
INTRODUCTION
Sunitinib a multi-target tyrosine kinase inhibitor (TKI) is currently approved by U.S. F.D.A. for treating advanced renal cell carcinoma, imatinib-resistant, or intolerant gastrointestinal stroma tumor (GIST), and advanced pancreatic neuroendocrine tumors [1]. Recent studies have reported that sunitinib controls c-KIT or non c-KIT mutated malignant melanoma (MM) [2–6]. However, sunitinib is not clinically widely used on solid tumors due to the low efficacy [6–9]. How to improve the efficacy of sunitinib in solid tumor remains a big challenge for oncologists’ world widely. Propranolol, a non-selective β-blocker, is widely used for hypertension. It has been found to be safe and effective in treating large infantile hemangioma [10, 11]. Follow-up studies suggest that it can also be used to treat the malignant tumors such as non-small-cell lung cancer, breast cancer, colorectal cancer, gastric cancer and neuroblastoma cancer [12–20]. Recently, propranolol has been found to inhibit MM proliferation in vitro and in vivo [21, 22] and prolong MM patient survival [23]. Propranolol shows synergistic effects with various chemotherapies and tumor targeting medications to obtain an anti-tumor effect, including a propranolol-TKI combination treatment [24–30]. However, the underlying mechanism of how propranolol enhanced the efficacy of TKIs remained unclear. This study ascertained that if propranolol would increase sunitinib efficacy in MM and aimed to explore potential mechanism.
RESULTS
Combined propranolol-sunitinib treatment (C-PST) inhibited cutaneous-acral MM proliferation
A cell viability assay demonstrated that sunitinib 1-20 μM and propranolol 25-200 μM significantly reduced the survival rate of two MM cell lines in a concentration and time dependent manner. The data at 24, 48, and 72 hours were shown in respectively (Figure 1. The greatest inhibition on cell proliferation in incubated MM cell lines was detected by using a combination of propranolol 50 μM and sunitinib 2.5 μM. The survival rate of A375 cell line and P8 cell line was 49.9±1.1% and 47.2±2.8% at 48 hours respectively (Figure 2B and 2E). This data demonstrated that the IC50 of sunitinib in propranolol-sunitinib combination was decreased by 53% and 65% respectively on A375 and P8 cell lines compared with single sunitinib treatment.
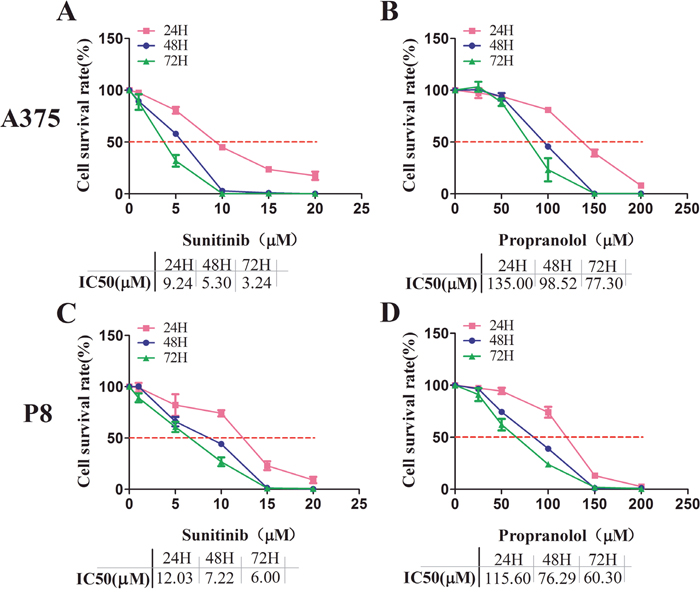
Figure 1: Sunitinib and propranolol effects on cell proliferation in A375 and P8 melanoma cell lines. (A-D) MTS assay measured cell viability after increasing suninitib (1, 5, 10, 15, 20 μM) and propranolol (25, 50, 100, 150, 200 μM) concentrations at 24, 48, and 72H MTS. Relative growth rate was calculated as the ratio of treated to untreated cells at each dose for each replicate. Sunitinib IC50 and propranolol, after incubation for 24, 48, and 72H, was 9.24, 5.3, 3.24μM, and 135, 98.52, 77.3 μM, respectively, in the A375 cell lines. It was 12.03, 7.22, 6.00 μM and 115.6, 76.29 60.3 μM in P8 cell lines respectively.
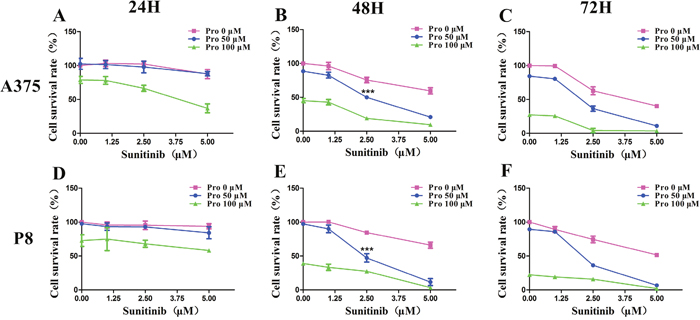
Figure 2: C-PST effects on cell proliferation in melanoma cell lines. (A-F) An MTS assay calculated survival rate from the combined treatment for sunitinib (1, 2.5, 5 μM) and propranolol (50, 100 μM) concentrations at 24, 48, 72H. Results are presented as mean ± SD. Significant differences were evaluated using one-way ANOVA, and the asterisk (***) indicates a significant difference compared the same concentration of sunitinib or propranolol alone with sunitinib 2.5 μM and propranolol 50 μM combination group using Dunnett’s multiple comparison test(P<0.001). Pro, propranolol.
