents 12 days after tumor implantation. The number of animals used in each experimental arm and P values between arms are shown. (B) SPECT-CT images were obtained 48 hours following the indicated treatments, which were performed 25 days following tumor implantation. Transverse, coronal, and sagittal images are shown and tumors indicated by the arrowheads. In this particular animal, two tumor nodules developed along the injection track and both showed tumor accumulation of 125I-labeled SSLs when TNF-α was co-administered.
Discussion
The major limitation for most chemotherapeutic agents is their toxicity toward normal tissues, which prohibits the use of doses high enough to eradicate all cancer cells. One approach to address this problem is to develop agents that are delivered to all cells but are preferentially toxic to tumor cells because of the abnormal signaling pathways. This strategy underlies the success of agents such as Gleevec (imatinib) and Iressa (gefitinib)[26, 27]. A second approach is to use agents that bind to extracellular molecules present at higher concentrations on the surface of tumor cells, such as Herceptin (trastuzumab) and Erbitux (cetuximab)[28, 29]. The third approach takes advantage of the abnormal vasculature present in tumors, allowing preferential accumulation of nanoparticles (the EPR effect)[6, 30]. Though all approaches have merit, the third has the advantage that virtually any drug, including a wealth of clinically approved agents, can in theory be made more effective by their incorporation into nanoparticles of appropriate sizes. The ability to use agents that are already clinically approved poses many practical advantages with respect to the performance of clinical trials and the duration of the drug approval process.
In this work, we have attempted to enhance the third approach through pharmacologic manipulation of the abnormal vasculature already present in tumors. We show that E2PR can dramatically increase the tumor:blood ratio of nanoparticles, as assessed by biodistribution, and we hypothesize that this increase is primarily responsible for the enhanced therapeutic response. It is worth noting that even a small difference in the intratumoral concentration of an agent can make a large difference in therapeutic effect[31]. In the studies described here, E2PR led to a tumor:blood ratio of more than 22-fold (Fig. 2A). However, it should be noted that TNF-α, C. novyi-NT infection, and doxorubicin-induced cancer cell apoptosis are all able to induce or enhance a host immune response that could have contributed to the observed therapeutic response.
We were particularly encouraged with the results in the GBM model. This tumor type in humans is highly recalcitrant to conventional therapies, leading to a dismal prognosis for patients with this disease. The blood-brain barrier is at least partly to blame for the limited efficacy of chemotherapy[32]. We found that TNF-α treatment could help breach the blood-brain barrier and result in major accumulations of 125I-labeled SSLs in the orthotopically implanted brain tumors as well as significantly prolong the survival of the tumor-bearing animals (Fig. 4). As the mouse cranial cavity is small, murine brain tumors are particularly difficult to treat as even a minimal amount of growth of a pre-existing tumor is lethal.
In sum, our results suggest a way to improve the therapeutic efficacy of conventional and novel drugs by incorporating them into nanoparticles and injecting them together with vascular-active agents such as TNF-α. The approach is versatile, as it should be practicable with a variety of nanoparticle formulations as well as with diverse chemical and radioactive agents. Different classes of agents with E2PR effect can be employed to enhance their tumor accumulation. In addition to those tested in this work, other vasoactive agents, including vascular endothelial growth factor (VEGF), are likely to have similar effect. An imaging-based companion test may be developed in the future to assess which vasoactive agent is most effective, as the vasculature of individual cancers may have varied sensitivity to a specific vascular-active agent. Future clinical trials can address whether this strategy is as efficacious in humans as it is in mice. To accelerate the translation of this approach, we have intentionally used TNF-α at doses either below or comparable to the maximum tolerated dose (MTD) as defined in a number of clinical trials [33].
Materials and Methods
Cell Lines
CT26 (CRL-2638) murine colorectal adenocarcinoma cells were purchased from the American Type Culture Collection (ATCC) and grown in McCoy’s 5A Medium (Invitrogen) supplemented with 10% Fetal Bovine Serum (FBS, HyClone) at 37°C with 5% CO2. GL261 glioma cells were kindly provided by Dr. Michael Lim (Johns Hopkins University, Baltimore) and maintained in DMEM media (ATCC) supplemented with 10% FBS.
Reagents
Bolton-Hunter reagent (BH, N-succinimidyl-3-(4-hydoxyphenyl)-propionate) and TNF-α (mouse, recombinant) were purchased from Sigma-Aldrich. Radioiodines (Sodium 125- or 131-iodide) were purchased from MP Biomedicals and Nordion, respectively. IODO-GEN was purchased from Pierce. Mouse monoclonal IgG1 isotype control antibody (ab18447) and CD20 antibody (ab8237) were purchased from Abcam. PEGylated liposomal doxorubicin (DOXIL®) was purchased from Tibotec Therapeutics. Hydrogenated Chicken Egg L-α-Phosphatidylcholine (HEPC), 1,2-Distearoyl-sn-Glycero-3-Phosphoethanolamine-N-[Methoxy(Polyethylene glycol)-2000] (DSPE-PEG2000) and Cholesterol (Chol) were purchased from Avanti Polar Lipids. C. novyi-NT spores were prepared as previously described [11].
Animal models
All animal experiments were overseen and approved by the Animal Welfare Committee of Johns Hopkins University, and were in compliance with the University standards. For the subcutaneous tumor model, female, six to eight week-old BALB/c mice (Harlan Breeders, ~20 g in weight) were used. Five million CT26 cells were injected subcutaneously into the right flank of each mouse and allowed to grow for ~10 days before randomization, group assignment, and treatment. C. novyi-NT spores were administered by a bolus tail vein injection of 300 million spores suspended in 0.2 mL of phosphate buffered saline, pH 7.5 (PBS). Cytotoxic anticancer agents were administered 16 hours later via the same route. TNF-α was reconstituted freshly before administration in doubly-distilled H2O at 100 µg/mL and diluted into 0.1% (w/v) BSA in PBS at a final concentration of 10 µg/mL. Cytotoxic agents were injected within a few minutes thereafter. Tumor volume was calculated as length x width2 x 0.5. For the orthotopic brain tumor model, female C57BL6 mice, 5-6 weeks of age, were purchased from the NCI-Frederick. Mice were anesthetized via intraperitoneal injection of 60 µL of a stock solution containing ketamine hydrochloride (75 mg/kg, Abbot Laboratories), xylazine (Xyla-ject®, 7.5 mg/kg, Phoenix Pharmaceutical), and ethanol (14.25%) in a sterile 0.9% NaCl solution. Following a 1-cm midline scalp incision, a 1-mm burr hole was placed over the right frontal bone, with its center 2 mm lateral to the sagittal suture and 1 mm anterior to the coronal suture. On a stereotactic frame, a sterile needle loaded with 20,000 GL261 cells was placed at a depth of 3 mm below the dura and the cells were injected slowly at a rate of 1 µL/minute. Afterwards, the animal was removed from the frame and the scalp incision closed with surgical staples. On day 12 post implantation of the tumor cells, a significant tumor was formed and 1 µg of mouse recombinant TNF-α or 100 µL of Doxil at 20 mg/kg, or both, were administered intravenously through the tail vein. Animals were monitored for potential side effects following drug administration. Animals were observed daily for any signs of deterioration, neurotoxicity, or movement disorders. They were inspected for signs of pain and distress, as per the Johns Hopkins Animal Care and Use Guidelines. If the symptoms persisted and resulted in debilitation, the moribund animals were euthanized. The brain and other organs were dissected and placed in formalin for further pathological studies. A single dose was administered for all therapeutic agents described above.