The formation of cell colony in the A375 cell line was reduced from 78±3.8% to 6±1.1% by C-PST compared with single sunitinib treatment measured by colony formation assay (Figure 3A and 3C). In P8 cell line, no cell colony was formed in C-PST treated group (Figure 3B, and Supplementary Figure 1). In the following experiment, the dosage of C-PST was fixed at propranolol 50 μM in combination with sunitinib 2.5 μM.
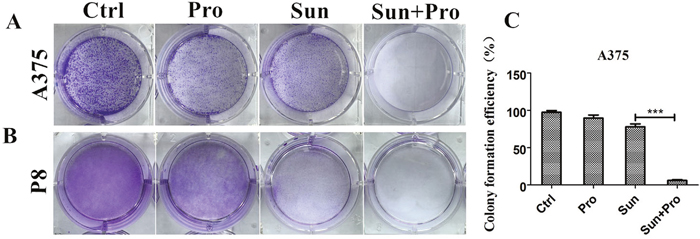
Figure 3: C-PST significantly reduced colony growth in A375 and P8 cell lines. A colony formation assay measured A375 cell (A) and P8 lines (B). Cell seed concentration was 2*103 and 1*103 cells. Cultivation lasted 7 and 9 days respectively. PBS and DMSO, in equal volumes, were mixed as negative controls and treated with propranolol, sunitinib, and the C-PST. (C) Colony formation efficiency is described in A. Error bars represent the standard deviation. Results are presented as mean ± SD, ***P<0.001.
C-PST induced G0/G1/S phase arrest in MM cell lines
Cell cycle progression was delayed by C-PST compared with the single group. After C-PST the cell amount was significantly increased in G0/G1 phase and decreased in S phase at 24, 48 hours in A375 (Figure 4A and 4B) and P8 (Figure 4C and 4D). No significantly changes were seen in the G2/M phase (P>0.05) (Supplementary Table 1). These findings suggested that the C-PST inhibited MM cell lines by arresting cell progression at the G0/G1 and S phases. The phospho-ERK1/2, phospho-Rb, Cyclin D1, and Cyclin E expression were significantly down-regulated in the A375 (Figure 5A) and P8 cell lines (Figure 5F) after C-PST treatment for 24 hours. Fold changes of the expression were calculated for ERK, p-ERK, Cyclin D1, Rb, p-Rb, and Cyclin E proteins treated by propranolol, sunitinib, C-PST and control group in (Figure 5B-5E) A375 cells and (Figure 5G, 5H) P8 cells. The expression of p-ERK, Cyclin D1, p-Rb, and Cyclin E proteins were statistically different between single sunitinib and C-PST group (P<0.001), meanwhile; the expression of ERK and Rb showed no significant statistical differences(P>0.05).
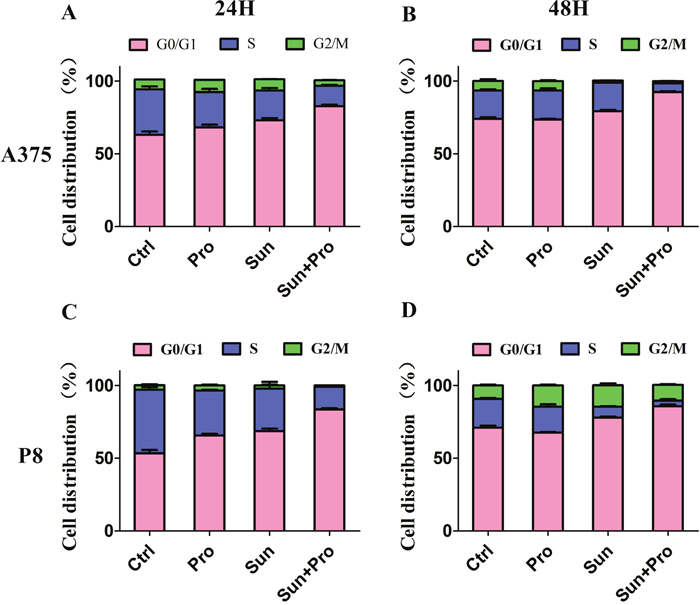
Figure 4: Cell-cycle analysis (G0/G1, S, and G2/M) of A375 and P8 cell lines. (A-D) Flow cytometric analysis of A375 and P8 human MM cells exposed to propranolol 50 μM, sunitinib 2.5 μM, and C-PST after 24 and 48H cultivation to establish cell percentages in G0/G1, S, and G2/M phases of the cell cycle. Data are presented as mean ± SD.
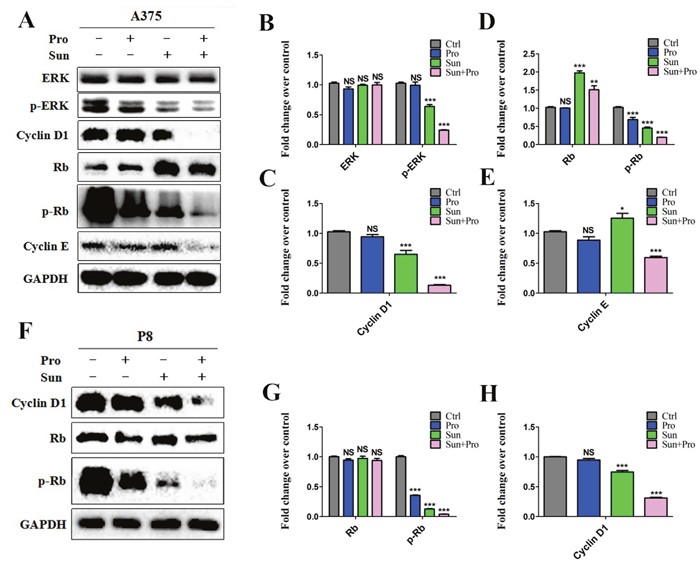
Figure 5: Cell cycle related western blot analysis of C-PST in MM cells. (A)(F) C-PST suppressed ERK1/2 and Rb phosphorlate levels. Cyclin D1 and Cyclin E expression levels were suppressed in both A375 and P8 cell lines. ERK, p-ERK, Cyclin D1, Rb, p-Rb, and Cyclin E protein fold levels changed in propranolol 50 μM, sunitinib 2.5 μM, the C-PST and the control groups in (B-E) A375 cells and (G, H) P8 cells. Results were determined by three independent experiments and normalized by total protein level. Data are presented as mean ± SD. *P<0.05, **P<0.01 and ***P<0.001 vs. Ctrl.
C-PST inhibited in vivo MM growth
A375 MM cell was engrafted into BALB/C nude mice. About 70-80% of mice developed solid tumors in 5-7 days. Mice were divided into five groups including: blank control (citrate buffer and PBS); low-dose sunitinib (40mg/ kg*day, Sun L); Sun L and propranolol (2mg/ kg*day) combination (C-PLST); high-dose sunitinib (80mg/ kg*day, Sun H); and propranolol (2mg/ kg*day). All mice were treated for two weeks. The average tumor size in C-PLST group was smaller than Sun L group (253.78.41±24.57mm3 vs. 477.41±48.11mm3, P<0.0001, Figure 6A, 6C). The tumor size was similar in C-PLST group and Sun H group at 14 days in A375 xenografts (253.78±24.57mm3 vs. 259.34±27.82 mm3, P>0.05). The wight of tumors were also significantly different between Sun L and C-PLST groups (0.346±0.019 g vs. 0.781±0.024 g, P<0.0001, Figure 6D). Meanwhile, the mice in all treatment groups were not lost body weight and got a slight increase (Figure 6E), which suggested the low toxicity of this combination of medicines. Meantime, in A375 xenografts (Figure 7), the Ki67 expression in C-PLST group was significantly lower than Sun L group (50.55±1.25% vs. 67.88±4.53 %, P<0.01), however, no difference from Sun H group (50.55±1.25% vs. 51.69±1.68 %, P>0.05). This result suggested C-PLST could significantly inhibit the proliferation of MM in vivo.
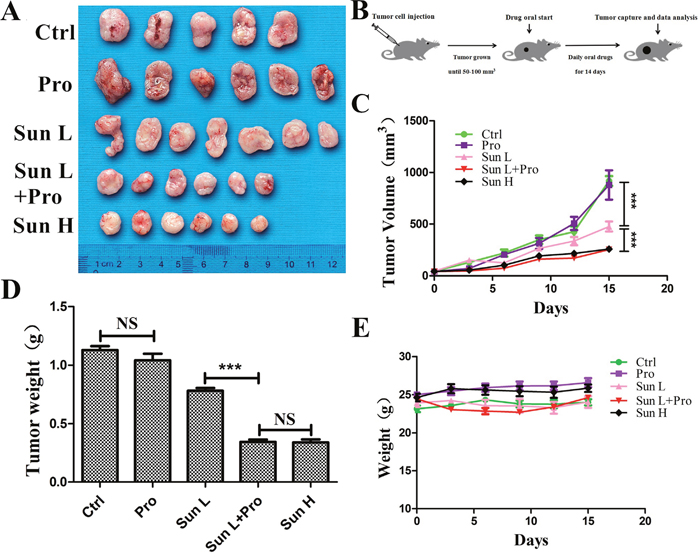
Figure 6: C-PST inhibited in vivo tumor development. (A) A375 untreated mice (PBS and citrate buffer mixed group) xenograft excised tumors and mice treated with propranolol 2mg/k*day: Pro, low-dose sunitinib 40mg/kg*day: Sun L, Sun L+Pro, high-dose sunitinib 80mg/kg*day: Sun H. (B) Schematic plan for the administration of control (PBS and citrate buffer mixed group), Pro, Sun L, Sun L+Pro or Sun H to tumor-bearing mice. (C) Tumor growth curves measured by average volume of 5 to 7 tumors for each group. (D) All the tumors were removed and measured by average weight for each group at day 15. (E) A375 model mice body weights were measured at the beginning of the test and every three days there after. Results are presented as mean ± SD, ***P<0.001.