Liposomase Antibody
Three peptides (JHU009A: CNVDLQQKLIEN; JHU009B: CYPEWGTKDENGNIRK; JHU009C: CDMAQMLRNLPVTE) were used to immunize the mice for generating antibodies against C. novyi-NT liposomase (A&G Pharmaceutical). After screening ~500 hybridoma clones by ELISA, one clone (JHU009-5F5) specific to the JHU009C peptide was eventually selected for the imaging study. The affinity and specificity of the JHU009-5F5 mAb were also confirmed by both ELISA and western blot analyses against purified liposomase protein [12].
Radioiodination of Antibodies
Typically, 20 µg of purified antibody in 100 µL of PBS was added to an iodogen-coated vial. Sodium 125- or 131-iodide was then added to the vial at 2 to 5 mCi in 2 to 5 µL of 0.1 M NaOH, pH 10. The reaction was then incubated for 10 minutes at room temperature before purification on a PBS-equilibrated Sephadex G-25 desalting column (Amersham Biosciences) to remove unincorporated radioiodine. The radiochemical yield was typically 30% to 40%. The radiochemical purity was at least 95% as determined by thin-layer chromatography. Antibodies were labeled within 24 hours of use and stored in PBS at 4°C after labeling and purification.
Preparation of Liposomes
A mixture of HEPC:Chol:DSPE-PEG2000 at a molar ratio of 50:45:5 was solubilized in chloroform, followed by drying to a thin film under rotary evaporation and then under vacuum for 2 hours. The film was hydrated with arginine solution (80 mmol/L) in 4-(2-hydroxyethyl)-piperazine-1-sulphonic acid (HEPES, 80 mmol/L, pH 8.0) and submerged in a 65°C sonication bath (Bransonic) to form Large Multilamellar Vesicles (MLVs). This lipid suspension was extruded 10 times through a double stack of 0.1 µm Nuclepore filters (Whatman) using a Lipex Thermobarrel Extruder (Northern Lipids). The resulting colloidal suspension of Single Unilamellar Vesicles (SUV) was dialyzed against 150 mmol/L phosphate buffer (pH 5.6) at 4°C to exchange the external milieu of the liposomes and then filter-sterilized. The mean size of the SUVs was ~100 nm in diameter and polydispersity index ~0.1 as determined by quasi-elastic light scattering using a Malvern Zetasizer 3000 (Malvern).
Radioiodination of Bolton-Hunter reagent
Bolton-Hunter reagent (BH, N-hydroxysuccinimide (NHS) ester of HPPA) was labeled with sodium 125- or 131-iodide by the chloramine-T method and purified by solvent extraction. Briefly, 50 µL of chloramine T (4 mg/mL in phosphate buffer) and 3.7 to 37 MBq (0.1–1.0 mCi) of 125I-NaI or 131I-NaI were added to 2 µL of BH freshly solubilized in anhydrous dioxin (0.5 mg/mL). Iodination was achieved by incubation at room temperature for approximately 15 sec and then 400 µL of 100 mmol/L phosphate buffer (pH 7.4) was added. The radiolabeled BH was immediately extracted with 500 µL of toluene and the organic phase was removed and transferred to a glass tube. For the encapsulation of the reagent into liposomes, the organic solvent was evaporated using a dry nitrogen stream before adding the liposome suspension.
Encapsulation of the Iodinated Reagents into the Liposomes
For the chemical entrapment of the iodinated BH, arginine-containing liposomes were incubated for 30 min at 37°C with 125I-BH. The labeling efficiency was determined by counting the liposome suspension before and after chromatography on a PD-10 column (GE Healthcare)[18]. The radiochemical yield was typically 50% to 70%.
Biodistribution Assay
CT26-bearing BALB/c mice were injected via the tail vein with 50 µCi of 125I-liposomes or 125I-IgG1. Three to four mice in each experimental arm were sacrificed by cervical dislocation at 48 hours post injection. The liver, spleen, kidneys, muscle, and tumor were quickly removed as was ~0.1 mL of blood. The organs and blood were weighed and their radioactivity was measured with an automated gamma counter (1282 Compugamma CS, Pharmacia/LKB Nuclear). The percent injected dose per gram of tissue (ID%/g) was calculated by comparison with samples of a standard dilution of the initial dose. All measurements were corrected for decay.