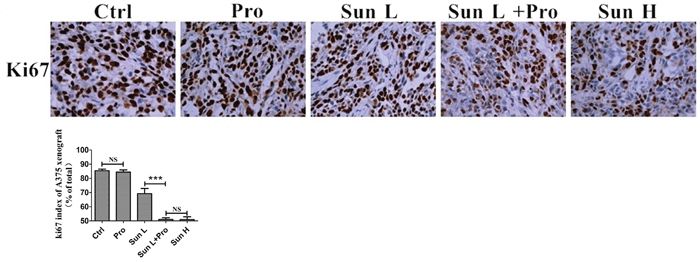
Figure 7: C-PLST inhibited xenograft tumor development. (A) An immunohistochemistry assay assessed Ki67. (B) The Ki67 index. Results are presented as mean ± SD, ***P<0.001.
DISCUSSION
Propranolol was commonly used for hypertension and arrhythmias; moreover, the anti-tumor effect has been widely reported recently [10–22]. Interestingly, propranolol also displayed synergistic effect on other anti-tumor drugs [24–30]. For instance, Wei, et al.[28] investigated the combined administration of propranolol with vemurafenib (a BRAF V600E inhibitor) in thyroid cancer. The two drugs effectively controlled the tumor growth by blocking the β2-AR, and then consequently suppressing the expression of p-Akt, p-mTOR, Bcl-2, cyclin D1, HK2 and GLUT1, which indicated that propranolol could enhance the efficacy of vemurafenib, a member of TKIs. However, whether propranolol could also enhance the efficacy of other TKIs, such as sunitinib (VEGF inhibitor) is still unclear.
In the present study, we found that propranolol enhanced sunitinib efficacy by inducing cell cycle arrest at G0/G1/S phase and inhibiting the MM proliferation in melanoma, rather than inducing tumor cell apoptosis (Supplementary Figure 2). Moreover, in A375 xenografts low-dose propranolol in combination with low-dose sunitinib achieved similar tumor control effect to high-dose sunitinib without obvious side effects. Immunohistochemistry of the tumor sections confirmed the enhanced anti-proliferation effect of propranolol on sunitinib measured by Ki67. Meantime, the expression of apoptosis-related proteins, including cleaved-PARP, bax, and bcl2 was not significantly different among propranolol, sunitinib or C-PST groups measured by WB assay (Supplementary Figure 2). Cell morphological changes were also observed (Supplementary Figure 3) which suggested no apoptosis had occurred by using C-PST. This finding highlighted a potential combination therapy of propranolol and sunitinib in the clinic.
In cancer adjuvant therapy, an increasing number of retrospective clinical studies demonstrated the potential benefit from β-blockers in some patients [19, 23, 31–35], such as prostate cancer, non-small-cell lung cancer, breast cancer, hepatocellular carcinoma, ovarian cancer and melanoma. However, inconsistent effectiveness was always found in using β-blockers, mainly due to the following four reasons [36]: 1. the duration time of using β-blockers, 2. the types of β-blocker and subtype of β-AR, 3. the different clinical end point, 4. different tumor heterogeneity and race diversity. Though it has many controversial reports, these findings suggest a role of propranolol in cancer treatment, especially the breast, melanoma and prostate cancer.
β-ARs belong to G protein-coupled receptors family which respond to Norepinephrine (NE) and epinephrine (E), which were widely studied to explore their role of tumor promoting. Liu et al. [37] verified that E promotes esophageal squamous cell carcinoma proliferation by β-AR induced MEK/ERK/COX-2 pathway. In addition, β1 or β2 selective antagonists could abolish HKESC-1 cell proliferation and down regulate phosphorylated ERK1/2 level along with the expression of COX-2. Liao, et al. [17] identified that propranolol promoted cell apoptosis and induced cell cycle arrest via the inhibition of β-ARs and NF- kB in gastric cancer, but the specific mechanism was not clear. Later, Zhang et al. [38] found Ras/Akt/NF-kB pathway was responsible for G1/S phase arrest induced by β2-AR blocker (ICI-118,551) in pancreatic cancer. Cyclin D1 and cyclin E were key proteins responsible for cell cycle, ICI-118,551 could inhibit the expression of cell cycle proteins, and it also has a better effect on cell proliferation [38]. These data suggested that, in different cancer cell lines, signal pathways of tumor behaviors could be different, which remind us of exploring other possible mechanisms.
Although there were many options for MM treatment, targeted therapy displayed promising efficacy in clinic, especially BRAF V600E inhibitors. However, other members of TKIs failed to achieve a satisfactory effect in MM such as sunitinib, a VEGF inhibitor. Combination therapy containing propranolol with TKIs may improve the efficacy of TKIs mentioned above [28]; however, the underline mechanism is still unclear. Until now, two reports demonstrated that propranolol could completely abolished the chronic stress-promoted tumor growth in mice MM model and colorectal cancer model through a β-AR dependent pathway and might enhance the sunitinib anti-tumor effects via β-adrenoceptor-cAMP-PKA signaling pathway [29, 30]. These findings provided strong evidence and possible mechanisms for our speculation in combination propranolol with TKIs, and suggested a tendency to extend to more drugs.
What’s more, a few reports showed other synergistic effects of propranolol in combination with other drugs, such as vinblastine, methotrexate, paclitaxel, etoposide, cyclophosphamide, and paclitaxel [24–27]. In triple-negative breast cancer models, Rico M, et al. [39] found that a combination of propranolol and metformin provides a new anti-tumor treatment by decreasing proliferation, mitochondrial activity, migration, and invasion. After examining the immunoregulatory function of propranolol, Ashrafi S et al [40] found that propranolol could play as an adjuvant to tumor vaccine to suppress tumor growth by modulating cytokine pattern in tumor microenvironment.
In conclusion, this study has provided a new treatment option of propranolol in combination with sunitinib for malignant melanoma control in addition to its primary anti-tumor function. Combined administration of propranolol with other TKIs or more anti-tumor drugs may also achieve enhanced anti-tumor effect, which worth further investigation.
MATERIALS AND METHODS
Cell lines and reagents
The A375 cell line derived from chronic sun induced damage (CSD) cutaneous MM, was obtained from American type culture collection (ATCC). A375 cell line was cultured in DMEM medium (BI, China) supplemented with 10% FBS(BI, China) at 37°C and 5% CO2 in tissue culture incubator.
Human MM primary cultures
Patient 8 cell line (P8) was derived from surgical resection samples of acral MM from a patient. Written informed consent was obtained from the patient. P8 cell line was cultured in DMEM medium (BI, China) supplemented with 10% FBS (Gibco, Life Technologies Australia) at 37°C and 5% CO2 in tissue culture incubator. MM specific antigens were detected via western blotting for CD31, CD63, CD166 and CD146 confirmed that the cells were melanocytes. Tumor tissues were mechanically dissociated with a small tissue chopper prior to sequential enzymatic digestion in 2 mg/ml Collagenase (Sigma-Aldrich, Schnelldorf, Germany) and 1 mg/ml Dispase I (Gibco/Invitrogen, Carlsbad, CA) in DMEM for 30min at 37°C. Cells were filtered (100 μm cell strainer) to obtain a single cell suspension and re-supplemented by phosphate-buffered saline, thereafter centrifuged at 1000 r.p.m. for 5 minutes. Pellet was re-suspended in DMEM with 10% FBS and then cultured in 75 cm3 plastic flasks (Themo Fisher Scientific, China).
Cell viability assays
The half-maximal inhibitory concentration (IC50) value and survival rate(%) were determined by MTS assay (Promega (Beijing) Biotech Co., Ltd. G111B). A375 and P8 were respectively plated in 96-well plates at a density of 3×103 and 5*103, treated with 1, 5, 10, 15, 20 μM sunitinib (sunitinib malate, S1042, Selleck, USA) and were treated with propranolol (propranolol hydrochloride, P0884, Sigma-Aldrich, U.S.A) for 24, 48 and 72 hours. Cells were incubated with 10% of MTS: PMS (20:1) mixed reagents 2-4 hours. Fluorescence of each plate was measured using a spectrophotometer at emission 490 nm (Spectra MAX Gemini EM, Molecular Devices).
Colony formation assays
For colony formation assays, A375 and P8 cells were seeded in a 6-well plate at a density of 2*103 and 1*103 cells per well and cultured for 7 and 9 days respectively. The dishes were photographed, and the total colony numbers and types of colonies on the plates were evaluated under a microscope. Colonies with > 50 cells were scored. Colony formation efficiency was determined by counting the combination treated group colonies stained with crystal violet solution.
Cell cycle analysis by flow cytometry
A375, P8 cell lines were cultured at a density of 50 percent in 60 mm2 culture dishes (Corning, China) and were treated with sunitinib, propranolol and combination therapy of sunitinb and propranolol after 24 and 48 hours. Cells were then harvested, washed, fixed overnight in 70 % ethanol at 4°C, after washed with PBS, digested with RNase A, stained with propidium iodide (PI). Cells were then processed for flow cytometry analysis by using BD FACSCanto II (Becton, Dickinson and Company, USA). Then, analysis and mapping by flowjo 10.
Western blot analysis
Western blot analysis was performed on cell extracts of A375 and P8 cell lines treated with sunitinib 2.5 μM, propranolol 50 μM and combination therapy of sunitinib and propranolol for 24, 48 and 72 hours. Cells were lysed in RIPA buffer (Beyotime, China) supplemented with phenylmethylsulphonyl fluoride (Sigma), fresh protease and phosphatase inhibitors (Sigma), and protein concentrations were determined by a BCA Protein Assay Kit (Thermo). Western blotting was performed as described previously. The protein boiled for 10 minimums in loading buffer before resolving by SDS-PAGE. After saturation in Tris-buffered saline supplemented with 5% milk or BSA, the membranes were incubated with antibodies overnight at 4°C. The blots were detected by an imaging system (Bio-Rad, USA). Antibodies specific for the following proteins were ERK1/2 (rabbit, 4695), phospho-p42/44-ERK (rabbit, 4370), Rb (mouse, 9309), phospho-ser807/811-Rb (rabbit, 8516), Cyclin D1 (rabbit, 2922), Parp (rabbit, 9532), purchased from Cell Signaling Technology, and Cyclin E (mouse, 4132, Santa Cruz). The antibodies specific for Bax (rabbit, 50599-2-Ig) and Bcl-2 (rabbit, 12789-1-AP), were purchased from Proteintech. The antibody specific for GAPDH (mouse, clone 6C5, MAB374) was purchased from Millipore. Quantification of the bands was done with Image J.