SPECT-CT Imaging
BALB/c mice bearing subcutaneous CT26 tumor or C57BL6 mice bearing orthotopic GL261 brain tumor were injected intravenously with 37.5 MBq (1 mCi) of either 125I-IgG1 or 125I-SSLs in saline. The mice were positioned on the X-SPECT (Gamma Medica-Ideas) gantry and scanned using two low-energy, high resolution pinhole collimators (Gamma Medica-Ideas) rotating through 360o in 6o increments for 40 seconds per increment. Immediately following SPECT acquisition, the mice were scanned by CT (X-SPECT) over a 4.6 cm field of view using a 600 mA, 50 kV beam. Data were reconstructed using the ordered subsets-expectation maximization algorithm. The SPECT and CT data were then coregistered using the instrument supplied software and displayed using AMIDE (http://amide.sourceforge.net/) or Amira software (Visage Imaging).
Statistical Analysis
The statistical significance of percent survival between different experimental arms was determined by Long-rank analysis.
ACKNOWLEDGEMENTS
We thank Evangeline Watson and James Fox for expert technical assistance. Support was provided by The Virginia and D.K. Ludwig Fund for Cancer Research, the Commonwealth Foundation, the Miracle Foundation, Maryland Cigarette Restitution Fund, Children’s Cancer Foundation, and the US National Institutes of Health grants CA062924, CA129825, CA92871 and NS052507.
CONFLICTS OF INTEREST STATEMENT
The Johns Hopkins University is considering filing a patent application based on some of the observations made in this work. Inventors include Yuan Qiao, Xin Huang, Sridhar Nimmagadda, Catherine A. Foss, Martin G. Pomper, Kenneth W. Kinzler, Luis A. Diaz, Jr, Bert Vogelstein, and Shibin Zhou
REFERENCES
1. Cotran RS: Inflammation: historical perspectives. In: Inflammation: basic principles and clinical correlates. Edited by Gallin JI, Snyderman, R., 3th edn. Philadelphia: Lippincott Williams & Wilkins 1999: 5-10.
2. Dvorak HF: Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med 1986, 315(26):1650-1659.
3. Jain RK, Duda DG: Vascular and Interstitial Biology of Tumors. In: ABELOFF’S CLINICAL ONCOLOGY. Edited by Abeloff MD, Armitage JO, Niederhuber JE, Kastan MB, McKenna WG, vol. 1, 4th edn. Philadelphia: Churchill Livingstone Elsevier; 2008: 105-124.
4. Ferrara N, Hillan KJ, Gerber HP, Novotny W: Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov 2004, 3(5):391-400.
5. Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E et al: Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004, 350(23):2335-2342.
6. Matsumura Y, Maeda H: A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res 1986, 46(12 Pt 1):6387-6392.
7. Maeda H, Fang J, Inutsuka T, Kitamoto Y: Vascular permeability enhancement in solid tumor: various factors, mechanisms involved and its implications. Int Immunopharmacol 2003, 3(3):319-328.
8. Drummond DC, Meyer O, Hong K, Kirpotin DB, Papahadjopoulos D: Optimizing liposomes for delivery of chemotherapeutic agents to solid tumors. Pharmacol Rev 1999, 51(4):691-743.
9. Torchilin VP: Micellar nanocarriers: pharmaceutical perspectives. Pharm Res 2007, 24(1):1-16.
10. Maeda H, Bharate GY, Daruwalla J: Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur J Pharm Biopharm 2009, 71(3):409-419.
11. Dang LH, Bettegowda C, Huso DL, Kinzler KW, Vogelstein B: Combination bacteriolytic therapy for the treatment of experimental tumors. Proc Natl Acad Sci U S A 2001, 27:27.
12. Cheong I, Huang X, Bettegowda C, Diaz LA, Jr., Kinzler KW, Zhou S, Vogelstein B: A bacterial protein enhances the release and efficacy of liposomal cancer drugs. Science 2006, 314(5803):1308-1311.