Xenografts of human MM
The five-six-week old NOD/SCID male mice (Hunan Silaike experimental Animals Inc, China) were injected subcutaneously with 1*106 living MM cells that was maintained in DMEM with 0.5% FBS and 15% Matrigel (BD Bioscience, Franklin Lakes, NJ) in the right flank. A solid tumor develops in A375 xenografts within one- two weeks.
In vivo combination therapy
The NOD/SCID mice with xenografts were randomly divided into five groups after the tumor volume reached 50-100 mm3. According to the previous studies [20–22], we choose the dose of propranolol 2mg/ kg*day in these studies, which is the lowest dose mentioned before and a good negative control in vivo. Meanwhile, mice received a daily oral of sunitinib at the dose of 40mg/ kg for two weeks, and combination therapy of sunitinib 40mg/ kg*day and propranolol 2mg/ kg*day. We set sunitinib 80mg/ kg*day group as the positive control, and the blank control was treated with the same volume of citrate buffer (0.1 mol/L, pH=3.5) and PBS. Tumor volume was measured by length (l), width (w), and height (h) twice a week with avernier caliper and tumor size was calculated by the modified ellipsoidal formula (Tumor volume =π/6× l × w × h). The mice were sacrificed at the end of two-week treatment. The xenografts were removed, and then snap-frozen in liquid nitrogen. Paraffin embedded tumor blocks were prepared for further analysis at the same time.
Immunohistochemical analysis
The slide was stained according to the manufacturer’s protocol. Briefly, the slide was baked at 60°C for 2 hours, dewaxed in turpentine and rehydrated in a graded ethanol series, and then treated with 3% hydrogen peroxide for 10 minimums to inhibit endogenous peroxidase. The slide was pretreated in a pressure cooker with Antigen Retrieval Solution(in 0.01 M sodium citrate buffer, pH 6.0) for 3 minimums. Tissue sections were then incubated with Peroxidase Blocking Solution (S2023, Dako) for 15 minimums and Protein Block (X0909, Dako) for 20 minimums. Primary antibody specific for Ki67 (anti-rabbit 1:400, ab16667, Abcam) were applied, and the slides were incubated overnight at 4°C. Signals were visualized rabbit HRP-conjugated secondary antibody (K4003, Dako) and a haematoxylin (MHS32, Sigma) counterstain.
Statistical procedures
Data were expressed as mean ± SD. One-way ANOVA and Dunnett's multiple comparison test were used to determine the statistical differences. A P value of less than 0.05 was considered statistically significant, with the analysis and mapping by Graphpad Prism software (GraphPad Software, Inc., version 5.0).
CONFLICTS OF INTEREST
The authors state no conflicts of interest.
FUNDING
This work was financially supported by Grant No. 81430075, 81403022, 81620108024, 81602399, 81673517 and 81773821 from the National Natural Science Foundation. The Strategy-Oriented Special Project of Central South University in China (ZLXD2017003).
HIGHLIGHTS
1. Sunitinib could inhibit the cells proliferation both in malignant melanoma cell line and acral melanoma cells derived from Chinese patient.
2. Using low-dose propranolol could enhance the anti-tumor effect of sunitinib to inhibit proliferation by inducing G0/G1/S phase arrest in malignant melanoma.
REFERENCES
1. Sutent S. Sunitinib (Sutent R). Quality, 2011; 1-21.
2. Minor DR, Kashani-Sabet M, Garrido M, O'Day SJ, Hamid O, Bastian BC. Sunitinib therapy for melanoma patients with KIT mutations. Clin Cancer Res. 2012; 18:1457-63.
3. Todd JR, Becker TM, Kefford RF, Rizos H. Secondary c-Kit mutations confer acquired resistance to RTK inhibitors in c-Kit mutant melanoma cells. Pigment Cell Melanoma Res. 2013; 26:518-26.
4. Santini D, Vincenzi B, Venditti O, Dell'Aquila E, Frezza AM, Silletta M, Guida FM, Grasso RF, Silvestris N, Lanzetta G, Tonini G. Sunitinib in malignant melanoma: a treatment option only for KIT-mutated patients? Future Oncol. 2013; 9:1809-11.
5. Valsecchi ME, Sato T. The potential role of sunitinib targeting melanomas. Expert Opin Investig Drugs. 2013, 22:1473-83.
6. Buchbinder EI, Sosman JA, Lawrence DP, McDermott DF, Ramaiya NH, Van den Abbeele AD, Linette GP, Giobbie-Hurder A, Hodi FS. Phase 2 study of sunitinib in patients with metastatic mucosal or acral melanoma. Cancer. 2015; 121:4007-15.
7. Decoster L, Vande Broek I, Neyns B, Majois F, Baurain JF, Rottey S, Rorive A, Anckaert E, De Mey J, De Brakeleer S, De Grève J. Biomarker analysis in a phase II study of sunitinib in patients with advanced melanoma. Anticancer Res. 2015, 35:6893-9.
8. Tannir NM, Figlin RA, Gore ME, Michaelson MD, Motzer RJ, Porta C, Rini BI, Hoang C, Lin X, Escudier B. Long-term response to sunitinib treatment in metastatic renal cell carcinoma: a pooled analysis of clinical trials. Clin Genitourin Cancer. 2017.
9. Wang Y, Jin K, Tan H, Zhang P, Yang Q, Wang W, Li J, Shao C, Xue L, Feng S, Chen M, Yu X, Chen J. Sunitinib is effective and tolerable in Chinese patients with advanced pancreatic neuroendocrine tumors: a multicenter retrospective study in China. Cancer Chemother Pharmacol. 2017.
10. Léauté-Labrèze C, Dumas de la Roque E, Hubiche T, Boralevi F, Thambo JB, Taïeb A. Propranolol for severe hemangiomas of infancy. N Engl J Med. 2008; 358:2649-51.
11. Léauté-Labrèze C, Hoeger P, Mazereeuw-Hautier J, Guibaud L, Baselga E, Posiunas G, Phillips RJ, Caceres H, Lopez Gutierrez JC, Ballona R, Friedlander SF, Powell J, Perek D, et al. A randomized, controlled trial of oral propranolol in infantile hemangioma. N Engl J Med. 2015; 372:735-46.
12. Yang EV, Sood AK, Chen M, Li Y, Eubank TD, Marsh CB, Jewell S, Flavahan NA, Morrison C, Yeh PE, Lemeshow S, Glaser R. Norepinephrine up-regulates the expression of vascular endothelial growth factor, matrix metalloproteinase (MMP)-2, and MMP-9 in nasopharyngeal carcinoma tumor cells. Cancer Res. 2006; 66:10357-64.
13. Wang H, Gomez DR, Liao Z. β-Blockers and metastasis in non-small-cell lung cancer. Expert Rev Anticancer Ther. 2013; 13:641-3.
14. Pasquier E, Ciccolini J, Carre M, Giacometti S, Fanciullino R, Pouchy C, Montero MP, Serdjebi C, Kavallaris M, André N. Propranolol potentiates the antiangiogenic effects and anti-tumor effcacy of chemotherapy agents: implication in breast cancer treatment. Oncotarget. 2011; 2:797-809. https://doi.org/10.18632/oncotarget.343.
15. Coelho M, Moz M, Correia G, Teixeira A, Medeiros R, Ribeiro L. Antiproliferative effects of β-blockers on human colorectal cancer cells. Oncol Rep. 2015; 33:2513-20.
16. Landen CN Jr, Lin YG, Armaiz Pena GN, Das PD, Arevalo JM, Kamat AA, Han LY, Jennings NB, Spannuth WA, Thaker PH, Lutgendorf SK, Savary CA, Sanguino AM, et al. Neuroendocrine modulation of signal transducer and activator of transcription-3 in ovarian cancer. Cancer Res. 2007; 67:10389-96.
17. Liao X, Che X, Zhao W, Zhang D, Bi T, Wang G. The β-adrenoceptor antagonist, propranolol, induces human gastric cancer cell apoptosis and cell cycle arrest via inhibiting nuclear factor kB signaling. Oncol Rep. 2010; 24:1669-76.
18. Lin X, Luo K, Lv Z, Huang J. β-adrenoceptor action on pancreatic cancer cell proliferation and tumor growth in mice. Hepatogastroenterology. 2012; 59:584-8.
19. Powe DG, Voss MJ, Zänker KS, Habashy HO, Green AR, Ellis IO, Entschladen F. β-blocker drug therapy reduces secondary cancer formation in breast cancer and improves cancer specific survival. Oncotarget. 2010; 1:628-38. https://doi.org/10.18632/oncotarget.101009.
20. Wolter JK, Wolter NE, Blanch A, Partridge T, Cheng L, Morgenstern DA, Podkowa M, Kaplan DR, Irwin MS. Anti-tumor activity of the β-adrenergic receptor antagonist propranolol in neuroblastoma. Oncotarget. 2014; 5:161-72. https://doi.org/10.18632/oncotarget.1083.
21. Wrobel LJ, Le Gal FA. Inhibition of human melanoma growth by a non-cardioselective β-blocker. J Invest Dermatol. 2015; 135:525-31.
22. Zhou C, Chen X, Zeng W, Peng C, Huang G, Li X, Ouyang Z, Luo Y, Xu X, Xu B, Wang W, He R, Zhang X, et al. Propranolol induced G0/G1/S phase arrest and apoptosis in melanoma cells via AKT/MAPK pathway. Oncotarget. 2016; 7:68314-27. https://doi.org/10.18632/oncotarget.11599.