13. Bettegowda C, Huang X, Lin J, Cheong I, Kohli M, Szabo SA, Zhang X, Diaz LA, Jr., Velculescu VE, Parmigiani G et al: The genome and transcriptomes of the anti-tumor agent Clostridium novyi-NT. Nat Biotechnol 2006, 24(12):1573-1580.
14. Coiffier B: Rituximab therapy in malignant lymphoma. Oncogene 2007, 26(25):3603-3613.
15. Jaglowski SM, Byrd JC: Rituximab in chronic lymphocytic leukemia. Semin Hematol, 47(2):156-169.
16. Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B: An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci U S A 1975, 72(9):3666-3670.
17. Brett J, Gerlach H, Nawroth P, Steinberg S, Godman G, Stern D: Tumor necrosis factor/cachectin increases permeability of endothelial cell monolayers by a mechanism involving regulatory G proteins. J Exp Med 1989, 169(6):1977-1991.
18. Mougin-Degraef M, Jestin E, Bruel D, Remaud-Le Saec P, Morandeau L, Faivre-Chauvet A, Barbet J: High-activity radio-iodine labeling of conventional and stealth liposomes. J Liposome Res 2006, 16(1):91-102.
19. Bolton AE, Hunter WM: The labelling of proteins to high specific radioactivities by conjugation to a 125I-containing acylating agent. Biochem J 1973, 133(3):529-539.
20. Plomp M, McCaffery JM, Cheong I, Huang X, Bettegowda C, Kinzler KW, Zhou S, Vogelstein B, Malkin AJ: Spore coat architecture of Clostridium novyi NT spores. J Bacteriol 2007, 189(17):6457-6468.
21. Schwartz EL: Antivascular actions of microtubule-binding drugs. Clin Cancer Res 2009, 15(8):2594-2601.
22. Tozer GM, Kanthou C, Parkins CS, Hill SA: The biology of the combretastatins as tumour vascular targeting agents. Int J Exp Pathol 2002, 83(1):21-38.
23. Holwell SE, Hill BT, Bibby MC: Anti-vascular effects of vinflunine in the MAC 15A transplantable adenocarcinoma model. Br J Cancer 2001, 84(2):290-295.
24. Schiller JH, Storer BE, Witt PL, Alberti D, Tombes MB, Arzoomanian R, Proctor RA, McCarthy D, Brown RR, Voss SD et al: Biological and clinical effects of intravenous tumor necrosis factor-alpha administered three times weekly. Cancer Res 1991, 51(6):1651-1658.
25. Ausman JI, Shapiro WR, Rall DP: Studies on the chemotherapy of experimental brain tumors: development of an experimental model. Cancer Res 1970, 30(9):2394-2400.
26. Druker BJ: Imatinib as a paradigm of targeted therapies. Adv Cancer Res 2004, 91:1-30.
27. Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y et al: Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med 2009, 361(10):947-957.
28. Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M et al: Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001, 344(11):783-792.
29. Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype C et al: Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med 2004, 351(4):337-345.
30. Seynhaeve AL, Hoving S, Schipper D, Vermeulen CE, de Wiel-Ambagtsheer G, van Tiel ST, Eggermont AM, Ten Hagen TL: Tumor necrosis factor alpha mediates homogeneous distribution of liposomes in murine melanoma that contributes to a better tumor response. Cancer Res 2007, 67(19):9455-9462.
31. Frei E, 3rd, Canellos GP: Dose: a critical factor in cancer chemotherapy. Am J Med 1980, 69(4):585-594.
32. Muldoon LL, Soussain C, Jahnke K, Johanson C, Siegal T, Smith QR, Hall WA, Hynynen K, Senter PD, Peereboom DM et al: Chemotherapy delivery issues in central nervous system malignancy: a reality check. J Clin Oncol 2007, 25(16):2295-2305.
33. Balkwill F: Tumour necrosis factor and cancer. Nat Rev Cancer 2009, 9(5):361-371.