23. De Giorgi V, Grazzini M, Gandini S, Benemei S, Lotti T, Marchionni N, Geppetti P. Treatment with β-blockers and reduced disease progression in patients with thick melanoma. Arch Intern Med. 2011; 171:779-81.
24. Banavali S, Pasquier E, Andre N. Targeted therapy with propranolol and metronomic chemotherapy combination: sustained complete response of a relapsing metastatic angiosarcoma. Ecancermedicalscience. 2015; 9:499.
25. Chow W, Amaya CN, Rains S, Chow M, Dickerson EB, Bryan BA. Growth attenuation of cutaneous angiosarcoma with propranolol-mediated β-blockade. JAMA Dermatol. 2015; 151:1226-9.
26. Daguzé J, Saint-Jean M, Peuvrel L, Cassagnau E, Quéreux G, Khammari A, Dréno B. Visceral metastatic angiosarcoma treated effectively with oral cyclophosphamide combined with propranolol. JAAD Case Rep. 2016; 2:497-9.
27. Pasquier E, André N, Street J, Chougule A, Rekhi B, Ghosh J, Philip DS, Meurer M, MacKenzie KL, Avallaris M, Banavali SD. Effective management of advanced angiosarcoma by the synergistic combination of propranolol and vinblastine-based metronomic chemotherapy: a bench to bedside study. EBioMedicine. 2016; 6:87-95.
28. Wei WJ, Shen CT, Song HJ, Qiu ZL, Luo QY. Propranolol sensitizes thyroid cancer cells to cytotoxic effect of vemurafenib. Oncol Rep. 2016; 36:1576-84.
29. Deng GH, Liu J, Zhang J, Wang Y, Peng XC, Wei YQ, Jiang Y. Exogenous norepinephrine attenuates the efficacy of sunitinib in a mouse cancer model. J Exp Clin Cancer Res. 2014; 33:21.
30. Liu J, Deng GH, Zhang J, Wang Y, Xia XY, Luo XM, Deng YT, He SS, Mao YY, Peng XC, Wei YQ, Jiang Y. The effect of chronic stress on anti-angiogenesis of sunitinib in colorectal cancer models. Psychoneuroendocrinology. 2015; 52:130-42.
31. Grytli HH, Fagerland MW, Fosså SD, Taskén KA, Håheim LL. Use of β-blockers is associated with prostate cancerspecifc survival in prostate cancer patients on androgen deprivation therapy. Prostate. 2013; 73:250-60.
32. Wang HM, Liao ZX, Komaki R, Welsh JW, O'Reilly MS, Chang JY, Zhuang Y, Levy LB, Lu C, Gomez DR. Improved survival outcomes with the incidental use of beta-blockers among patients with non-small-cell lung cancer treated with definitive radiation therapy. Ann Oncol. 2013; 24:1312-9.
33. Barron TI, Connolly RM, Sharp L, Bennett K, Visvanathan K. Beta blockers and breast cancer mortality: a population based study. J Clin Oncol. 2011. 29:2635-44.
34. Nkontchou G, Aout M, Mahmoudi A, Roulot D, Bourcier V, Grando-Lemaire V, Ganne-Carrie N, Trinchet JC, Vicaut E, Beaugrand M. Effect of longterm propranolol treatment on hepatocellular carcinoma incidence in patients with HCV-associated cirrhosis. Cancer Prev Res. 2012; 5:1007-14.
35. Lemeshow S, Sorensen HT, Phillips G, Yang EV, Antonsen S, Riis AH, Lesinski GB, Jackson R, Glaser R. β-Blockers and survival among Danish patients with malignant melanoma: a population-based cohort study. Cancer Epidemiol Biomarkers Prev. 2011; 20:2273-9.
36. He RH, He YJ, Tang YJ, Zhou HH, McLeod HL, Liu J. The potential anticancer effect of beta-blockers and the genetic variations involved in the interindividual difference. Pharmacogenomics. 2016; 17:74-9.
37. Liu X, Wu WK, Yu L, Sung JJ, Srivastava G, Zhang ST, Cho CH. Epinephrine stimulates esophageal squamous-cell carcinoma cell proliferation via β-adrenoceptor-dependent transactivation of extracellular signal-regulated kinase/cyclooxygenase-2 pathway. J Cell Biochem. 2008; 105:53–60.
38. Zhang D, Ma Q, Wang Z, Zhang M, Guo K, Wang F, Wu E. β2-adrenoceptor blockage induces G1/S phase arrest and apoptosis in pancreatic cancer cells via Ras/Akt/NF-κB pathway. Mol Cancer. 2011; 10:146.
39. Rico M, Baglioni M, Bondarenko M, Laluce NC, Rozados V, André N, Carré M, Scharovsky OG, Menacho Márquez M. Metformin and propranolol combination prevents cancer progression and metastasis in different breast cancer models. Oncotarget. 2017; 8:2874-89. https://doi.org/10.18632/oncotarget.13760.
40. Ashrafi S, Shapouri R, Mahdavi M. Immunological consequences of immunization with tumor lysate vaccine and propranolol as an adjuvant: a study on cytokine profiles in breast tumor microenvironment. Immunol Lett. 2017; 181:63-70.